Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease

 ,
,
Abstract
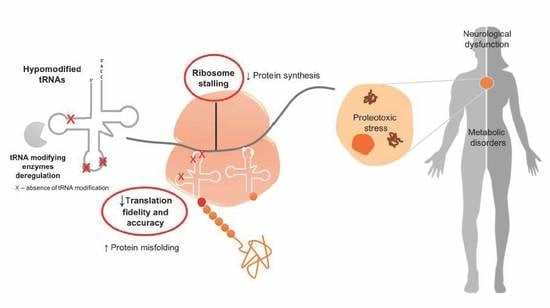
1. Introduction
2. Proteome Disruption in Yeast upon U34 Hypomodification
3. Deregulation of tRNA Modifications in Protein Conformational Diseases
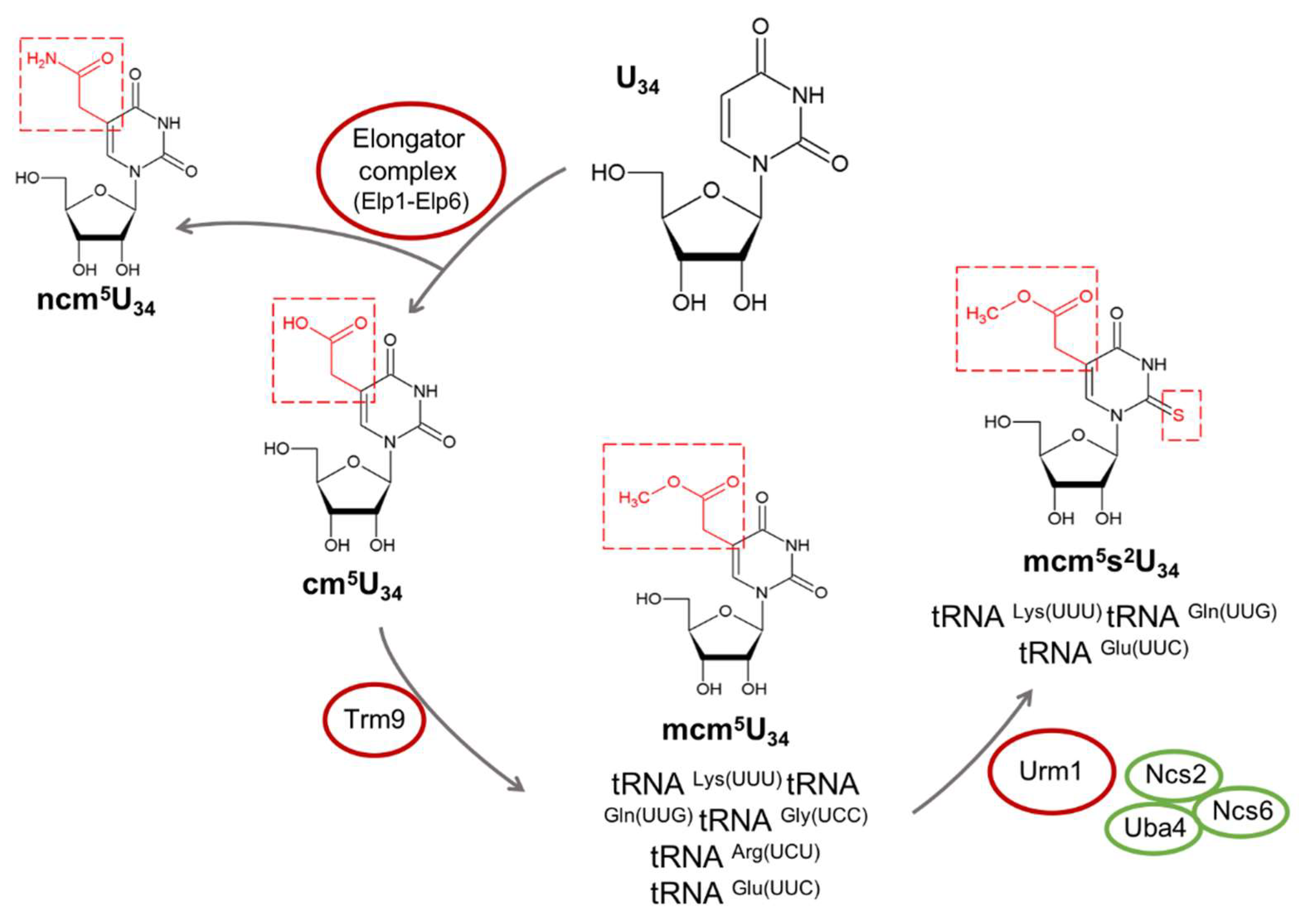
3.1. Role of Elongator Complex, mcm5, ncm5, and mcm5s2 Modifications in Neurological Disorders
3.2. Impact of Other Anticodon Modifications in Neurological and Metabolic Diseases
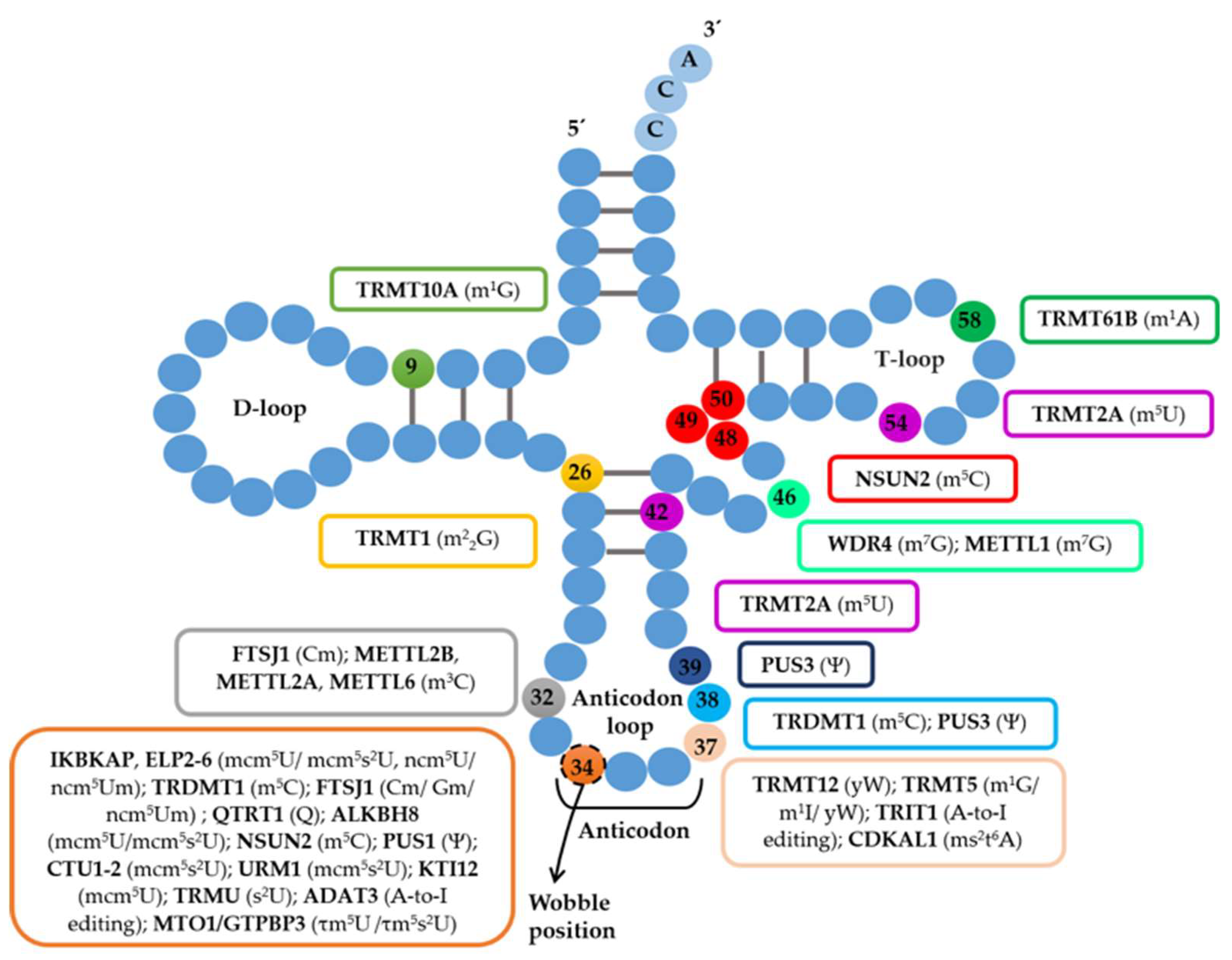
3.3. tRNA-Modifying Enzymes that Catalyze tRNA Modifications outside the Anticodon Are Also Associated with Disease
4. tRNA Hypomodification and Generation of tRNA-Derived Fragments
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Phizicky, E.M.; Hopper, A.K. tRNA biology charges to the front. Genes Dev. 2010, 24, 1832–1860. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.C. Codon—Anticodon pairing: The wobble hypothesis. J. Mol. Biol. 1966, 19, 548–555. [Google Scholar] [CrossRef]
- Phizicky, E.M.; Alfonzo, J.D. Do all modifications benefit all tRNAs? FEBS Lett. 2010, 584, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; MacHnicka, M.A.; Purta, E.; Pitkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Yarham, J.W.; Lamichhane, T.N.; Pyle, A.; Mattijssen, S.; Baruffini, E.; Bruni, F.; Donnini, C.; Vassilev, A.; He, L.; Blakely, E.L.; et al. Defective i6A37 modification of mitochondrial and cytosolic tRNAs results from pathogenic mutations in TRIT1 and its substrate tRNA. PLoS Genet. 2014, 16, e1004424. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Suzuki, T.; Saito, A.; Wei, F.-Y.; Ikeuchi, Y.; Numata, T.; Tanaka, R.; Yamane, Y.; Yamamoto, T.; Goto, T.; Kishita, Y.; et al. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res. 2018, 46, 1565–1583. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, N.; Rodnina, M.V. tRNA wobble modifications and protein homeostasis. Translation 2016, 4, e1143076. [Google Scholar] [CrossRef] [PubMed]
- Tuorto, F.; Lyko, F. Genome recoding by tRNA modifications. Open Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lünse, C.E.; Mörl, M. Trna modifications: Impact on structure and thermal adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Väre, V.Y.P.; Eruysal, E.R.; Narendran, A.; Sarachan, K.L.; Agris, P.F. Chemical and conformational diversity of modified nucleosides affects tRNA structure and function. Biomolecules 2017, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Karlsborn, T.; Tükenmez, H.; Mahmud, A.K.M.F.; Xu, F.; Xu, H.; Byström, A.S. Elongator, a conserved complex required for wobble uridine modifications in eukaryotes. RNA Biol. 2014, 11, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Babu, I.R.; Su, D.; Yin, S.; Begley, T.J.; Dedon, P.C. Trm9-catalyzed tRNA modifications regulate global protein expression by Codon-Biased translation. PLoS Genet. 2015, 11, e1005706. [Google Scholar] [CrossRef] [PubMed]
- Songe-Moller, L.; van den Born, E.; Leihne, V.; Vagbo, C.B.; Kristoffersen, T.; Krokan, H.E.; Kirpekar, F.; Falnes, P.O.; Klungland, A. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell. Biol. 2010, 30, 1814–1827. [Google Scholar] [CrossRef] [PubMed]
