Possible Involvement of 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
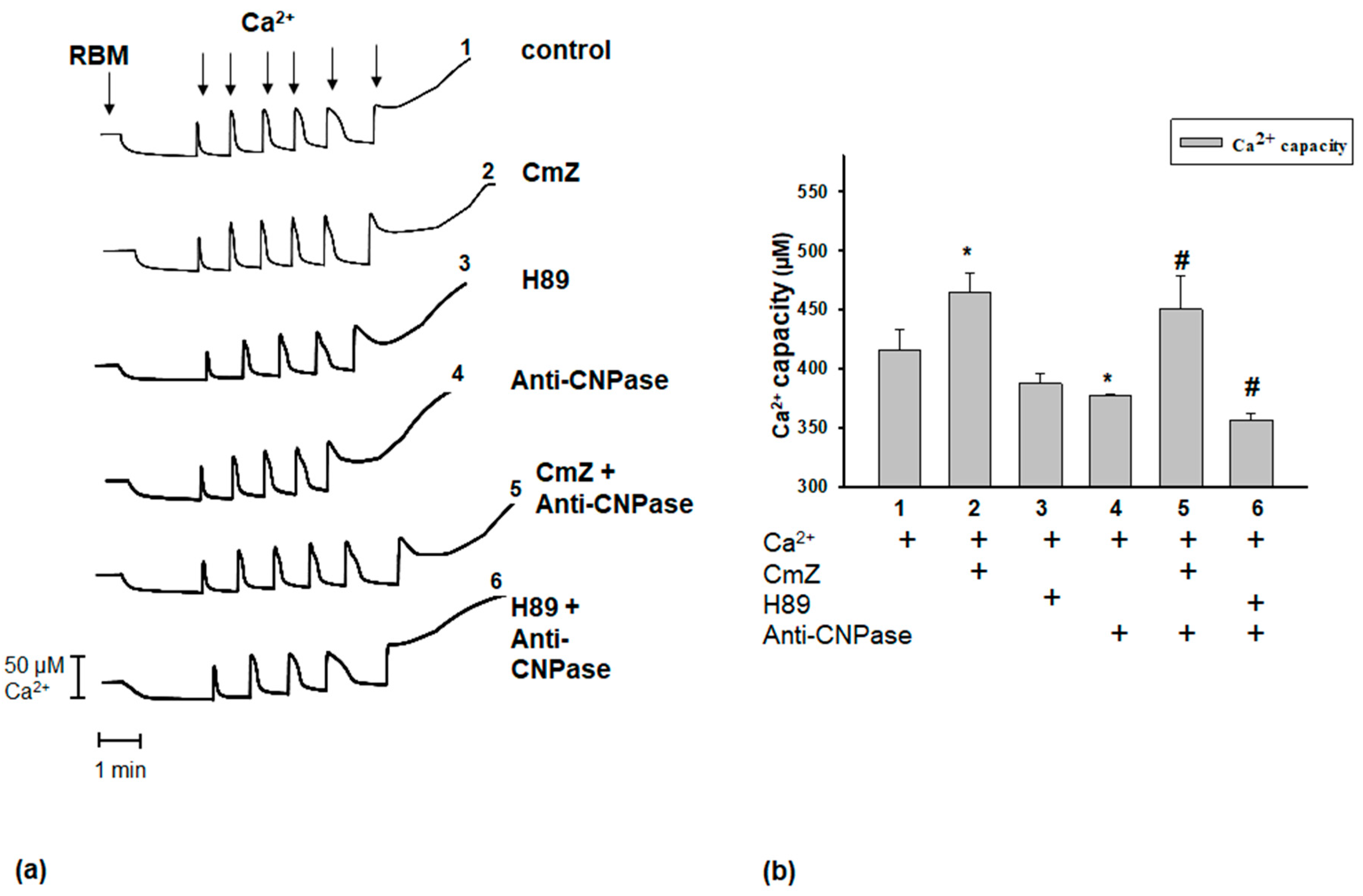
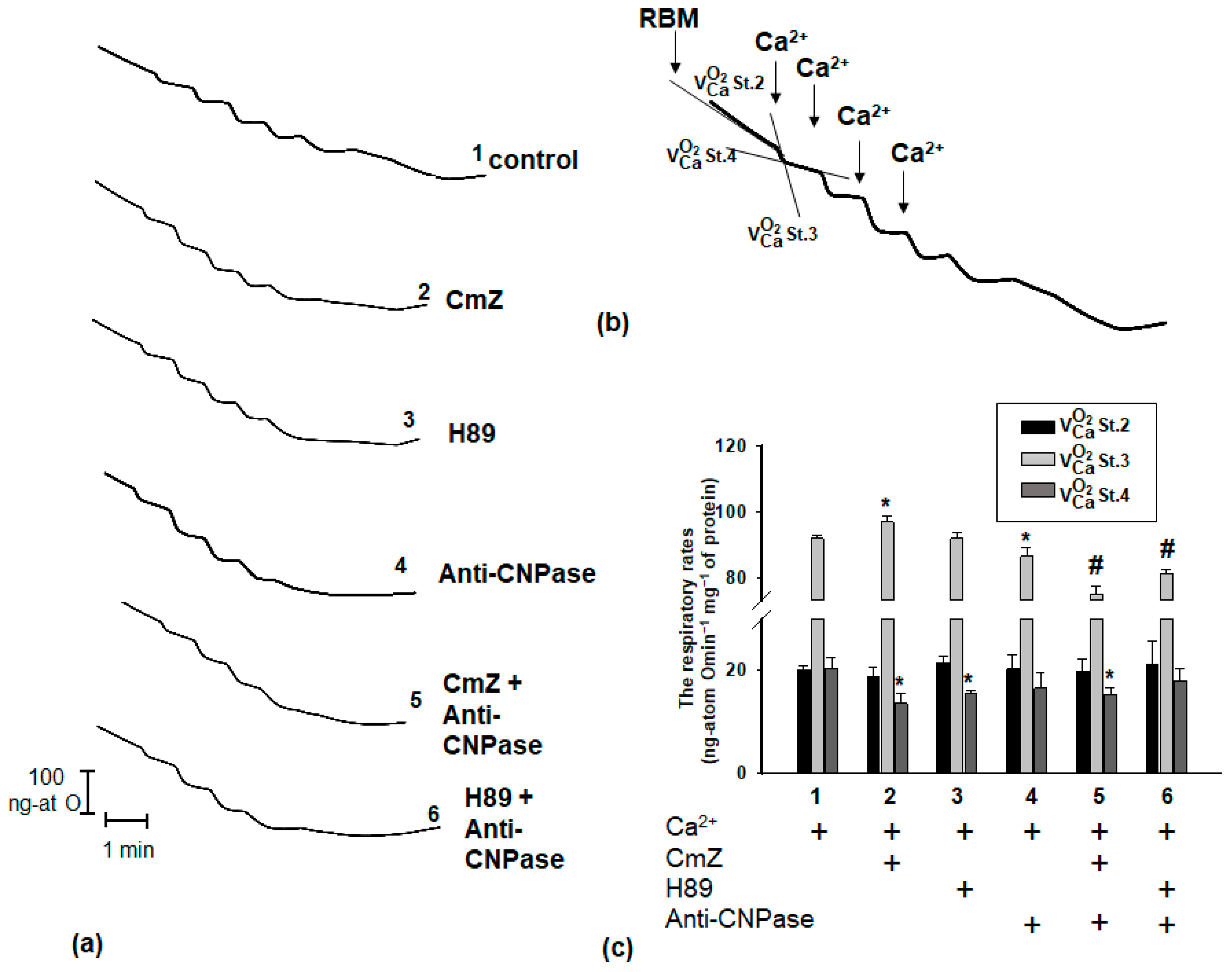
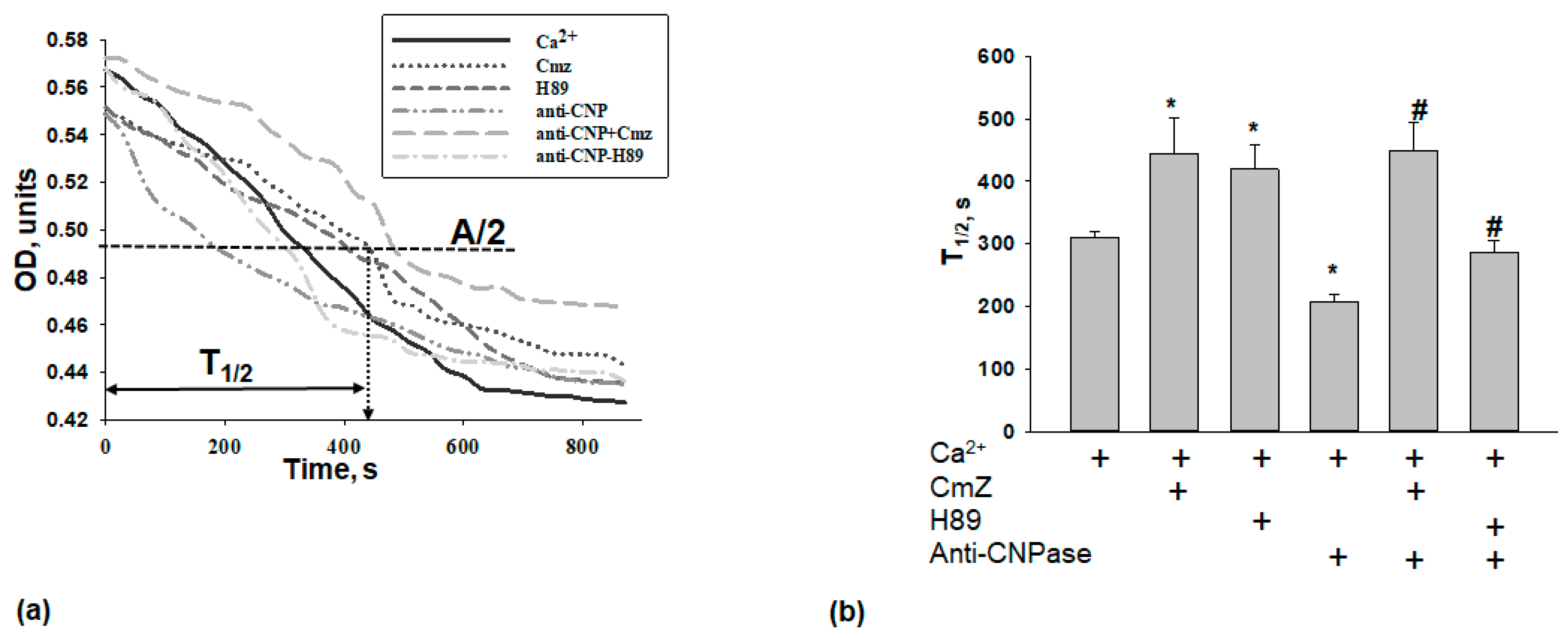
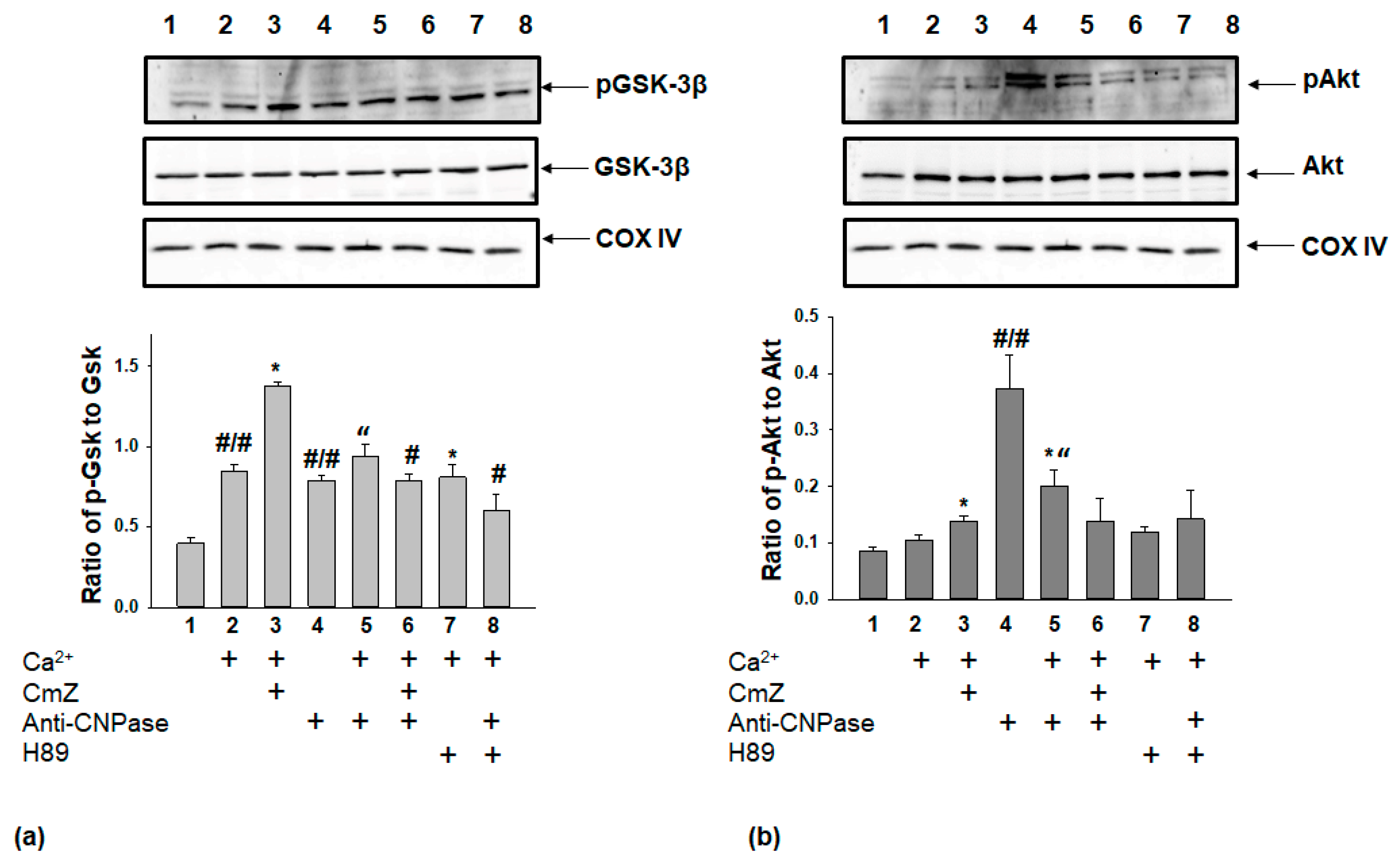
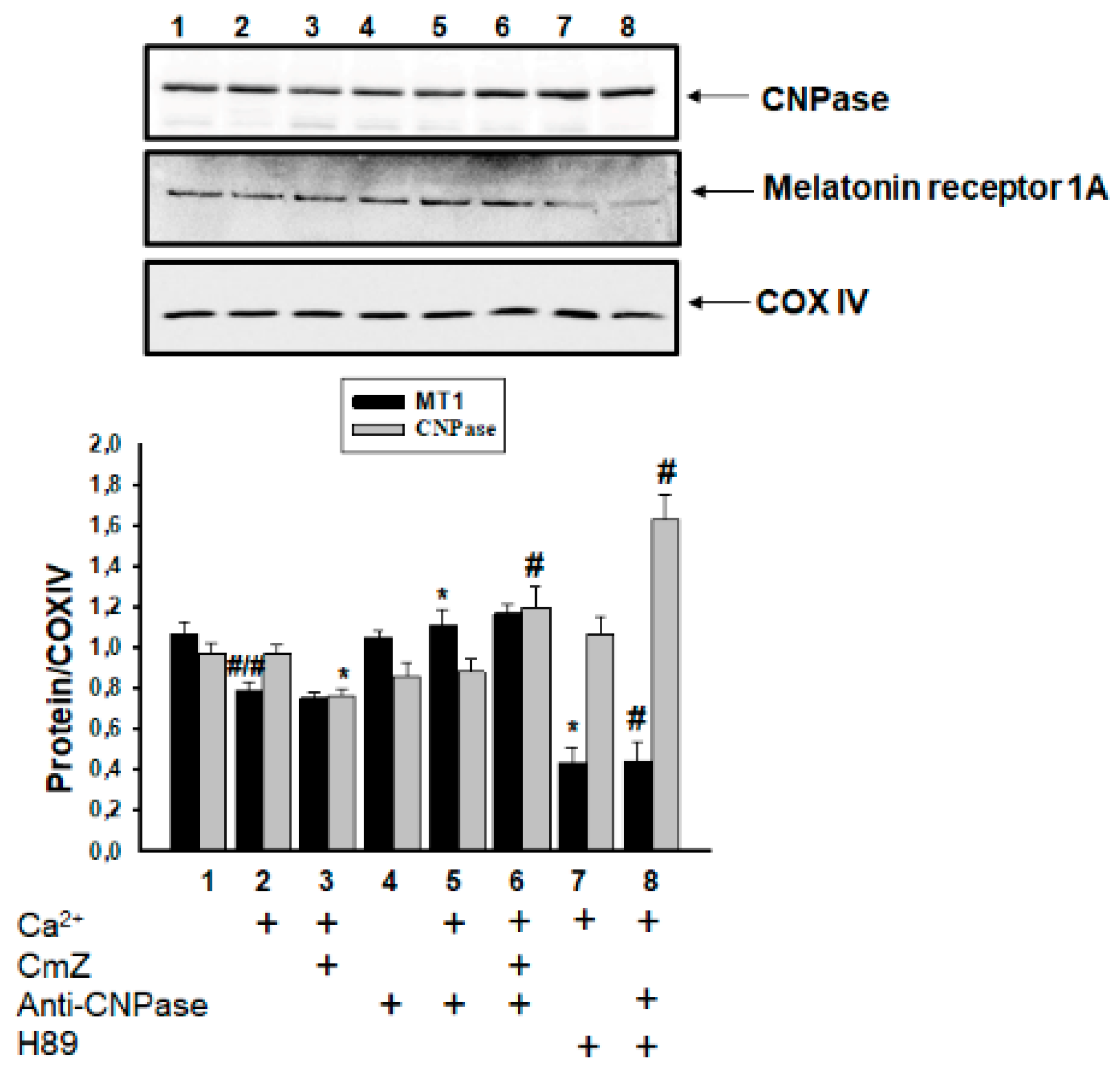
2. Results
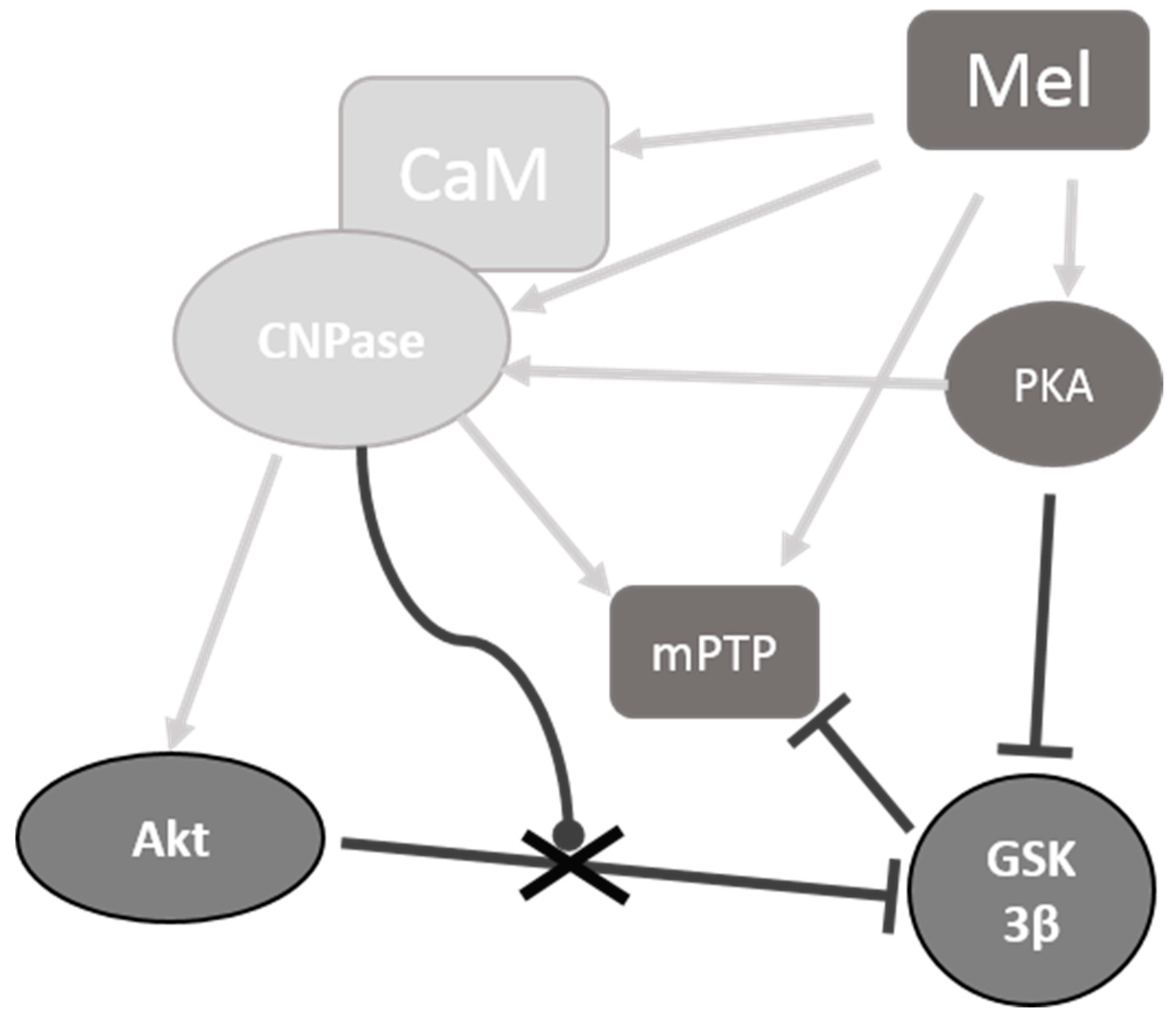
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Rat Brain Mitochondria
4.3. Evaluation of Mitochondrial Functions
4.4. Sample Preparation
4.5. Electrophoresis and Immunoblotting of Mitochondrial Proteins
4.6. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Lim, S.; Smith, K.R.; Lim, S.T.; Tian, R.; Lu, J.; Tan, M. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell Biosci. 2016, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Sato, M.; Umezawa, Y. Fluorescent indicators for Akt/protein kinase B and dynamics of Akt activity visualized in living cells. J. Biol. Chem. 2003, 278, 30945–30951. [Google Scholar] [CrossRef] [PubMed]
- Majewski, M.; Nieborowska-Skorska, M.; Salomoni, P.; Slupianek, A.; Reiss, K.; Trotta, R.; Calabretta, B.; Skorski, T. Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 1999, 59, 2815–2819. [Google Scholar] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Yang, J.Y.; Deng, W.; Chen, Y.; Fan, W.; Baldwin, K.M.; Jope, R.S.; Wallace, D.C.; Wang, P.H. Impaired translocation and activation of mitochondrial Akt1 mitigated mitochondrial oxidative phosphorylation Complex V activity in diabetic myocardium. J. Mol. Cell Cardiol. 2013, 59, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Sato, M.; Kondo, S.; Takashima, A.; Noguchi, K.; Takahashi, M.; Ishiguro, K.; Imahori, K. Different localization of tau protein kinase I/glycogen synthase kinase-3 beta from glycogen synthase kinase-3 alpha in cerebellum mitochondria. J. Biochem. 1995, 118, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Bijur, G.N.; Jope, R.S. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport 2003, 14, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Miura, T.; Miki, T.; Sakamoto, J.; Tanno, M.; Kobayashi, H.; Ikeda, Y.; Ohori, K.; Takahashi, A.; Shimamoto, K. Erythropoietin affords additional cardioprotection to preconditioned hearts by enhanced phosphorylation of glycogen synthase kinase-3 beta. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H748–H755. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wang, H.; Mueller, R.A.; Norfleet, E.A.; Xu, Z. Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3beta and mitochondrial permeability transition pore. Eur. J. Pharmacol. 2009, 604, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabo, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Lim, S.Y.; Ong, S.G.; Davidson, S.M.; Yellon, D.M. Mitochondrial cyclophilin-D as a critical mediator of ischaemic preconditioning. Cardiovasc. Res. 2010, 88, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.; Lesnefsky, E.J.; Stowe, D.F. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 2010, 13, 279–347. [Google Scholar] [CrossRef] [PubMed]
- Swulius, M.T.; Waxham, M.N. Ca(2+)/calmodulin-dependent protein kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Neugebauer, U.; Burck, J.; Myllykoski, M.; Baumgartel, P.; Popp, J.; Kursula, P. Charge isomers of myelin basic protein: Structure and interactions with membranes, nucleotide analogues, and calmodulin. PLoS ONE 2011, 6, e19915. [Google Scholar] [CrossRef] [PubMed]
- Bamm, V.V.; De Avila, M.; Smith, G.S.; Ahmed, M.A.; Harauz, G. Structured functional domains of myelin basic protein: Cross talk between actin polymerization and Ca(2+)-dependent calmodulin interaction. Biophys. J. 2011, 101, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Majava, V.; Wang, C.; Myllykoski, M.; Kangas, S.M.; Kang, S.U.; Hayashi, N.; Baumgartel, P.; Heape, A.M.; Lubec, G.; Kursula, P. Structural analysis of the complex between calmodulin and full-length myelin basic protein, an intrinsically disordered molecule. Amino Acids 2010, 39, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Izadi, M.; Hou, W.; Qualmann, B.; Kessels, M.M. Direct effects of Ca(2+)/calmodulin on actin filament formation. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Krestinina, O.; Galvita, A.; Grachev, D.; Baburina, Y.; Stricker, R.; Evtodienko, Y.; Reiser, G. Ca2+-dependent permeability transition regulation in rat brain mitochondria by 2′,3′-cyclic nucleotides and 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Am. J. Physiol. Cell Physiol. 2009, 296, C1428–C1439. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; O’Neill, R.C.; Park, M.W.; Gravel, M.; Braun, P.E. Mitochondrial localization of CNP2 is regulated by phosphorylation of the N-terminal targeting signal by PKC: Implications of a mitochondrial function for CNP2 in glial and non-glial cells. Mol. Cell. Neurosci. 2006, 31, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Raasakka, A.; Myllykoski, M.; Laulumaa, S.; Lehtimaki, M.; Hartlein, M.; Moulin, M.; Kursula, I.; Kursula, P. Determinants of ligand binding and catalytic activity in the myelin enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Sci. Rep. 2015, 5, 16520. [Google Scholar] [CrossRef] [PubMed]
- Baburina, Y.L.; Gordeeva, A.E.; Moshkov, D.A.; Krestinina, O.V.; Azarashvili, A.A.; Odinokova, I.V.; Azarashvili, T.S. Interaction of myelin basic protein and 2′,3′-cyclic nucleotide phosphodiesterase with mitochondria. Biochemistry 2014, 79, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Myllykoski, M.; Seidel, L.; Muruganandam, G.; Raasakka, A.; Torda, A.E.; Kursula, P. Structural and functional evolution of 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Brain Res. 2015, 1641, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Krestinina, O.V.; Odinokova, I.; Baburina, Y.L.; Azarashvili, T.S. Detection of Protein Kinase A and C Target Proteins in Rat Brain Mitochondria. Biol. Membrany 2017, 34, 42–47. [Google Scholar] [CrossRef]
