Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
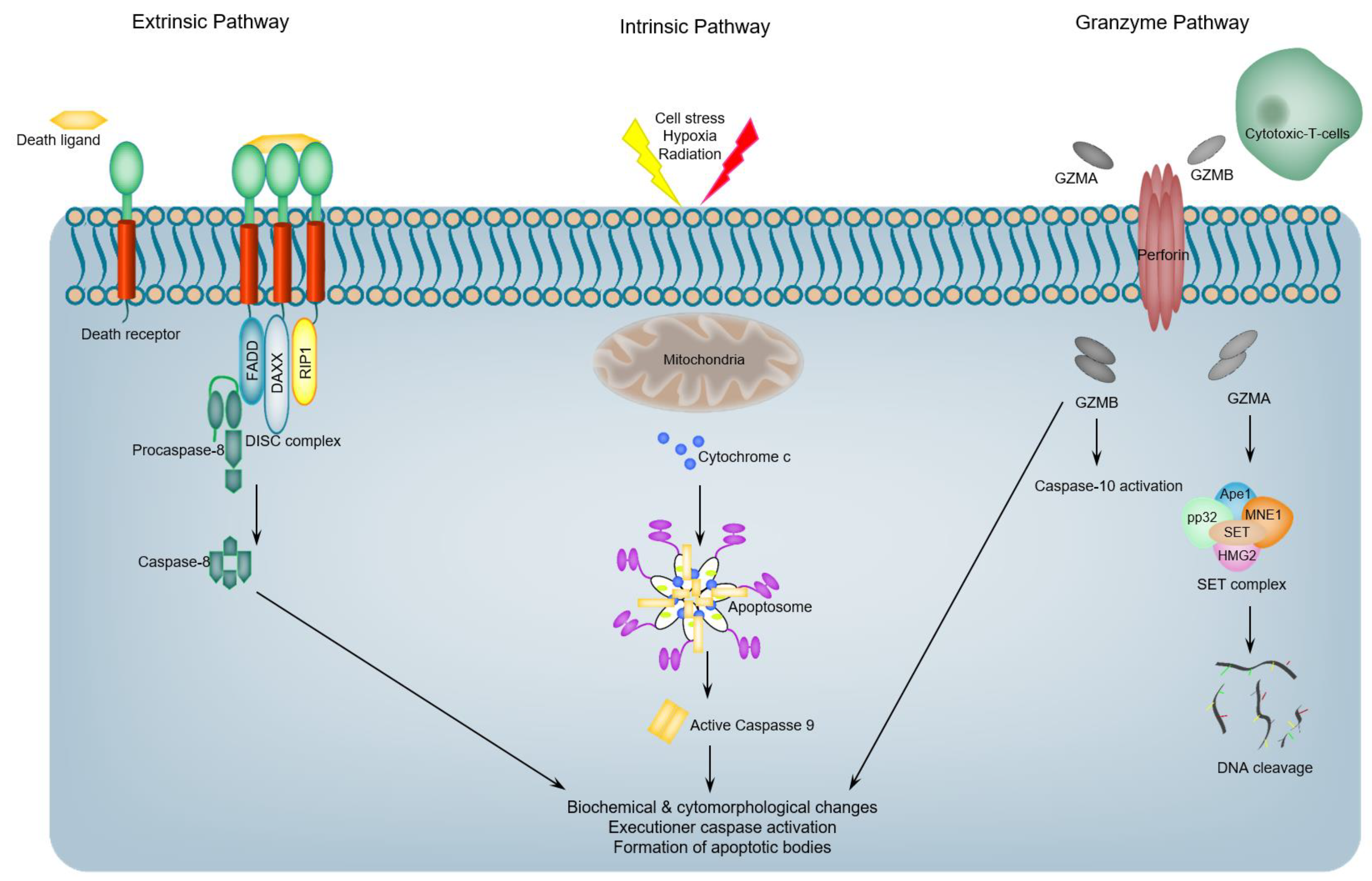
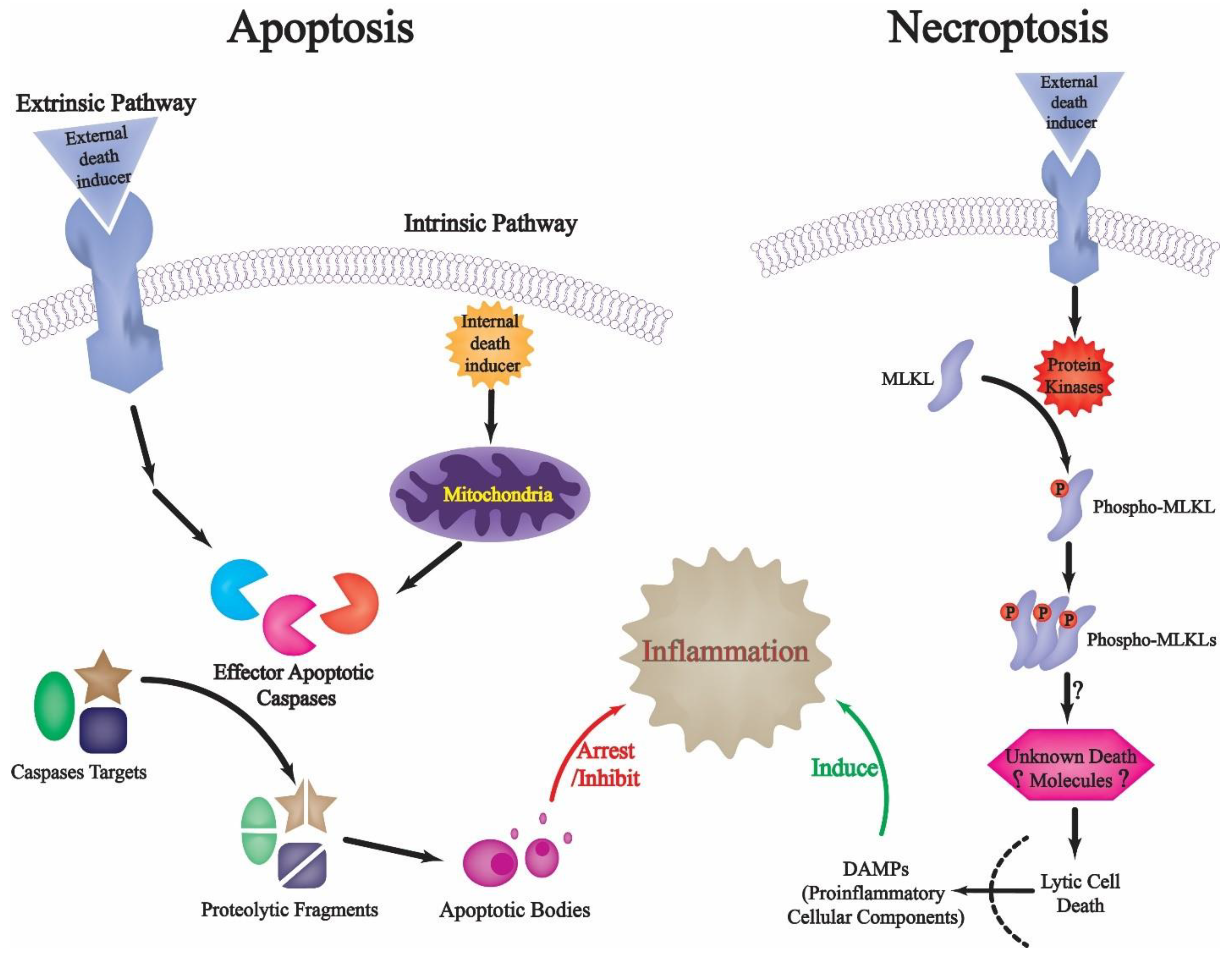
1. Introduction: An Overview of Programmed Apoptotic Cell Death
2. Restricted Proteolysis and Apoptotic Cell Death: Regulating Death and Preventing Inflammation-Related Danger