- Rezgui, V.A.N.; Tyagi, K.; Ranjan, N.; Konevega, A.L.; Mittelstaet, J.; Rodnina, M.V.; Peter, M.; Pedrioli, A.P.G. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc. Natl. Acad. Sci. USA 2013, 110, 12289–12294. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Ciftci, A.; Funk, J.; Bruch, A.; Butter, F.; Schaffrath, R. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucl Acids Res. 2016, 44, 10946–10959. [Google Scholar] [CrossRef] [PubMed]
- Nedialkova, D.D.; Leidel, S.A. Optimization of Codon Translation Rates via tRNA modifications maintains proteome integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Goffena, J.; Lefcort, F.; Zhang, Y.; Lehrmann, E.; Chaverra, M.; Felig, J.; Walters, J.; Buksch, R.; Becker, K.G.; George, L. Elongator and codon bias regulate protein levels in mammalian peripheral neurons. Nat. Commun. 2018, 9, 889. [Google Scholar] [CrossRef] [PubMed]
- Bento-Abreu, A.; Jager, G.; Swinnen, B.; Rué, L.; Hendrickx, S.; Jones, A.; Staats, K.A.; Taes, I.; Eykens, C.; Nonneman, A.; et al. Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum. Mol. Genet. 2018, 27, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Fakruddin, M.; Wei, F.Y.; Suzuki, T.; Asano, K.; Kaieda, T.; Omori, A.; Izumi, R.; Fujimura, A.; Kaitsuka, T.; Miyata, K.; et al. Defective mitochondrial tRNA taurine modification activates global proteostress and leads to mitochondrial disease. Cell Rep. 2018, 22, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.W.; Jong, C.J.; Ito, T.; Azuma, J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014, 46, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Cang, X.; Peng, Y.; Li, R.; Zhang, Z.; Li, F.; Fan, Q.; Guan, A.S.; Fischel-Ghosian, N.; Zhao, X.; Guan, M.X. Biochemical evidence for a nuclear modifier allele (A10S) in TRMU (Methylaminomethyl-2-thiouridylatemethyltransferase) related to mitochondrial trna modification in the phenotypic manifestation of deafness associated 12S rRNA Mutation. J. Biol. Chem. 2017, 292, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.-X.; Yan, Q.; Li, X.; Bykhovskaya, Y.; Gallo-Teran, J.; Hajek, P.; Umeda, N.; Zhao, H.; Garrido, G.; Mengesha, E.; Suzuki, T.; et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006, 79, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Brzezicha, B.; Schmidt, M.; Makałowska, I.; Jarmołowski, A.; Pieńkowska, J.; Szweykowska-Kulińska, Z. Identification of human tRNA: m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA(CAA)Leu. Nucleic Acids Res. 2006, 34, 6034–6043. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.P.; Shaw, M.; Weiner, C.L.; Hobson, L.; Stark, Z.; Rose, K.; Kalscheuer, V.M.; Gecz, J.; Phizicky, E.M. Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum. Mutat. 2015, 36, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Xing, L.; Gong, P.; Zhang, K.; Gao, X.; Zheng, Z.; Zhou, J.; Guo, Y.; Guo, S.; Zhang, F. Positive association of the FTSJ1 gene polymorphisms with nonsyndromic X-linked mental retardation in young Chinese male subjects. J. Hum. Genet. 2008, 53, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, M.; Pathak, C. Queuosine modification of tRNA: Its divergent role in cellular machinery. Biosci. Rep. 2009, 30, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Zaborske, J.M.; DuMont, V.L.B.; Wallace, E.W.J.; Pan, T.; Aquadro, C.F.; Drummond, D.A. A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biol. 2014, 12, e1002015. [Google Scholar] [CrossRef] [PubMed]