- Hardeland, R.; Poeggeler, B. Non-vertebrate melatonin. J. Pineal Res. 2003, 34, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Coto-Montes, A.; Tomas-Zapico, C.; Escames, G.; Leon, J.; Rodriguez-Colunga, M.J.; Tolivia, D.; Acuna-Castroviejo, D. Specific binding of melatonin to purified cell nuclei from mammary gland of swiss mice: Day-night variations and effect of continuous light. J. Pineal Res. 2003, 34, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Drew, J.E.; Barrett, P.; Mercer, J.G.; Moar, K.M.; Canet, E.; Delagrange, P.; Morgan, P.J. Localization of the melatonin-related receptor in the rodent brain and peripheral tissues. J. Neuroendocrinol. 2001, 13, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Pozo, D.; Garcia-Maurino, S.; Guerrero, J.M.; Calvo, J.R. mRNA expression of nuclear receptor RZR/RORalpha, melatonin membrane receptor MT, and hydroxindole-O-methyltransferase in different populations of human immune cells. J. Pineal Res. 2004, 37, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef] [PubMed]
- Lochner, A.; Marais, E.; Huisamen, B. Melatonin and cardioprotection against ischaemia/reperfusion injury: What’s new? A review. J. Pineal Res. 2018, 65, e12490. [Google Scholar] [CrossRef] [PubMed]
- Baburina, Y.; Odinokova, I.; Azarashvili, T.; Akatov, V.; Lemasters, J.J.; Krestinina, O. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase as a messenger of protection of the mitochondrial function during melatonin treatment in aging. Bba-Biomembranes 2017, 1859, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Myllykoski, M.; Itoh, K.; Kangas, S.M.; Heape, A.M.; Kang, S.U.; Lubec, G.; Kursula, I.; Kursula, P. The N-terminal domain of the myelin enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase: Direct molecular interaction with the calcium sensor calmodulin. J. Neurochem. 2012, 123, 515–524. [Google Scholar] [CrossRef] [PubMed]