3. Proteases Are Essential Elements of Apoptosis
4. Caspases: Activation, Specificity, Function and Regulation
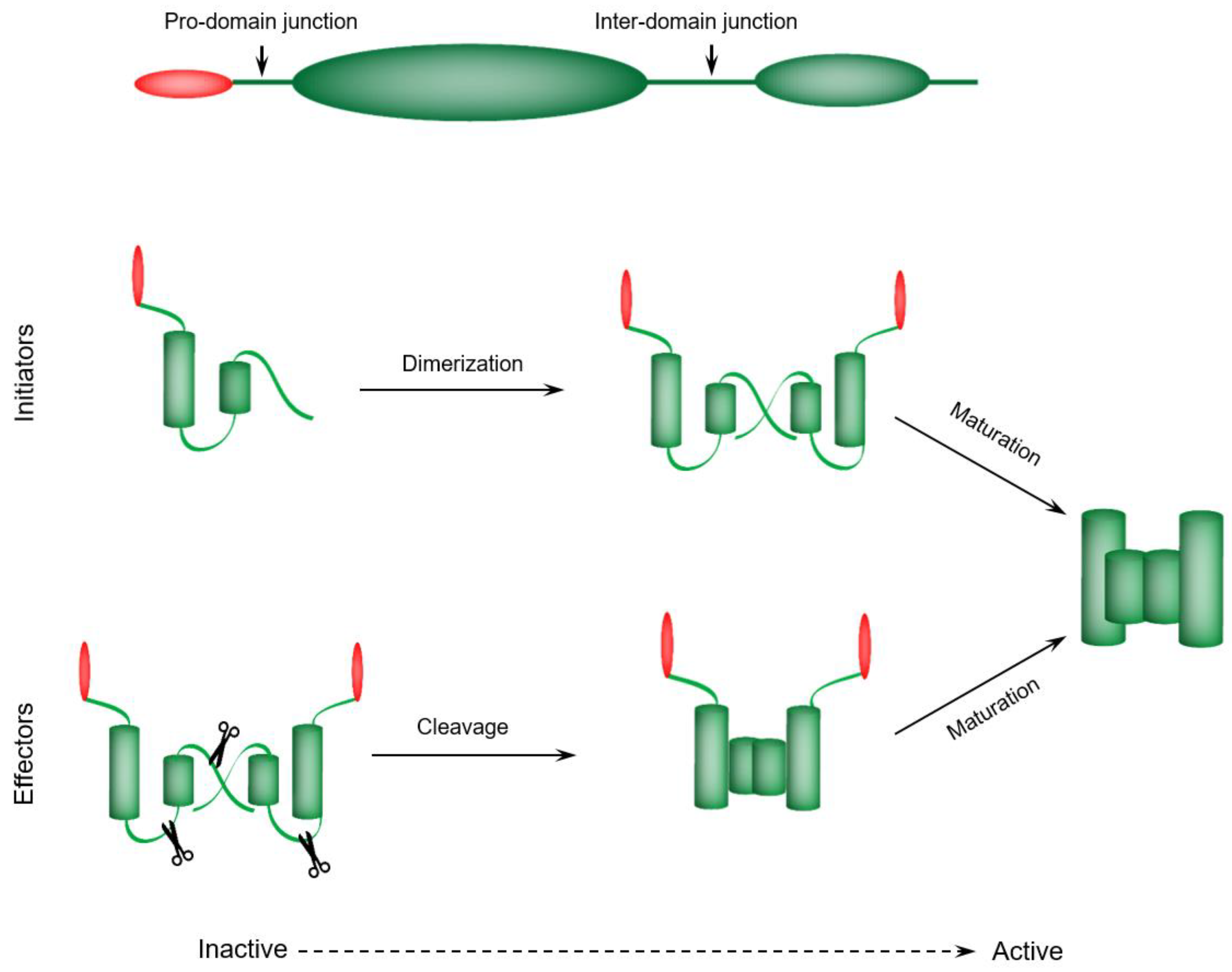
4.1. Initiator Caspases—Dimerization-Dependent Activation
4.2. Executioner Caspases—Proteolytic Cleavage-Dependent Activation
4.3. Caspases Maturation Events
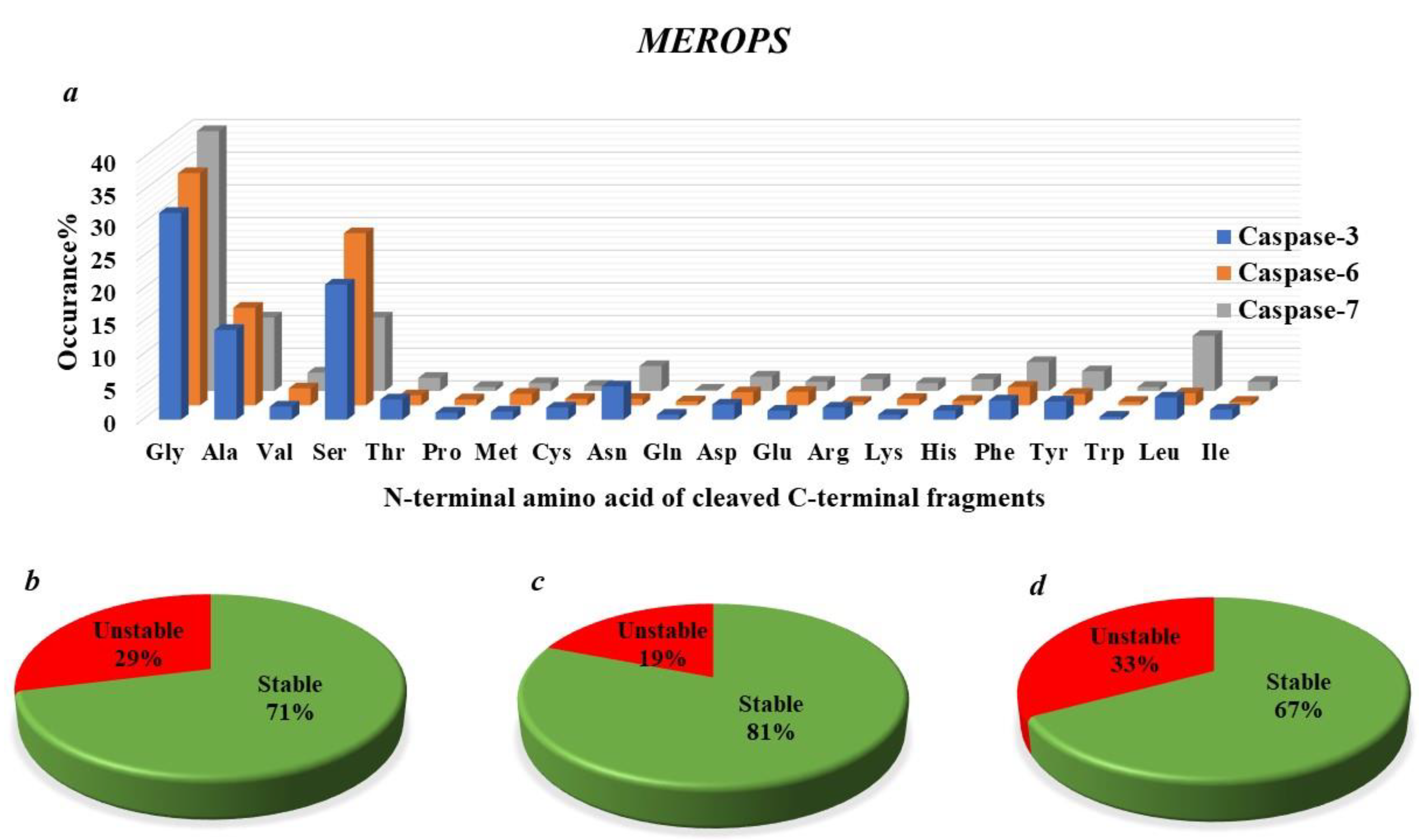
