Transcriptomic Analysis of Betula halophila in Response to Salt Stress




Abstract
1. Introduction
2. Results
2.1. Transcriptome Sequencing and Assembly
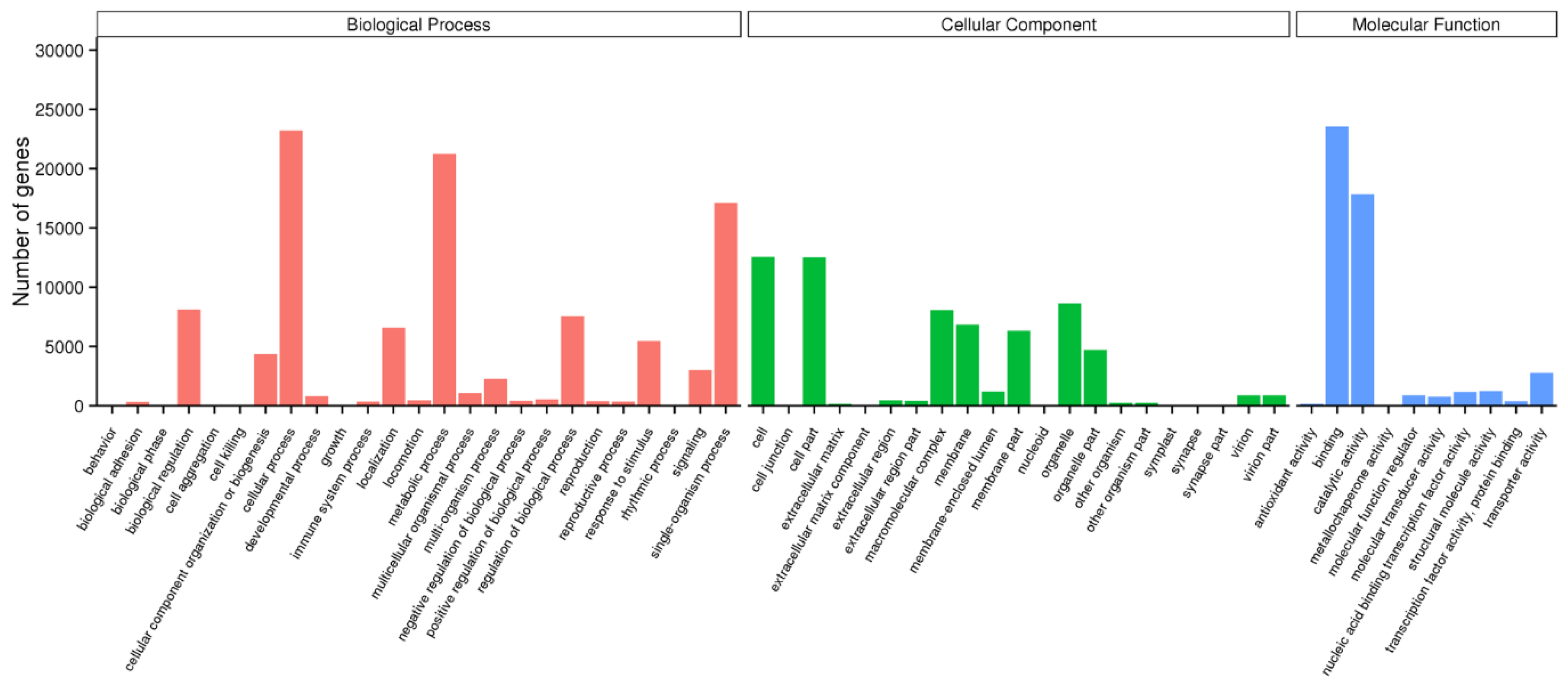
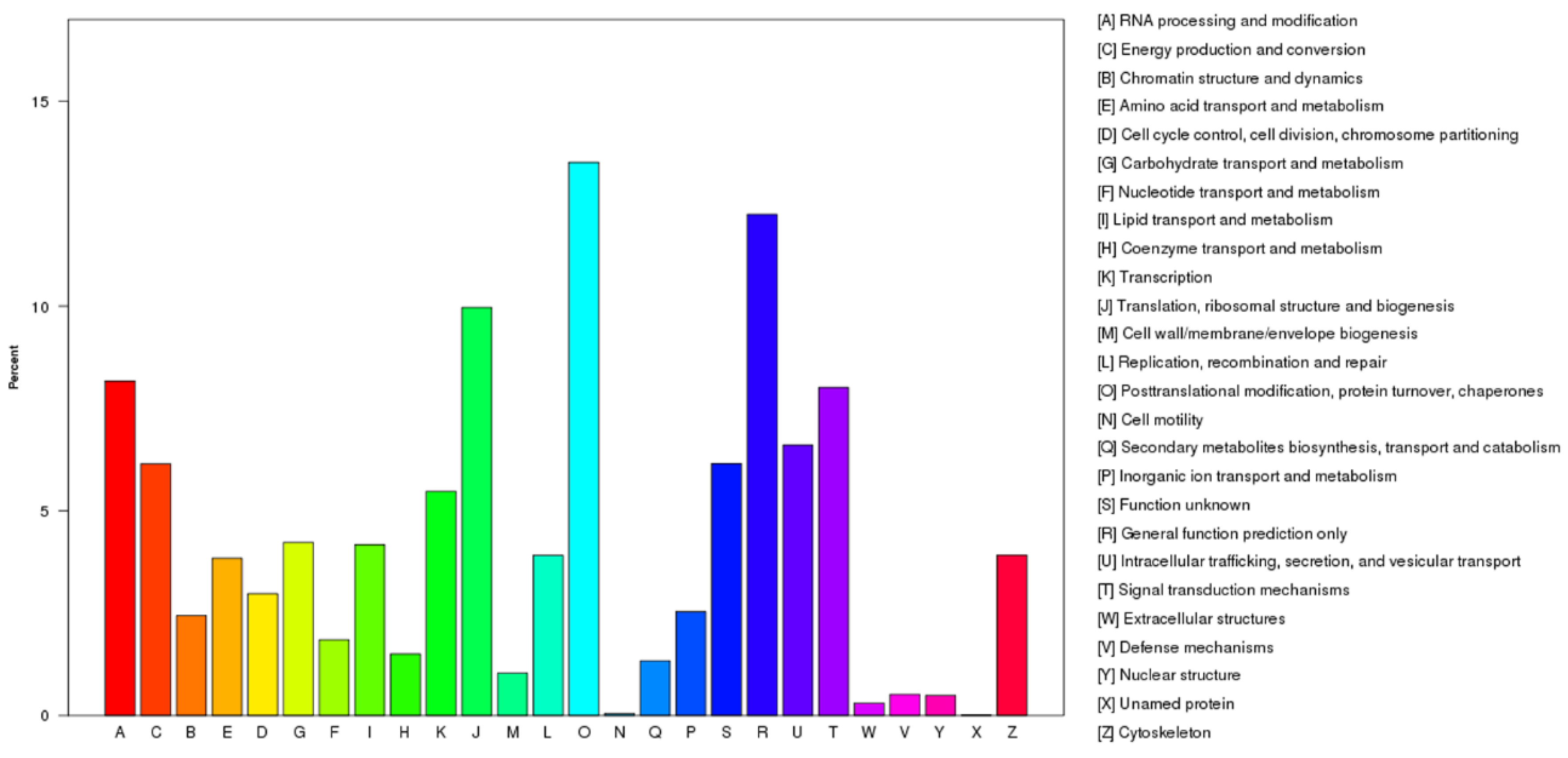
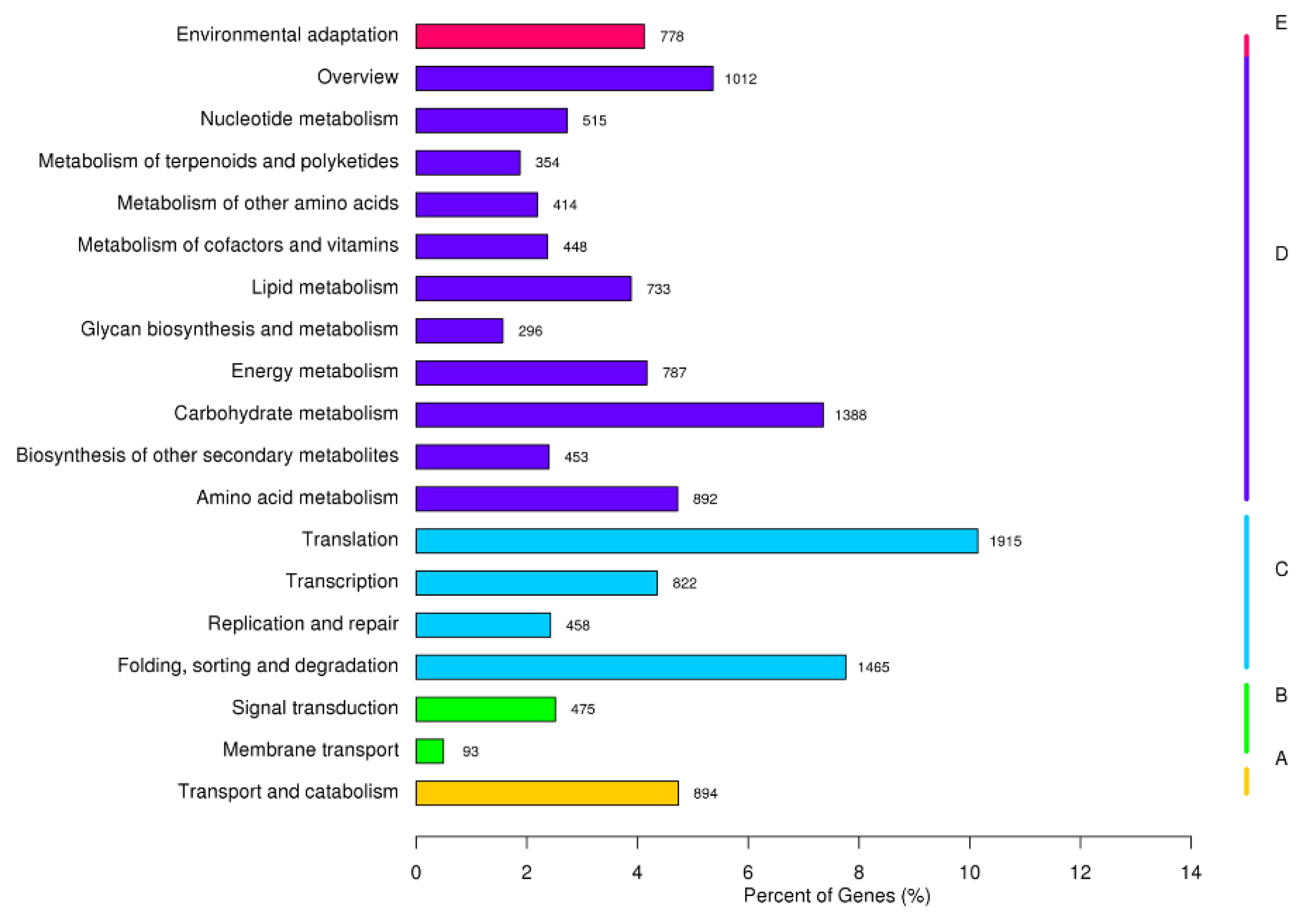
2.2. Functional Annotation and Classification of the Unigenes
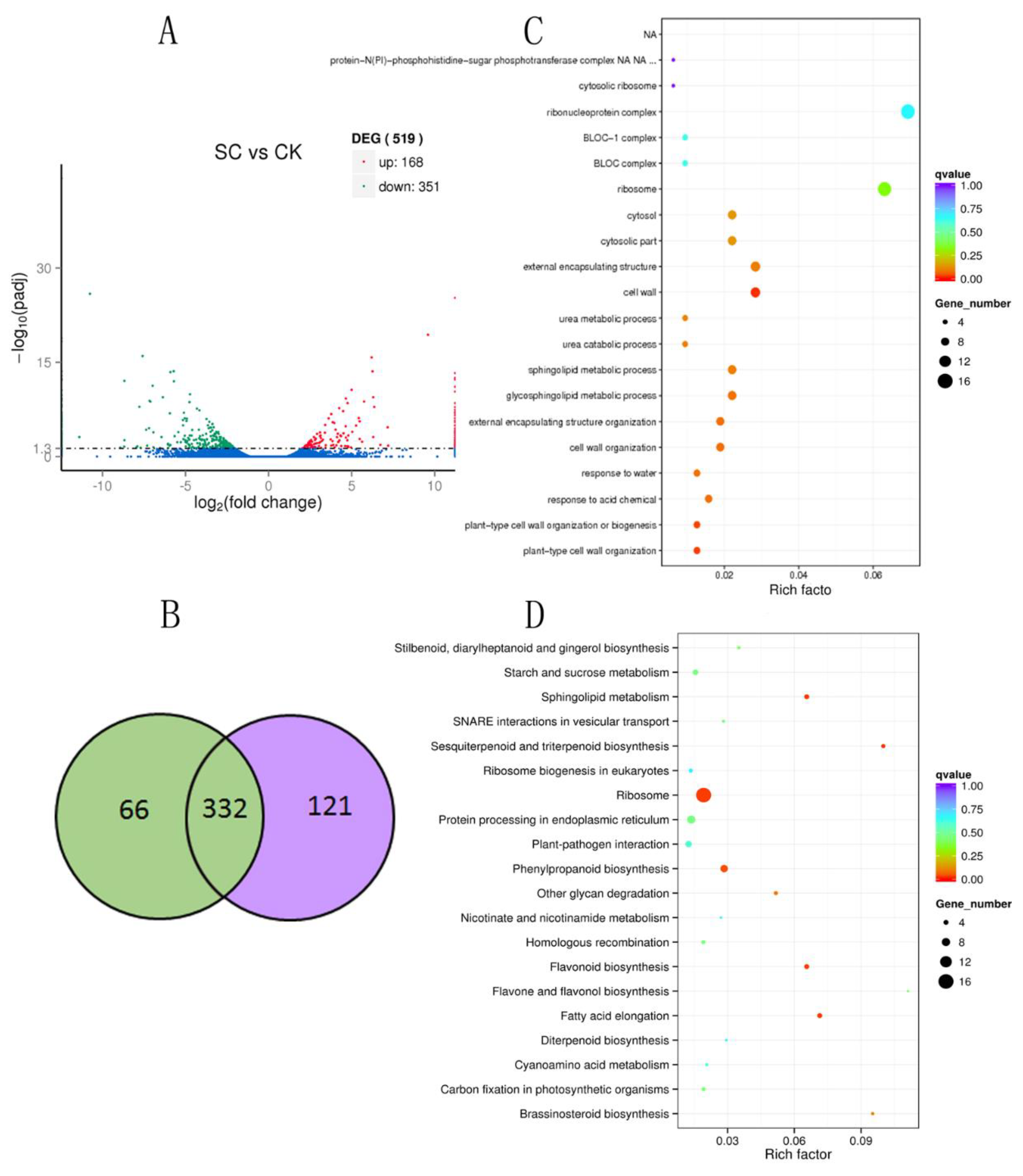
2.3. Differential Expression Genes in B. halophila Response to Salt
2.4. GO category Enrichment of DEGs Under Salt Stress
2.5. KEGG Enrichment of DEGs under Salt Stress
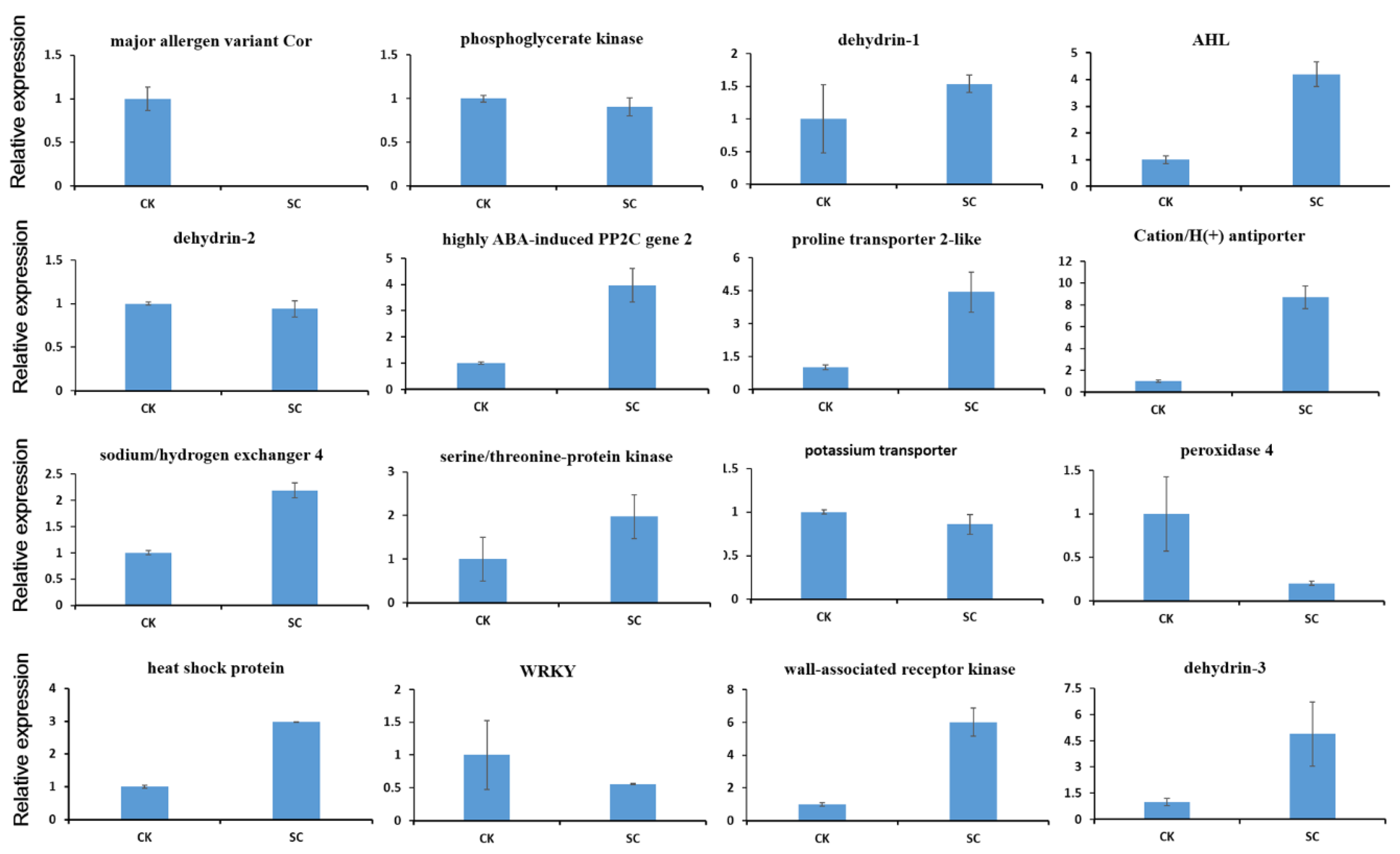
2.6. qRT-PCR Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Salt stress Treatment
4.3. Library Construction and Sequencing for RNA-seq
4.4. Transcriptome Assembly and Bioinformatics Analysis
4.5. Quantitative RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHL | AT-Hook Motif Nuclear Localized gene |
| BLAST | the Basic Local Alignment Search Tool |
| DEGs | differentially expressed genes |
| DHNs | dehydrins |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOG | Eukaryotic Orthologous Groups |
| LEA | Late Embryogenesis Abundant |
| Nr | NCBI non-redundant protein sequence database |
| Nt | NCBI nucleotide sequences |
| ORFs | Open reading frames |
| Pfam | Protein family |
| SOS | Salt Overly Sensitive |
| SwissProt | Swiss-prot sequence data bases |
References