- De Faria Poloni, J.; Feltes, B.C.; Bonatto, D. Melatonin as a central molecule connecting neural development and calcium signaling. Funct. Integr. Genom. 2011, 11, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Reiser, G.; Kunzelmann, U.; Steinhilber, G.; Binmoller, F.J. Generation of a monoclonal antibody against the myelin protein CNP (2′,3′-cyclic nucleotide 3′-phosphodiesterase) suitable for biochemical and for immunohistochemical investigations of CNP. Neurochem. Res. 1994, 19, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Krestinina, O.; Azarashvili, T.; Baburina, Y.; Galvita, A.; Grachev, D.; Stricker, R.; Reiser, G. In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 2015, 80, 41–50. [Google Scholar] [CrossRef] [PubMed]
- De Monasterio-Schrader, P.; Jahn, O.; Tenzer, S.; Wichert, S.P.; Patzig, J.; Werner, H.B. Systematic approaches to central nervous system myelin. Cell. Mol. Life Sci. 2012, 69, 2879–2894. [Google Scholar] [CrossRef] [PubMed]
- Lappe-Siefke, C.; Goebbels, S.; Gravel, M.; Nicksch, E.; Lee, J.; Braun, P.E.; Griffiths, I.R.; Nave, K.A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 2003, 33, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Majava, V.; Greffier, A.; Hayes, R.L.; Kursula, P.; Wang, K.K. Collapsin response mediator protein-2 is a calmodulin-binding protein. Cell. Mol. Life Sci. 2009, 66, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ottens, A.K.; Sadasivan, S.; Kobeissy, F.H.; Fang, T.; Hayes, R.L.; Wang, K.K. Calpain-mediated collapsin response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. J. Neurotrauma 2007, 24, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Berggard, T.; Arrigoni, G.; Olsson, O.; Fex, M.; Linse, S.; James, P. 140 mouse brain proteins identified by Ca2+-calmodulin affinity chromatography and tandem mass spectrometry. J. Proteome Res. 2006, 5, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Schulman, H.; Greengard, P. Stimulation of brain membrane protein phosphorylation by calcium and an endogenous heat-stable protein. Nature 1978, 271, 478–479. [Google Scholar] [CrossRef] [PubMed]