4.4. Caspases Specificity
4.5. Caspases Regulation
4.6. Does the N-End Rule Pathway Regulate Caspase-Generated Proteolytic Fragments during Apoptosis?
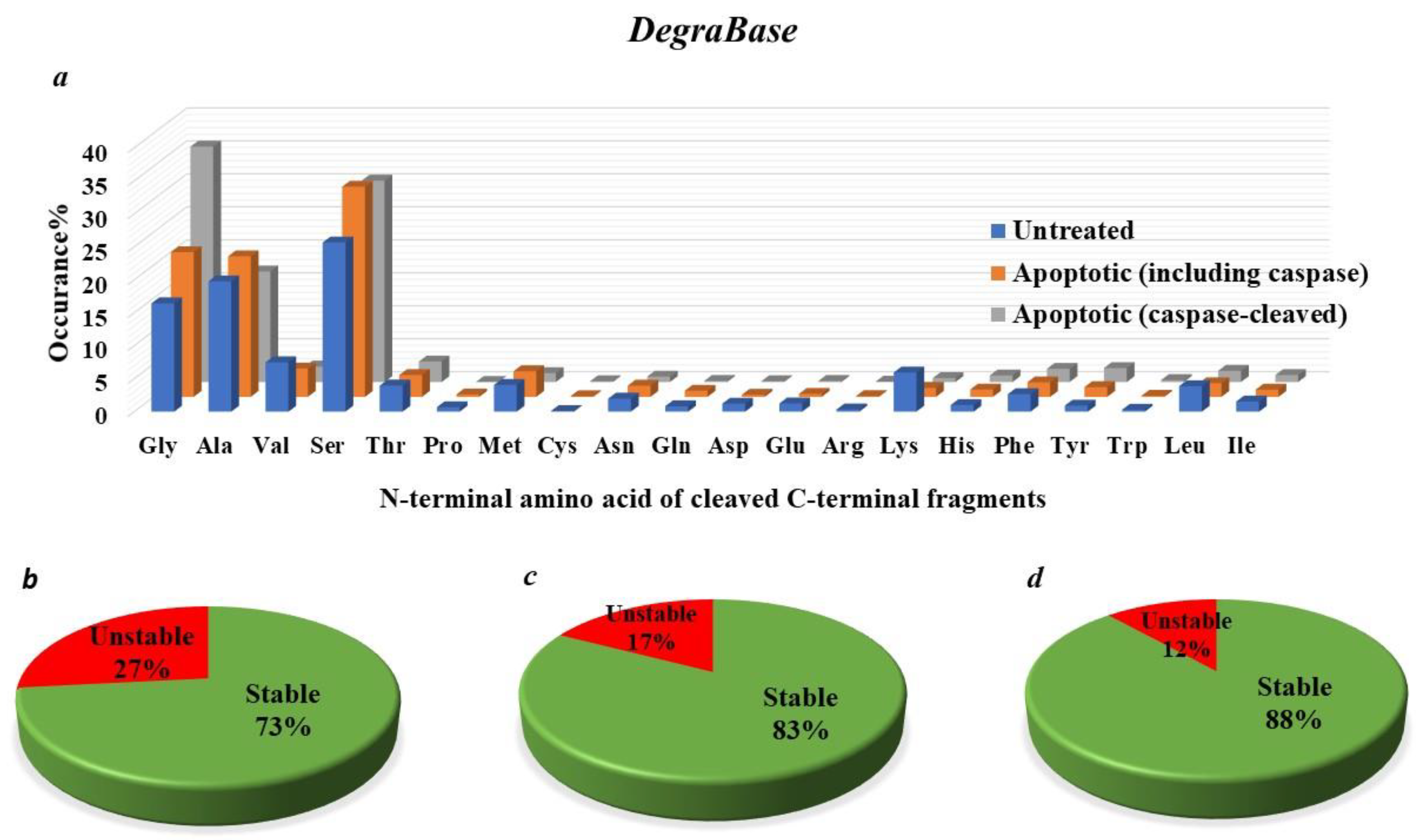
4.7. How Many Protease-Generated Proteolytic Fragments Are Predicted to Be Putative Targets of the N-End Rule Degradation Pathways?