- Rafels-Ybern, À.; Torres, A.G.; Grau-Bove, X.; Ruiz-Trillo, I.; de Pouplana, L.R. Codon adaptation to tRNAs with Inosine modification at position 34 is widespread among Eukaryotes and present in two Bacterial phyla. RNA Biol. 2018, 15, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.G.; Piñeyro, D.; Filonava, L.; Stracker, T.H.; Batlle, E.; de Pouplana, L.R. A-to-I editing on tRNAs: Biochemical, biological and evolutionary implications. FEBS Lett. 2014, 588, 4279–4286. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Miyauchi, K.; Harada, T.; Okita, R.; Takeshita, E.; Komaki, H.; Fujioka, K.; Yagasaki, H.; Goto, Y.I.; Yanaka, K.; et al. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018, 9, 1895. [Google Scholar] [CrossRef] [PubMed]
- Edvardson, S.; Prunetti, L.; Arraf, A.; Haas, D.; Bacusmo, J.M.; Hu, J.F.; Ta-Shma, A.; Dedon, P.C.; de Crécy-Lagard, V.; Elpeleg, O. TRNA N6-adenosine threonylcarbamoyltransferase defect due to KAE1/TCS3 (OSGEP) mutation manifest by neurodegeneration and renal tubulopathy. Eur. J. Hum. Genet. 2017, 25, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.J.; Bruckner, R.J.; Paulo, J.A.; Kazak, L.; Long, J.Z.; Mina, A.I.; Deng, Z.; LeClair, K.B.; Hall, J.A.; Hong, S.; et al. Cdkal1, a type 2 diabetes susceptibility gene, regulates mitochondrial function in adipose tissue. Mol. MeTable 2017, 6, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Arimbasseri, A.G.; Rijal, K.; Iben, J.R.; Wei, F.Y.; Tomizawa, K.; Maraia, R.J. Lack of tRNA-i6A modification causes mitochondrial-like metabolic deficiency in S. pombe by limiting activity of cytosolic tRNATyr, not mito-tRNA. RNA 2016, 22, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Kapur, M.; Ackerman, S.L. MRNA translation gone awry: Translation fidelity and neurological disease. Trends Genet. 2018, 34, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Motorin, Y. Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol. 2017, 14, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Thüring, K.; Schmid, K.; Keller, P.; Helm, M. Analysis of RNA modifications by liquid chromatography–tandem mass spectrometry. Methods 2016, 107, 48–56. [Google Scholar] [CrossRef] [PubMed]
- McGlincy, N.J.; Ingolia, N.T. Transcriptome-wide measurement of translation by ribosome profiling. Methods 2017, 126, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Pereira, P.M.; Varanda, A.S.; Carvalho, J.; Azevedo, M.; Mateus, D.D.; Mendes, N.; Oliveira, P.; Trindade, F.; Pinto, M.T.; et al. Codon misreading tRNAs promote tumor growth in mice. RNA Biol. 2018, 15, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.M.; Kon, Y.; Hauke, A.C.; Ruiz, B.Y.; Fields, S.; Phizicky, E.M. Conditional accumulation of toxic tRNAs to cause amino acid misincorporation. Nucleic Acids Res. 2018, 46, 7831–7843. [Google Scholar] [CrossRef] [PubMed]