- Hatase, O.; Doi, A.; Itano, T.; Matsui, H.; Ohmura, Y. A direct evidence of the localization of mitochondrial calmodulin. Biochem. Biophys. Res. Commun. 1985, 132, 63–66. [Google Scholar] [CrossRef]
- Lee, J.; Gravel, M.; Zhang, R.; Thibault, P.; Braun, P.E. Process outgrowth in oligodendrocytes is mediated by CNP, a novel microtubule assembly myelin protein. J. Cell Biol. 2005, 170, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Jan, C.R.; Tseng, C.J. Calmidazolium-induced rises in cytosolic calcium concentrations in Madin Darby canine kidney cells. Toxicol. Appl. Pharmacol. 2000, 162, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Krestinina, O.; Odinokova, I.; Evtodienko, Y.; Reiser, G. Physiological Ca2+ level and Ca2+-induced Permeability Transition Pore control protein phosphorylation in rat brain mitochondria. Cell Calcium 2003, 34, 253–259. [Google Scholar] [CrossRef]
- Nishihara, M.; Miura, T.; Miki, T.; Tanno, M.; Yano, T.; Naitoh, K.; Ohori, K.; Hotta, H.; Terashima, Y.; Shimamoto, K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J. Mol. Cell. Cardiol. 2007, 43, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Mo, Y.B.; Bu, X.; Wang, J.J.; Bai, J.; Zhang, J.W.; Cheng, A.B.; Ma, J.H.; Wang, Y.W.; Xie, Y.X. Erinacine Facilitates the Opening of the Mitochondrial Permeability Transition Pore Through the Inhibition of the PI3K/Akt/GSK-3beta Signaling Pathway in Human Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2018, 50, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3beta. Diabetes 2007, 56, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Baburina, Y.; Azarashvili, T.; Grachev, D.; Krestinina, O.; Galvita, A.; Stricker, R.; Reiser, G. Mitochondrial 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) interacts with mPTP modulators and functional complexes (I-V) coupled with release of