5. Protein Degradation by Ubiquitin Proteasome System (UPS) and the N-End Rule Degradation Pathway
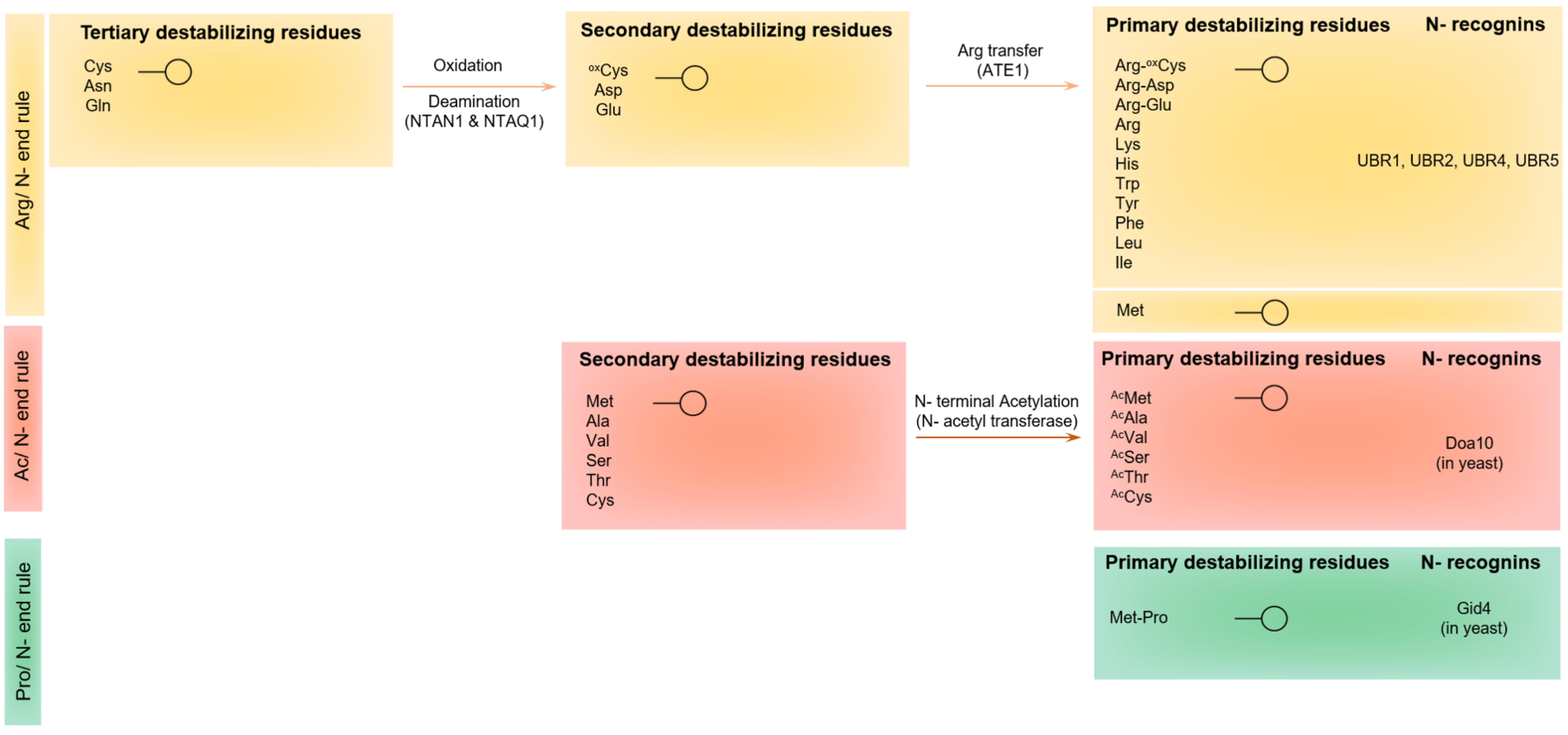
5.1. Protein Recognition by the UBR-Box E3 Ubiquitin Ligases of the Arg-N-End Rule Pathway
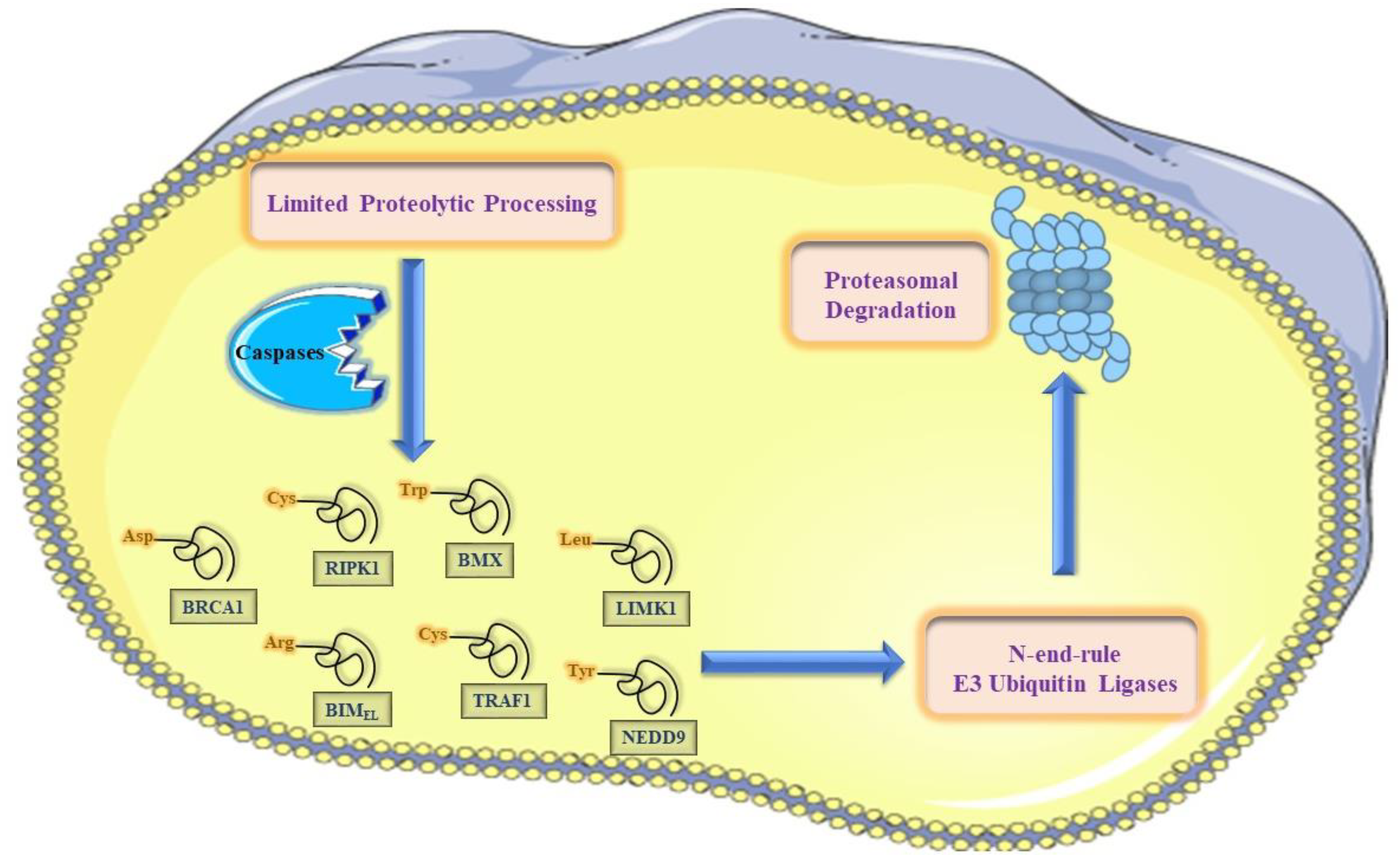
5.2. The N-End-Rule Pathway vis-à-vis Caspase-Generated Proteolytic Fragments: The Beginning Determines the End
6. The N-End Rule Pathway and Regulation of Apoptosis
7. The N-End Rule Degradation Machinery Counteracts Pro-Apoptotic Cell Death Signaling
8. The N-End Rule Pathway Can Mediate Pro-Apoptotic Signaling
9. The N-End Rule-Mediated Degradation of a Caspase-Generated Fragment Can Be Modulated by Phosphorylation
10. Mutual Repression between the N-End Rule Pathway and Apoptotic Activated Proteolytic Machinery
11. Concluding Remarks and Future Directions
11.1. The N-End Rule Degradation Pathways and the Non-Apoptotic Functions of Caspases: Is There A Link?
11.2. Beyond the N-End Rule: The N-End-Code beyond the Mere Identity of the N-Terminal Amino Acid Residue
11.3. Might the Arg/N-End Rule Pathway Serve as A Modulator of Apoptosis?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thompson, C.B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, X. Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Boil. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Nijhawan, D.; Honarpour, N.; Wang, X. Apoptosis in neural development and disease. Annu. Rev. Neurosci. 2000, 23, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Utz, P.; Anderson, P. Life and death decisions: Regulation of apoptosis by proteolysis of signaling molecules. Cell Death Differ. 2000, 7, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Boil. 2013, 5, a008672. [Google Scholar] [CrossRef] [PubMed]
- Shimbo, K.; Hsu, G.W.; Nguyen, H.; Mahrus, S.; Trinidad, J.C.; Burlingame, A.L.; Wells, J.A. Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. USA 2012, 109, 12432–12437. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Seaman, J.E.; Agard, N.; Hsu, G.W.; Julien, O.; Mahrus, S.; Nguyen, H.; Shimbo, K.; Yoshihara, H.A.; Zhuang, M. The degrabase: A database of proteolysis in healthy and apoptotic human cells. Mol. Cell. Proteom. 2013, 12, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Piatkov, K.I.; Brower, C.S.; Varshavsky, A. The n-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. USA 2012, 109, E1839–E1847. [Google Scholar] [CrossRef] [PubMed]
- Dizin, E.; Ray, H.; Suau, F.; Voeltzel, T.; Dalla Venezia, N. Caspase-dependent brca1 cleavage facilitates chemotherapy-induced apoptosis. Apoptosis 2008, 13, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhou, Q. Caspase cleavage of bimel triggers a positive feedback amplification of apoptotic signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.-J.; Yuan, J. Cleavage of bid by caspase 8 mediates the mitochondrial damage in the fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Cao, X.; Deng, X.; May, W.S. Cleavage of bax to p18 bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 bax. Blood 2003, 102, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Gamas, P.; Marchetti, S.; Puissant, A.; Grosso, S.; Jacquel, A.; Colosetti, P.; Pasquet, J.; Mahon, F.; Cassuto, J.; Auberger, P. Inhibition of imatinib-mediated apoptosis by the caspase-cleaved form of the tyrosine kinase lyn in chronic myelogenous leukemia cells. Leukemia 2009, 23, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Giaime, E.; Sunyach, C.; Herrant, M.; Grosso, S.; Auberger, P.; McLean, P.J.; Checler, F.; Da Costa, C.A. Caspase-3-derived c-terminal product of synphilin-1 displays antiapoptotic function via modulation of the p53-dependent cell death pathway. J. Boil. Chem. 2006, 281, 11515–11522. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Sordet, O.; Droin, N.; Munsch, B.; Haugg, M.; Van de Craen, M.; Vandenabeele, P.; Solary, E. Caspase-induced proteolysis of the cyclin-dependent kinase inhibitor p27kip1 mediates its anti-apoptotic activity. Oncogene 1999, 18, 4839–4847. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Michod, D.; Walicki, J.; Murphy, B.M.; Kasibhatla, S.; Martin, S.J.; Widmann, C. Partial cleavage of rasgap by caspases is required for cell survival in mild stress conditions. Mol. Cell. Boil. 2004, 24, 10425–10436. [Google Scholar] [CrossRef] [PubMed]