- Svejstrup, J.Q. Elongator complex: How many roles does it play? Curr. Opin. Cell Biol. 2007, 19, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Chan, C.T.Y.; Dyavaiah, M.; Rooney, J.P.; Dedon, P.C.; Begley, T.J. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol. 2012, 9, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Begley, U.; Dyavaiah, M.; Patil, A.; Rooney, J.P.; DiRenzo, D.; Young, C.M.; Conklin, D.S.; Zitomer, R.S.; Begley, T.J. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell. 2007, 28, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Vázquez, J.; Vargas-Pérez, I.; Sansó, M.; Buhne, K.; Carmona, M.; Paulo, E.; Hermand, D.; Rodríguez-Gabriel, M.; Ayté, J.; Leidel, S.; et al. Modification of tRNALys UUU by elongator is essential for efficient translation of stress mRNAs. PLoS Genet. 2013, 9, e1003647. [Google Scholar] [CrossRef] [PubMed]
- Klassen, R.; Grunewald, P.; Thüring, K.L.; Eichler, C.; Helm, M.; Schaffrath, R. Loss of anticodon wobble uridine modifications affects tRNALysfunction and protein levels in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0119261. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Dufey, E.; Urra, H.; Hetz, C. ER. proteostasis addiction in cancer biology: Novel concepts. Semin. Cancer Biol. 2015, 33, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Momoi, M.Y.; Tominaga, K.; Momoi, T.; Nihei, K.; Yanagisawa, M.; Kagawa, Y.; Ohta, S. A point mutation in the mitochondrial tRNALeu(UUR) gene in melas (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun. 1990, 173, 816–822. [Google Scholar] [CrossRef]
- Torres, A.G.; Batlle, E.; de Pouplana, L.R. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.L.; Lemmens, R.; Miskiewicz, K.; Broom, W.J.; Hansen, V.K.; van Vught, P.W.J.; Landers, J.E.; Sapp, P.; van den Bosch, L.; Knight, J.; et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 2009, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.S.; Srivastava, S.; Farwell, K.D.; Lu, H.M.; Zeng, W.; Lu, H.; Chao, E.C.; Fatemi, A. ELP2 is a novel gene implicated in neurodevelopmental disabilities. Am. J. Med. Genet. Part A 2015, 167, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Abbasi-Moheb, L.; Mertel, S.; Gonsior, M.; Nouri-Vahid, L.; Kahrizi, K.; Cirak, S.; Wieczorek, D.; Motazacker, M.M.; Esmaeeli-Nieh, S.; Cremer, K.; et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Davarniya, B.; Hu, H.; Kahrizi, K.; Musante, L.; Fattahi, Z.; Hosseini, M.; Maqsoud, F.; Farajollahi, R.; Wienker, T.F.; Ropers, H.H.; et al. The role of a novel TRMT1 gene mutation and rare GRM1 gene defect in intellectual disability in two azeri families. PLoS ONE 2015, 10, e0129631. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Q.; Sun, B.; Xiong, Z.P.; Shu, Y.; Zhou, H.H.; Zhang, W.; Xiong, J.; Li, Q. The dysregulation of tRNAs and tRNA derivatives in cancer. J. Exp. Clin. Cancer Res. 2018, 37, 101. [Google Scholar] [CrossRef] [PubMed]
- Rapino, F.; Delaunay, S.; Zhou, Z.; Chariot, A.; Close, P. tRNA modification: is cancer having a wobble? Trends Cancer 2017, 3, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Rafiq, M.A.; Noor, A.; Hussain, S.; Flores, J.V.; Rupp, V.; Vincent, A.K.; Malli, R.; Ali, G.; Khan, F.S.; et al. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Lee, J.H.; Lee, J.E.; Blanco, S.; Nickerson, E.; Gabrie, S.; Frye, M.; Al-Gazali, L.; Gleeson, J.G. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 2012, 49, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Watt, F.M. The RNA methyltransferase misu (NSun2) mediates myc-induced proliferation and is upregulated in tumors. Curr. Biol. 2006, 16, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhu, G.; Zeng, H.; Xu, Q.; Holzmann, K. High tRNA transferase NSUN2 gene expression is associated with poor prognosis in head and neck squamous carcinoma. Cancer Investig. 2018, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083. [Google Scholar] [CrossRef] [PubMed]