- Flowers, T.J. Improving crop salt tolerance. J. Exp. Bot. 2004, 55, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.C.; Peng, Z.Y.; Li, C.L.; Li, F.; Liu, C.; Xia, G.M. Proteomic analysis on a high salt tolerance introgression strain of Triticumaestivum/Thinopyrumponticum. Proteomics 2008, 8, 1470–1489. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Chakraborty, U. Salt tolerance mechanisms in Salt Tolerant Grasses (STGs) and their prospects in cereal crop improvement. Bot. Stud. 2014, 55, 31. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Huang, B. Mechanism of Salinity Tolerance in Plants: Physiological, Biochemical, and Molecular Characterization. Int. J. Genom. 2014, 2014, 701596. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Pardo, J.M.; Batelli, G.; Van Oosten, M.J.; Bressan, R.A.; Li, X. The Salt Overly Sensitive (SOS) pathway: Established and emerging roles. Mol. Plant 2013, 6, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Barragán, V.; Leidi, E.O.; Andrés, Z.; Rubio, L.; De Luca, A.; Fernández, J.A.; Cubero, B.; Pardo, J.M. Ion exchangers NHX1 and NHX2 mediate active potassium uptake into vacuoles to regulate cell turgor and stomatal function in Arabidopsis. Plant Cell 2012, 24, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Dugasa, M.T.; Cao, F.; Ibrahim, W.; Wu, F. Genotypic difference in physiological and biochemical characteristics in response to single and combined stresses of drought and salinity between the two wheat genotypes (Triticumaestivum) differing in salt tolerance. Physiol. Plant 2018. [Google Scholar] [CrossRef] [PubMed]