- Soskine, M.; Tawfik, D.S. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 2010, 11, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The n-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef] [PubMed]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Boil. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Maiuri, M.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 214074. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Younes, A.B.; Maiuri, M.C.; Lavandero, S. Senescence, apoptosis or autophagy? Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. Faseb J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Boil. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, E.N.; Chai, J.; Shi, Y. Oligomerization and activation of caspase-9, induced by apaf-1 card. Proc. Natl. Acad. Sci. USA 2002, 99, 4197–4202. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef]
- Yeh, W.-C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.-B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V. Requirement for casper (c-flip) in regulation of death receptor–induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Chang, C.-C.; Elia, A.; Fang, M.; Wakeham, A.; Okada, H.; Calzascia, T.; Jang, Y.; You-Ten, A. Specific ablation of the apoptotic functions of cytochrome c reveals a differential requirement for cytochrome c and apaf-1 in apoptosis. Cell 2005, 121, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Green, D.R. Protease activation during apoptosis: Death by a thousand cuts? Cell 1995, 82, 349–352. [Google Scholar] [CrossRef]
- Goping, S.; Barry, M.; Liston, P.; Sawchuk, T.; Constantinescu, G.; Michalak, K.M.; Shostak, I.; Roberts, D.L.; Hunter, A.M.; Korneluk, R. Granzyme b-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity 2003, 18, 355–365. [Google Scholar] [CrossRef]
- Hiebert, P.R.; Granville, D.J. Granzyme b in injury, inflammation, and repair. Trends Mol. Med. 2012, 18, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Buzza, M.S.; Zamurs, L.; Sun, J.; Bird, C.H.; Smith, A.I.; Trapani, J.A.; Froelich, C.J.; Nice, E.C.; Bird, P.I. Extracellular matrix remodeling by human granzyme b via cleavage of vitronectin, fibronectin and laminin. J. Boil. Chem. 2005, 280, 23549–23558. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, S.; Lolkema, M.; Matzinger, P. Natural adjuvants: Endogenous activators of dendritic cells. Nat. Med. 1999, 5, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G.; Sher, A. Cooperation of toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein hmgb1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Shaham, S.; Ledoux, S.; Ellis, H.M.; Horvitz, H.R. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell 1993, 75, 641–652. [Google Scholar] [CrossRef]
- Miura, M.; Zhu, H.; Rotello, R.; Hartwieg, E.A.; Yuan, J. Induction of apoptosis in fibroblasts by il-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell 1993, 75, 653–660. [Google Scholar] [CrossRef]
- Sarin, A.; Adams, D.H.; Henkart, P.A. Protease inhibitors selectively block t cell receptor-triggered programmed cell death in a murine t cell hybridoma and activated peripheral t cells. J. Exp. Med. 1993, 178, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Miura, M.; Bergeron, L.; Zhu, H.; Yuan, J. Ich-1, an ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 1994, 78, 739–750. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly (adp-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Neamati, N.; Fernandez, A.; Wright, S.; Kiefer, J.; McConkey, D.J. Degradation of lamin b1 precedes oligonucleosomal DNA fragmentation in apoptotic thymocytes and isolated thymocyte nuclei. J. Immunol. 1995, 154, 3788–3795. [Google Scholar] [PubMed]