- Pavon-Eternod, M.; Gomes, S.; Rosner, M.R.; Pan, T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 2013, 19, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, S.; Rapino, F.; Tharun, L.; Zhou, Z.; Heukamp, L.; Termathe, M.; Shostak, K.; Klevernic, I.; Florin, A.; Desmecht, H.; et al. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J. Exp. Med. 2016, 213, 2503–2523. [Google Scholar] [CrossRef] [PubMed]
- Lefler, S.; Cohen, M.A.; Kantor, G.; Cheishvili, D.; Even, A.; Birger, A.; Turetsky, T.; Gil, Y.; Even-Ram, S.; Aizenman, E.; et al. Familial dysautonomia (FD) human embryonic stem cell derived PNS neurons reveal that synaptic vesicular and neuronal transport genes are directly or indirectly affected by IKBKAP downregulation. PLoS ONE 2015, 10, e0138807. [Google Scholar] [CrossRef] [PubMed]
- Naumanen, T.; Johansen, L.D.; Coffey, E.T.; Kallunki, T. Loss-of-function of IKAP/ELP1: Could neuronal migration defect underlie familial dysautonomia? Cell Adh. Migr. 2008, 2, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Karlsborn, T.; Tükenmez, H.; Chen, C.; Byström, A.S. Familial dysautonomia (FD) patients have reduced levels of the modified wobble nucleoside mcm5s2U in tRNA. Biochem. Biophys. Res. Commun. 2014, 454, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Kataoka, N.; Miyauchi, K.; Ohe, K.; Iida, K.; Yoshida, S.; Nojima, T.; Okuno, Y.; Onogi, H.; Usui, T.; et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc. Natl. Acad. Sci. USA 2015, 112, 2764–2769. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tuck, S.; Byström, A.S. Defects in tRNA modification associated with neurological and developmental dysfunctions in Caenorhabditis elegans elongator mutants. PLoS Genet. 2009, 5, e1000561. [Google Scholar] [CrossRef] [PubMed]
- Chaverra, M.; George, L.; Mergy, M.; Waller, H.; Kujawa, K.; Murnion, C.; Sharples, E.; Thorne, J.; Podgajny, N.; Grindeland, A.; et al. The familial dysautonomia disease gene IKBKAP is required in the developing and adult mouse central nervous system. Dis. Model. Mech. 2017, 10, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.J.; Shen, L.; Jang, C.W.; Falnes, P.; Zhang, Y. Ikbkap/Elp1 deficiency causes male infertility by disrupting meiotic progression. PLoS Genet. 2013, 9, e1003516. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, M.; Mullen, B.; Wojtas, A.; Heckman, M.G.; Diehl, N.N.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.Y.R.; et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Laguesse, S.; Creppe, C.; Nedialkova, D.D.; Prévot, P.P.; Borgs, L.; Huysseune, S.; Franco, B.; Duysens, G.; Krusy, N.; Lee, G.; et al. A dynamic unfolded protein response contributes to the control of cortical neurogenesis. Dev. Cell 2015, 35, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Taskesen, E.; Mishra, A.; van der Sluis, S.; Ferrari, R.; Veldink, J.H.; van Es, M.A.; Smit, A.B.; Posthuma, D.; Pijnenburg, Y. Susceptible genes and disease mechanisms identified in frontotemporal dementia and frontotemporal dementia with amyotrophic lateral sclerosis by DNA-methylation and GWAS. Sci. Rep. 2017, 7, 8899. [Google Scholar] [CrossRef] [PubMed]