- James, R.A.; Blake, C.; Byrt, C.S.; Munns, R. Major genes for Na+ exclusion, Nax1 and Nax2 (wheatHKT1;4 and HKT1;5), decrease Na+ accumulation in bread wheat leaves under saline and waterlogged conditions. J. Exp. Bot. 2011, 62, 2939–2947. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.W.; Chen, X.; Mei, Y. Function and regulation of phospholipid signalling in plants. Biochem. J. 2009, 421, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Zhao, Q.Y.; Ma, C.L.; Zhang, Z.H.; Cao, H.L.; Kong, Y.M.; Yue, C.; Hao, X.Y.; Chen, L.; Ma, J.Q.; et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genom. 2013, 14, 415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cai, M.; Yu, X.; Wang, L.; Guo, C.; Ming, R.; Zhang, J. Transcriptome dynamics of Camellia sinensis in response to continuous salinity and drought stress. Tree Genet. Genomes 2017, 13, 78. [Google Scholar] [CrossRef]
- Zeng, A.; Chen, P.; Korth, K.L.; Ping, J.; Thomas, J.; Wu, C.; Srivastava, S.; Pereira, A.; Hancock, F.; Brye, K.; et al. RNA sequencing analysis of salt tolerance in soybean (Glycine max). Genomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, S.; Zhao, Q.; Wang, T.; Yang, C.; Diaz, C.; Sun, G.; Dai, S. Physiological and Proteomic Analysis of Salinity Tolerance in Puccinelliatenuiflora. J. Proteome Res. 2011, 10, 3852–3870. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.S.; Jian, D.Y.; Fu, C.Z.; Yu, F.Z.; Xue, W.D. Research on introduction and salt tolerance of Betulahalophila. J. Gansu Agric. Univ. 2011, 5, 101–105. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.; Lu, J.; Yang, Y.; Zhang, X.; Wan, D.; Liu, J. Transcriptome differences between two sister desert poplar species under salt stress. BMC Genom. 2014, 15, 337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome Analysis of Salt Stress Responsiveness in the Seedlings of Dongxiang Wild Rice (OryzarufipogonGriff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef]
- Wang, W.S.; Zhao, X.Q.; Li, M.; Huang, L.Y.; Xu, J.L.; Zhang, F.; Cui, Y.R.; Fu, B.Y.; Li, Z.K. Complex molecular mechanisms underlying seedling salt tolerance in rice revealed by comparative transcriptome and metabolomic profiling. J. Exp. Bot. 2016, 67, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Villarino, G.H.; Hu, Q.; Scanlon, M.J.; Mueller, L.; Bombarely, A.; Mattson, N.S. Dissecting Tissue-Specific Transcriptomic Responses from Leaf and Roots under Salt Stress in Petunia hybrida Mitchell. Genes 2017, 8, 195. [Google Scholar] [CrossRef] [PubMed]
- Villarino, G.H.; Bombarely, A.; Giovannoni, J.J.; Scanlon, M.J.; Mattson, N.S. Transcriptomic analysis of Petunia hybrida in response to salt stress using high throughput RNA sequencing. PLoS ONE 2014, 9, e94651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.B.; Zeng, Y.L.; Lan, H.Y.; Zhang, F.C. Physiological response of Betulahalophila (Betaluceae) to salt stress. Acta Bot. Yunnanica 2009, 31, 260–264. [Google Scholar] [CrossRef]