- Voelkel-Johnson, C.; Entingh, A.J.; Wold, W.; Gooding, L.R.; Laster, S.M. Activation of intracellular proteases is an early event in tnf-induced apoptosis. J. Immunol. 1995, 154, 1707–1716. [Google Scholar] [PubMed]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Boil. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D. Noncaspase proteases in apoptosis. Leukemia 2000, 14, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Alnemri, E.S.; Livingston, D.J.; Nicholson, D.W.; Salvesen, G.; Thornberry, N.A.; Wong, W.W.; Yuan, J. Human ice/ced-3 protease nomenclature. Cell 1996, 87, 171. [Google Scholar] [CrossRef]
- Callus, B.; Vaux, D. Caspase inhibitors: Viral, cellular and chemical. Cell Death Differ. 2007, 14, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the tnf receptors, fas/apo1, and dr3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004, 384, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Boil. 2007, 8, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Wu, Q.; Shiozaki, E.; Srinivasula, S.M.; Alnemri, E.S.; Shi, Y. Crystal structure of a procaspase-7 zymogen: Mechanisms of activation and substrate binding. Cell 2001, 107, 399–407. [Google Scholar] [CrossRef]
- Riedl, S.J.; Fuentes-Prior, P.; Renatus, M.; Kairies, N.; Krapp, S.; Huber, R.; Salvesen, G.S.; Bode, W. Structural basis for the activation of human procaspase-7. Proc. Natl. Acad. Sci. USA 2001, 98, 14790–14795. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Fitzgerald, P.; Green, D.R.; Salvesen, G.S. Role of proteolysis in caspase-8 activation and stabilization. Biochemistry 2007, 46, 4398–4407. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-B.; Oh, G.-S.; Scandella, E.; Bolinger, B.; Ludewig, B.; Kovalenko, A.; Wallach, D. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J. Immunol. 2008, 181, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P. A combinatorial approach defines specificities of members of the caspase family and granzyme b functional relationships established for key mediators of apoptosis. J. Boil. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef]
- Timmer, J.; Salvesen, G. Caspase substrates. Cell Death Differ. 2007, 14, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Renatus, M.; Meldal, M.; Salvesen, G.S. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 2000, 350, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Ryan, C.A.; Salvesen, G.S. Reprieval from execution: The molecular basis of caspase inhibition. Trends Biochem. Sci. 2002, 27, 94–101. [Google Scholar] [CrossRef]
- Blankenship, J.W.; Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Kirkpatrick, D.S.; Izrael-Tomasevic, A.; Phu, L.; Arnott, D.; Aghajan, M.; Zobel, K. Ubiquitin binding modulates iap antagonist-stimulated proteasomal degradation of c-iap1 and c-iap2. Biochem. J. 2009, 417, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Darding, M.; Miasari, M.; Santoro, M.M.; Zender, L.; Xue, W.; Tenev, T.; Da Fonseca, P.C.; Zvelebil, M.; Bujnicki, J.M. Iaps contain an evolutionarily conserved ubiquitin-binding domain that regulates nf-κb as well as cell survival and oncogenesis. Nat. Cell Boil. 2008, 10, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Schile, A.J.; García-Fernández, M.; Steller, H. Regulation of apoptosis by xiap ubiquitin-ligase activity. Genes Dev. 2008, 22, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why xiap is the black sheep of the family. Embo Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Tawa, P.; Hell, K.; Giroux, A.; Grimm, E.; Han, Y.; Nicholson, D.; Xanthoudakis, S. Catalytic activity of caspase-3 is required for its degradation: Stabilization of the active complex by synthetic inhibitors. Cell Death Differ. 2004, 11, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. Dff, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Boil. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Yuen, K.; Tang, H.; Fung, M. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Fung, M.C.; Hardwick, J.M. In vivo caspasetracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci. Rep. 2015, 5, 9015. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.A.; Hickman, J.A.; Chachar, M.; Polster, B.M.; Brandt, T.A.; Fannjiang, Y.; Ivanovska, I.; Basañez, G.; Kinnally, K.W.; Zimmerberg, J.; et al. Proapoptotic n-truncated bcl-xl protein activates endogenous mitochondrial channels in living synaptic terminals. Proc. Natl. Acad. Sci. USA 2004, 101, 13590–13595. [Google Scholar] [CrossRef] [PubMed]