- Strug, L.J.; Clarke, T.; Chiang, T.; Chien, M.; Baskurt, Z.; Li, W.; Dorfman, R.; Bali, B.; Wirrell, E.; Kugler, S.L.; et al. Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4). Eur. J. Hum. Genet. 2009, 17, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Reinthaler, E.M.; Lal, D.; Jurkowski, W.; Feucht, M.; Steinböck, H.; Gruber-Sedlmayr, U.; Ronen, G.M.; Geldner, J.; Haberlandt, E.; Neophytou, B.; et al. Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 2014, 55, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Ramser, J.; Winnepenninckx, B.; Lenski, C.; Errijgers, V.; Platzer, M.; Schwartz, C.E.; Meindl, A.; Kooy, R.F. A splice site mutation in the methyltransferase gene FTSJ1 in Xp11.23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9). J. Med. Genet. 2004, 41, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Dewe, J.M.; Fuller, B.L.; Lentini, J.M.; Kellner, S.M.; Fu, D. TRMT1-catalyzed tRNA modifications are required for redox homeostasis to ensure proper cellular proliferation and oxidative stress survival. Mol. Cell Biol. 2017, 37, e00214-17. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Genin, A.; Lambert, N.; Désir, J.; Pirson, I.; Abdulkarim, B.; Simonis, N.; Drielsma, A.; Marselli, L.; Marchetti, P.; et al. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Gillis, D.; Krishnamohan, A.; Yaacov, B.; Shaag, A.; Jackman, J.E.; Elpeleg, O. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J. Med. Genet. 2014, 51, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Yew, T.W.; McCreight, L.; Colclough, K.; Ellard, S.; Pearson, E.R. tRNA methyltransferase homologue gene TRMT10A mutation in young adult-onset diabetes with intellectual disability, microcephaly and epilepsy. Diabet. Med. 2016, 33, e21–e25. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, M.; Ramsey, K.; Grebe, T.; Schrauwen, I.; Szelinger, S.; Huentelman, M.; Craig, D.; Narayanan, V. Case report: Compound heterozygous nonsense mutations in TRMT10A are associated with microcephaly, delayed development, and periventricular white matter hyperintensities. F1000Research 2015, 4, 912. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Toivonen, S.; Villamil, E.D.; Atta, M.; Ravanat, J.-L.; Demine, S.; Schiavo, A.A.; Pachera, N.; Deglasse, J.-P.; Jonas, J.-C.; Balboa, D.; Otonkoski, T.; Pearson, E.R.; et al. Pancreatic β-cell tRNA hypomethylation and fragmentation link TRMT10A deficiency with diabetes. Nucleic Acids Res. 2018, 46, 10302–10318. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Han, L.; Faqeih, E.; Ewida, N.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 2016, 135, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Hijazi, H.; Al-Dosari, M.S.; Shaheen, R.; Hashem, A.; Aldahmesh, M.A.; Mohamed, J.Y.; Kentab, A.; Salih, M.a.; Awaji, A.; et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J. Med. Genet. 2013, 50, 425–430. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Saleh, M.A.; Hashem, A.; Al-Owain, M.; Al Asmari, A.; Rabei, H.; Abdelraouf, H.; Hashem, M.; Alazami, A.M.; Patel, N.; et al. ADAT3-related intellectual disability: Further delineation of the phenotype. Am. J. Med. Genet. Part A. 2016, 170, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.