- Liu, Y.; Song, Q.; Li, D.; Yang, X.; Li, D. Multifunctional Roles of Plant Dehydrins in Response to Environmental Stresses. Front. Plant Sci. 2017, 8, 1018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Lee, S.C.; Kim, J.Y.; Kim, S.J.; Aye, S.S.; Kim, S.R. Over-expression of dehydrin gene, OsDhn1, improves drought and salt stress tolerance through scavenging of reactive oxygen species in rice (Oryza sativa L.). J. Plant Biol. 2014, 57, 383–393. [Google Scholar] [CrossRef]
- Brini, F.; Hanin, M.; Lumbreras, V.; Amara, I.; Khoudi, H.; Hassairi, A.; Pagès, M.; Masmoudi, K. Overexpression of wheat dehydrin DHN5 enhances tolerance to salt and osmotic stress in Arabidopsis thaliana. Plant Cell Rep. 2007, 26, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Roy Choudhury, A.; Sengupta, D.N. Transgenic tobacco plants overexpressing the heterologous lea gene Rab16A from rice during high salt and water deficit display enhanced tolerance to salinity stress. Plant Cell Rep. 2007, 26, 1839–1859. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Targolli, J.; Huang, X.; Wu, R. Wheat LEA genes, PMA80 and PMA1959, enhance dehydration tolerance of transgenic rice (Oryza sativa L.). Mol. Breed. 2002, 10, 71–82. [Google Scholar] [CrossRef]
- Perdiguero, P.; Collada, C.; Soto, A. Novel dehydrins lacking complete K-segments in Pinaceae. The exception rather than the rule. Front. Plant Sci. 2014, 5, 682. [Google Scholar] [CrossRef] [PubMed]
- Close, T.J. Dehydrins: A commonalty in the response of plants to dehydration and low temperature. Physiol. Plant 1997, 100, 291–296. [Google Scholar] [CrossRef]