- Huesmann, G.R.; Clayton, D.F. Dynamic role of postsynaptic caspase-3 and birc4 in zebra finch song-response habituation. Neuron 2006, 52, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, Y.; Gibbs-Bar, L.; Kalifa, Y.; Feinstein-Rotkopf, Y.; Arama, E. Gradients of a ubiquitin e3 ligase inhibitor and a caspase inhibitor determine differentiation or death in spermatids. Dev. Cell 2010, 19, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. Iaps: From caspase inhibitors to modulators of nf-κb, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Dissmeyer, N.; Rivas, S.; Graciet, E. Life and death of proteins after protease cleavage: protein degradation by the N-end rule pathway. New Phytol. 2018, 218, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. Merops: The peptidase database. Nucleic Acids Res. 1999, 27, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Gonda, D.K.; Bachmair, A.; Wünning, I.; Tobias, J.W.; Lane, W.S.; Varshavsky, A. Universality and structure of the n-end rule. J. Boil. Chem. 1989, 264, 16700–16712. [Google Scholar]
- Varshavsky, A. The ubiquitin system, an immense realm. Annu. Rev. Biochem. 2012, 81, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Naujokat, C.; Hoffmann, S. Role and function of the 26s proteasome in proliferation and apoptosis. Lab. Investig. 2002, 82, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Tam, S.W.; Theodoras, A.M.; Beer-Romero, P.; Del Sal, G.; Chau, V.; Yew, P.R.; Draetta, G.F.; Rolfe, M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.M.; Aranda-Orgilles, B.; Strikoudis, A.; Apostolou, E.; Loizou, E.; Moran-Crusio, K.; Farnsworth, C.L.; Koller, A.A.; Dasgupta, R.; Silva, J.C.; et al. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell 2012, 11, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005, 12, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Wells, R.; Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: The pest hypothesis. Science 1986, 234, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Rogers, S.W. Pest sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef]
- Campanero, M.R.; Flemington, E.K. Regulation of e2f through ubiquitin–proteasome-dependent degradation: Stabilization by the prb tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2221–2226. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, R.J.; Wu, P.; Varshavsky, A. Heat-inducible degron: A method for constructing temperature-sensitive mutants. Science 1994, 263, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, J.; Linskens, M.; Sadis, S.; Rubin, D.M.; Futcher, B.; Finley, D. P34cdc28-mediated control of cln3 cyclin degradation. Mol. Cell. Boil. 1995, 15, 731–741. [Google Scholar] [CrossRef]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21cip1 and p27kip1 cdk ‘inhibitors’ are essential activators of cyclin d-dependent kinases in murine fibroblasts. Embo J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, K.; Kubota, Y.; Kato, J.-y. Degradation of the cyclin-dependent-kinase inhibitor p27kip1 is instigated by jab1. Nature 1999, 398, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.A.; Levine, A.J. Nuclear export is required for degradation of endogenous p53 by mdm2 and human papillomavirus e6. Mol. Cell. Boil. 1998, 18, 7288–7293. [Google Scholar] [CrossRef]
- Tobias, J.W.; Shrader, T.E.; Rocap, G.; Varshavsky, A. The n-end rule in bacteria. Science 1991, 254, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.J.; Lee, S.C.; Isa, N.M.; Gramuglia, S.; Fukao, T.; Bassel, G.W.; Correia, C.S.; Corbineau, F.; Theodoulou, F.L.; Bailey-Serres, J.; et al. Homeostatic response to hypoxia is regulated by the n-end rule pathway in plants. Nature 2011, 479, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Shemorry, A.; Hwang, C.