-Y.; Tomizawa, K. Functional loss of Cdkal1, a novel tRNA modification enzyme, causes the development of type 2 diabetes. Endocr. J. 2011, 58, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Suzuki, T.; Watanabe, S.; Kimura, S.; Kaitsuka, T.; Fujimura, A.; Matsui, H.; Atta, M.; Michiue, H.; Fontecave, M.; et al. Deficit of tRNA Lys modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Investig. 2011, 121, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; McDonald, J.; Cuyugan, L.; Aldrich, J.; Kurdoglu, A.; Adkins, J.; Serrano, G.; Beach, T.G.; Craig, D.W.; Valla, J.; et al. Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 2015, 36, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Richter, U.; Evans, M.E.; Clark, W.C.; Marttinen, P.; Shoubridge, E.A.; Suomalainen, A.; Wredenberg, A.; Wedell, A.; Pan, T.; Battersby, B.J. RNA modification landscape of the human mitochondrial tRNALysregulates protein synthesis. Nat. Commun. 2018, 9, 3966. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Kurowski, A.; Nichols, J.; Watt, F.M.; Benitah, S.A.; Frye, M. The RNA-methyltransferase misu (NSun2) poises epidermal stem cells to differentiate. PLoS Genet. 2011, 7, e1002403. [Google Scholar] [CrossRef] [PubMed]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Franke, B.; Vermeulen, S.H.H.M.; Steegers-Theunissen, R.P.M.; Coenen, M.J.; Schijvenaars, M.M.V.A.P.; Scheffer, H.; den Heijer, M.; Blom, H.J. An association study of 45 folate-related genes in spina bifida: Involvement of Cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birth Defects Res. Part A Clin. Mol. Teratol. 2009, 85, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Sharkia, M.M.R.; Zalan, A.; Jabareen-Masri, A.; Zahalka, H. A new case confirming and expanding the phenotype spectrum of ADAT3-related intellectual disability syndrome. Eur. J. Med. Genet. 2018, S1769-7212, 30574–30583. [Google Scholar] [CrossRef] [PubMed]
- Chaleshtori, M.N.A.R.S.; Miyake, N.; Ahmadvand, M.; Bashti, O.; Matsumoto, N. A novel 8-bp duplication in ADAT3 causes mild intellectual disability. Hum. Gen. Var. 2018, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Boutoual, R.; Meseguer, S.; Villarroya, M.; Martín-Hernández, E.; Errami, M.; Martín, M.A.; Casado, M.; Armengod, M.E. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARγ-UCP2-AMPK axis. Sci. Rep. 2018, 8, 1163. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Lalli, S.; Monsurrò, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; la Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Arragain, S.; Handelman, S.K.; Forouhar, F.; Wei, F.Y.; Tomizawa, K.; Hunt, J.F.; Douki, T.; Fontecave, M.; Mulliez, E.; Atta, M. Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J. Biol. Chem. 2010, 285, 28425–28433. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Benítez, D.; Eggers, C.; Glavic, A. Modulation of the proteostasis machinery to overcome stress caused by diminished levels of t6a-modified tRNAs in drosophila. Biomolecules 2017, 7, 25. [Google Scholar] [CrossRef]