- Liu, C.C.; Li, C.M.; Liu, B.G.; Ge, S.J.; Dong, X.M.; Li, W.; Zhu, H.; Wang, B.; Yang, C. Genome-wide identification and characterization of a dehydrin gene family in poplar (Populustrichocarpa). Plant Mol. Biol. Rep. 2012, 30, 848–859. [Google Scholar] [CrossRef]
- Du, H.; Liu, H.; Xiong, L. Endogenous auxin and jasmonic acid levels are differentially modulated by abiotic stresses in rice. Front. Plant Sci. 2013, 4, 397. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Oh, C.J.; Park, Y.C.; Lee, Y.; Choe, S.; An, C.S.; Choi, S.B. Comprehensive analysis of AHL homologous genes encoding AT-hook motif nuclear localized protein in rice. BMB Rep. 2011, 44, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, A.; Furumoto, T.; Ishida, S.; Takahashi, Y. AGF1, an AT-hook protein, is necessary for the negative feedback of AtGA3ox1 encoding GA3-oxidase. Plant Physiol. 2007, 143, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Endt, D.V.; Silva, M.S.; Kijne, J.W.; Pasquali, G.; Memelink, J. Identification of a bipartite jasmonate-responsive promoter element in the Catharanthusroseus ORCA3 transcription factor gene that interacts specifically with AT-hook DNA-binding proteins. Plant Physiol. 2007, 144, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Street, I.H.; Shah, P.K.; Smith, A.M.; Avery, N.; Neff, M.M. The AT-hook-containing proteins SOB3/AHL29 and ESC/AHL27 are negative modulators of hypocotyl growth in Arabidopsis. Plant J. 2008, 54, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Kim, Y.; Breeze, E.; Koo, J.C.; Woo, H.R.; Ryu, J.S.; Park, D.H.; Beynon, J.; Tabrett, A.; Buchanan-Wollaston, V.; et al. Overexpression of a chromatin architecture-controlling AT-hook protein extends leaf longevity and increases the post-harvest storage life of plants. Plant J. 2007, 52, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zou, Y.; Feng, N. Overexpression of AHL20 negatively regulates defenses in Arabidopsis. J. Int. Plant Biol. 2010, 52, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Lu, Q.; Wilson, I.W.; Qiu, D. Genome-wide identification and characterization of the SPL gene family in Ziziphusjujuba. Gene 2017, 627, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Shao, F.; Macmillan, C.; Wilson, I.W.; Van der Merwe, K.; Hussey, S.G.; Myburg, A.A.; Dong, X.; Qiu, D. Genomewide analysis of the lateral organ boundaries domain gene family in Eucalyptus grandis reveals members that differentially impact secondary growth. Plant Biotechnol. J. 2018, 16, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta, C.(T.)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC Content(%) |
|---|---|---|---|---|---|---|---|
| CK_1 | 68,348,352 | 66,834,236 | 10.03G | 0.03 | 96.5 | 94.17 | 46.88 |
| CK_2 | 75,684,144 | 73,359,338 | 11G | 0.03 | 96.48 | 94.14 | 47.4 |
| CK_3 | 70,510,750 | 69,082,978 | 10.36G | 0.03 | 96.25 | 93.83 | 47.29 |
| SC_1 | 86,101,536 | 84,155,542 | 12.62G | 0.03 | 96.54 | 94.23 | 47.28 |