apoptotic factors. Neurochem. Int. 2015, 90, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Hasemann, C.A.; Istvan, E.S.; Uyeda, K.; Deisenhofer, J. The crystal structure of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase reveals distinct domain homologies. Structure 1996, 4, 1017–1029. [Google Scholar] [CrossRef]
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Von Gall, C.; Stehle, J.H.; Weaver, D.R. Mammalian melatonin receptors: Molecular biology and signal transduction. Cell Tissue Res. 2002, 309, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Sprinkle, T.J. 2′,3′-cyclic nucleotide 3′-phosphodiesterase, an oligodendrocyte-Schwann cell and myelin-associated enzyme of the nervous system. Crit. Rev. Neurobiol. 1989, 4, 235–301. [Google Scholar] [PubMed]
- Verrier, J.D.; Exo, J.L.; Jackson, T.C.; Ren, J.; Gillespie, D.G.; Dubey, R.K.; Kochanek, P.M.; Jackson, E.K. Expression of the 2′,3′-cAMP-adenosine pathway in astrocytes and microglia. J. Neurochem. 2011, 118, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.K.; Gillespie, D.G.; Mi, Z.; Cheng, D.; Bansal, R.; Janesko-Feldman, K.; Kochanek, P.M. Role of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the renal 2′,3′-cAMP-adenosine pathway. Am. J. Physiol. Ren. Physiol. 2014, 307, F14–F24. [Google Scholar] [CrossRef] [PubMed]
- Raasakka, A.; Kursula, P. The myelin membrane-associated enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase: On a highway to structure and function. Neurosci. Bull. 2014, 30, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Gravel, M.; Gao, E.; Hervouet-Zeiber, C.; Parsons, V.; Braun, P.E. Transcriptional regulation of 2′,3′-cyclic nucleotide 3′-phosphodiesterase gene expression by cyclic AMP in C6 cells. J. Neurochem. 2000, 75, 1940–1950. [Google Scholar] [CrossRef] [PubMed]
- Krestinina, O.V.; Kruglov, A.G.; Grachev, D.E.; Baburina, Y.L.; Evtodienko, Y.V.; Moshkov, D.A.; Santalova, I.M.; Azarashvili, T.S. Age-Related Changes of Mitochondrial Functions under the Conditions of Ca2+-Induced Opening of Permeability Transition Pore. Biol. Membrany 2010, 27, 177–183. [Google Scholar]