- Chujo, T.; Suzuki, T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 2012, 18, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Santos, M. Discovery and function of transfer RNA-derived fragments and their role in disease. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.V.; Cordero-Espinoza, L.; Oeztuerk-Winder, F.; Andersson-Rolf, A.; Selmi, T.; Blanco, S.; Tailor, J.; Dietmann, S.; Frye, M. Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Rep. 2017, 8, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, N.; Cieśla, M.; Ngoc, P.C.T.; Lang, S.; Arora, S.; Dimitriou, M.; Pimková, K.; Sommarin, M.N.E.; Munita, R.; Lubas, M.; et al. Pseudouridylation of tRNA-derived fragments steers translational control in stem cells. Cell 2018, 173, 1204–1216. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
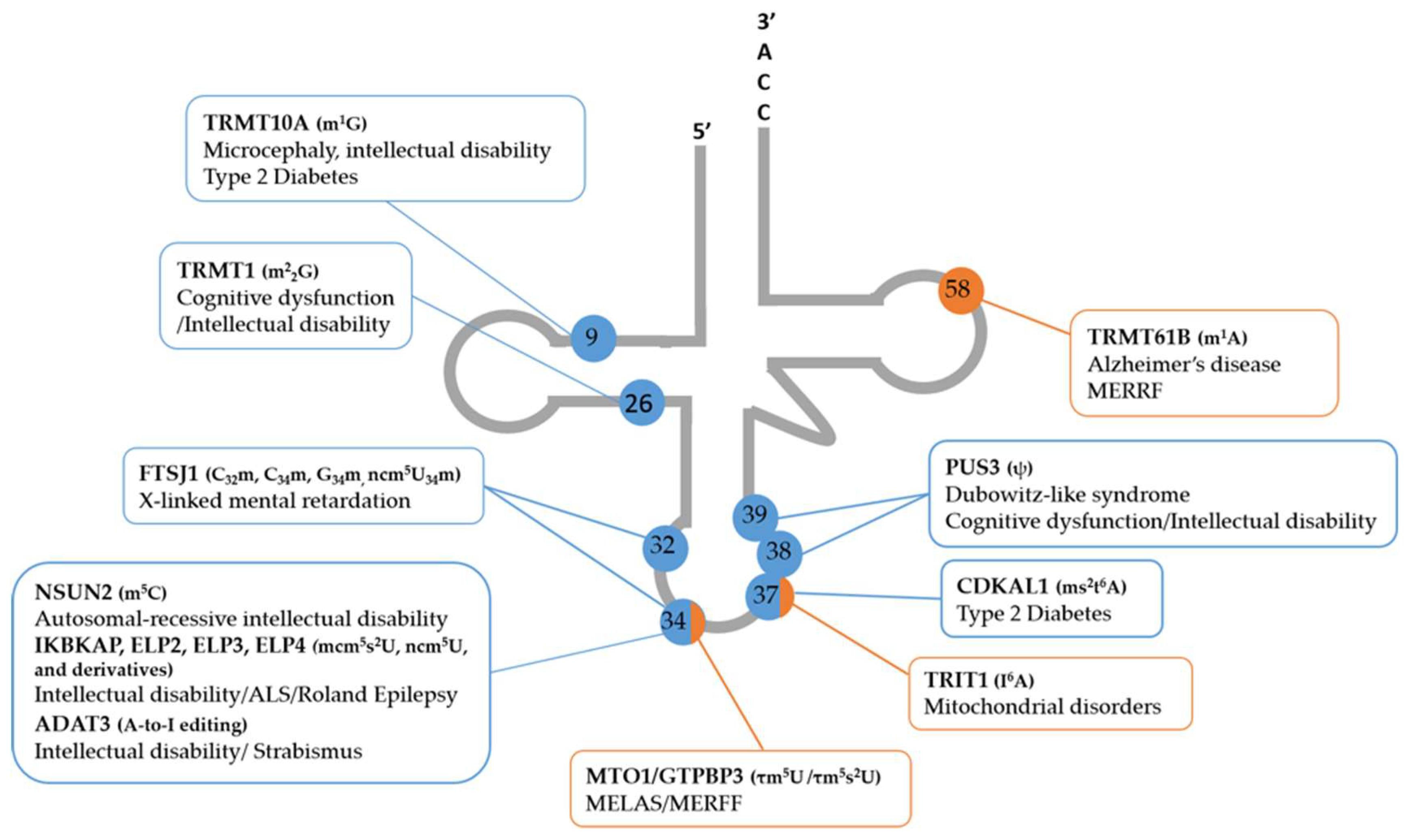{kind=link}
| tRNA ModEnz | tRNA Modifications | Neurological and Metabolic Disorders | tRNAs Deregulated | References |
|---|---|---|---|---|
| IKBKAP (ELP1) | mcm5U34, mcm5s2U34, ncm5U34, ncm5Um34 | Familial dysautonomia (FD) | tRNAGln (UUG) tRNALys (UUU) | [19,66,67,68,69] |
| ELP2 | Neurodevelopmental disabilities | Several | [54,75,76] | |
| ELP3 | Amyotrophic lateral sclerosis (ALS) | tRNAGln (UUG) tRNALys (UUU) tRNAGlu (UUC) | [20,53] | |
| ELP4 | Rolandic epilepsy | Several | [77,78] | |
| FTSJ1 | mC32, mC34, mG34, ncm5mU34 | Nonsyndromic X-linked intellectual disability | tRNALeu tRNAPhe tRNATrp | [27,28,79] |
| TRMT1 | m2,2G26 | Autosomal-recessive intellectual disability; | Several | [56,80] |
| NSUN2 | m5C34, m5C48, m5C49, m5C50 | Autosomal-recessive intellectual disability; Dubowitz-like syndrome | tRNALeu (CAA) tRNAGly (GCC) | [55,59,60] |
| TRMT10A (RG9MTD2) | m1G9 | Microcephaly, epilepsy, intellectual disability, type 2 diabetes | Several | [81,82,83,84,85] |
| PUS3 | ψU38, ψU39 | Autosomal-recessive intellectual disability | tRNAPhe | [86] |
| ADAT3 | A-to-I editing | Intellectual disability | tRNAAla, Pro, Thr tRNAVal, Ser, Arg tRNALeu, Ile | [87,88] |
| CDKAL1 | ms2t6A37 | Type 2 diabetes | tRNALys (UUU) | [35,89,90] |
| TRMT61B | m1A58 | Alzheimer´s disease MERRF | mt-tRNALeu (UUR) mt-tRNALys (UCN) mt-tRNASer | [91,92] |
| MTO1 | τm5U34 | MELAS | mt-tRNALeu(UUR) | [8,21,22] |
| GTPBP3 | τm5s2U34 | MERRF | Mt-tRNALys | [8,22] |
| TRIT1 | I6A37 | Mitochondrial disorders | Mt-tRNASer(UCN) | [7] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, M.; Francisco, S.; Varanda, A.S.; Santos, M.; Santos, M.A.S.; Soares, A.R. Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease. Int. J. Mol. Sci. 2018, 19, 3738. https://doi.org/10.3390/ijms19123738
Pereira M, Francisco S, Varanda AS, Santos M, Santos MAS, Soares AR. Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease. International Journal of Molecular Sciences. 2018; 19(12):3738. https://doi.org/10.3390/ijms19123738
Chicago/Turabian StylePereira, Marisa, Stephany Francisco, Ana Sofia Varanda, Mafalda Santos, Manuel A. S. Santos, and Ana Raquel Soares. 2018. "Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease" International Journal of Molecular Sciences 19, no. 12: 3738. https://doi.org/10.3390/ijms19123738
APA StylePereira, M., Francisco, S., Varanda, A. S., Santos, M., Santos, M. A. S., & Soares, A. R. (2018). Impact of tRNA Modifications and tRNA-Modifying Enzymes on Proteostasis and Human Disease. International Journal of Molecular Sciences, 19(12), 3738. https://doi.org/10.3390/ijms19123738