| SC_2 | 77,402,824 | 75,577,004 | 11.34G | 0.03 | 96.49 | 94.16 | 47.33 |
| SC_3 | 84,837,142 | 82,931,756 | 12.44G | 0.03 | 96.45 | 94.12 | 47.29 |
| Number of Unigenes | Percentage (%) | |
|---|---|---|
| Annotated in NR | 51,105 | 43.64 |
| Annotated in NT | 45,933 | 39.22 |
| Annotated in KO | 18,876 | 16.12 |
| Annotated in SwissProt | 40,624 | 34.69 |
| Annotated in PFAM | 40,661 | 34.72 |
| Annotated in GO | 41,116 | 35.11 |
| Annotated in KOG | 15,572 | 13.29 |
| Annotated in all Databases | 8973 | 7.66 |
| Annotated in at least one Database | 64,551 | 55.12 |
| Total Unigenes | 117,091 | 100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, F.; Zhang, L.; Wilson, I.W.; Qiu, D. Transcriptomic Analysis of Betula halophila in Response to Salt Stress. Int. J. Mol. Sci. 2018, 19, 3412. https://doi.org/10.3390/ijms19113412
Shao F, Zhang L, Wilson IW, Qiu D. Transcriptomic Analysis of Betula halophila in Response to Salt Stress. International Journal of Molecular Sciences. 2018; 19(11):3412. https://doi.org/10.3390/ijms19113412
Chicago/Turabian StyleShao, Fenjuan, Lisha Zhang, Iain W. Wilson, and Deyou Qiu. 2018. "Transcriptomic Analysis of Betula halophila in Response to Salt Stress" International Journal of Molecular Sciences 19, no. 11: 3412. https://doi.org/10.3390/ijms19113412
APA StyleShao, F., Zhang, L., Wilson, I. W., & Qiu, D. (2018). Transcriptomic Analysis of Betula halophila in Response to Salt Stress. International Journal of Molecular Sciences, 19(11), 3412. https://doi.org/10.3390/ijms19113412