- Azarashvili, T.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Yurkov, I.; Papadopoulos, V.; Reiser, G. The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeability transition pore opening in rat brain mitochondria. Cell Calcium 2007, 42, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Stricker, R.; Lottspeich, F.; Reiser, G. The myelin protein CNP (2′,3′-cyclic nucleotide 3′-phosphodiesterase): Immunoaffinity purification of CNP from pig and rat brain using a monoclonal antibody and phosphorylation of CNP by cyclic nucleotide-dependent protein kinases. Biol. Chem. Hoppe Seyler 1994, 375, 205–209. [Google Scholar] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baburina, Y.; Odinokova, I.; Azarashvili, T.; Akatov, V.; Sotnikova, L.; Krestinina, O. Possible Involvement of 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore. Int. J. Mol. Sci. 2018, 19, 3499. https://doi.org/10.3390/ijms19113499
Baburina Y, Odinokova I, Azarashvili T, Akatov V, Sotnikova L, Krestinina O. Possible Involvement of 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore. International Journal of Molecular Sciences. 2018; 19(11):3499. https://doi.org/10.3390/ijms19113499
Chicago/Turabian StyleBaburina, Yulia, Irina Odinokova, Tamara Azarashvili, Vladimir Akatov, Linda Sotnikova, and Olga Krestinina. 2018. "Possible Involvement of 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore" International Journal of Molecular Sciences 19, no. 11: 3499. https://doi.org/10.3390/ijms19113499
APA StyleBaburina, Y., Odinokova, I., Azarashvili, T., Akatov, V., Sotnikova, L., & Krestinina, O. (2018). Possible Involvement of 2′,3′-Cyclic Nucleotide-3′-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore. International Journal of Molecular Sciences, 19(11), 3499. https://doi.org/10.3390/ijms19113499