-S.; Varshavsky, A. Control of protein quality and stoichiometries by n-terminal acetylation and the n-end rule pathway. Mol. Cell 2013, 50, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Uhlmann, F.; Nasmyth, K.; Varshavsky, A. Degradation of a cohesin subunit by the n-end rule pathway is essential for chromosome stability. Nature 2001, 410, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Piatkov, K.I.; Colnaghi, L.; Békés, M.; Varshavsky, A.; Huang, T.T. The auto-generated fragment of the usp1 deubiquitylase is a physiological substrate of the n-end rule pathway. Mol. Cell 2012, 48, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Ditzel, M.; Wilson, R.; Tenev, T.; Zachariou, A.; Paul, A.; Deas, E.; Meier, P. Degradation of diap1 by the n-end rule pathway is essential for regulating apoptosis. Nat. Cell Boil. 2003, 5, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Kashina, A.S.; Davydov, I.V.; Hu, R.-G.; An, J.Y.; Seo, J.W.; Du, F.; Varshavsky, A. An essential role of n-terminal arginylation in cardiovascular development. Science 2002, 297, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Turner, G.C.; Du, F.; Varshavsky, A. Peptides accelerate their uptake by activating a ubiquitin-dependent proteolytic pathway. Nature 2000, 405, 579–583. [Google Scholar] [PubMed]
- Chen, S.-J.; Wu, X.; Wadas, B.; Oh, J.-H.; Varshavsky, A. An n-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 2017, 355, eaal3655. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.-S.; Shemorry, A.; Varshavsky, A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science 2010, 327, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Mulder, L.C.; Iwamatsu, A.; Lee, M.J.; Davydov, I.V.; Varshavsky, A.; Muesing, M.; Kwon, Y.T. A family of mammalian e3 ubiquitin ligases that contain the ubr box motif and recognize n-degrons. Mol. Cell. Boil. 2005, 25, 7120–7136. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.; Fahlman, R. The-N-end rule: The beginning determines the end. Protein Pept. Lett. 2016, 23, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, T.; Zakrzewska, A.; Dudgeon, D.D.; Jiang, Y.; Lazo, J.S.; Kwon, Y.T. The substrate recognition domains of the n-end rule pathway. J. Boil. Chem. 2009, 284, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Webster, A.; Du, F.; Piatkov, K.; Ghislain, M.; Varshavsky, A. Substrate-sinding sites of ubr1, the ubiquitin ligase of the n-end rule pathway. J. Boil. Chem. 2008, 283, 24011–24028. [Google Scholar] [CrossRef] [PubMed]
- Erbse, A.; Schmidt, R.; Bornemann, T.; Schneider-Mergener, J.; Mogk, A.; Zahn, R.; Dougan, D.; Bukau, B. Clps is an essential component of the n-end rule pathway in Escherichia coli. Nature 2006, 439, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, V.J.; Kralik, S.M.; Albrecht, R.; Spall, S.K.; Truscott, K.N.; Dougan, D.A.; Zeth, K. Structural basis of n-end rule substrate recognition in escherichia coli by the clpap adaptor protein clps. Embo Rep. 2009, 10, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Román-Hernández, G.; Grant, R.A.; Sauer, R.T.; Baker, T.A. Molecular basis of substrate selection by the n-end rule adaptor protein clps. Proc. Natl. Acad. Sci. USA 2009, 106, 8888–8893. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Jeong, B.-C.; Joo, Y.J.; Lee, M.-R.; Kim, J.; Eck, M.J.; Song, H.K. Structural basis for the recognition of n-end rule substrates by the ubr box of ubiquitin ligases. Nat. Struct. Mol. Boil. 2010, 17, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Matta-Camacho, E.; Kozlov, G.; Li, F.F.; Gehring, K. Structural basis of substrate recognition and specificity in the n-end rule pathway. Nat. Struct. Mol. Boil. 2010, 17, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.M.; Kwon, Y.T. The molecular principles of n-end rule recognition. Nat. Struct. Mol. Boil. 2010, 17, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Bachmair, A.; Varshavsky, A. The degradation signal in a short-lived protein. Cell 1989, 56, 1019–1032. [Google Scholar] [CrossRef]
- Kim, H.-K.; Kim, R.-R.; Oh, J.-H.; Cho, H.; Varshavsky, A.; Hwang, C.-S. The N-terminal methionine of cellular proteins as a degradation signal. Cell 2014, 156, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Fahlman, R.P. Phosphorylation impacts n-end rule degradation of the proteolytically activated form of bmx kinase. J. Boil. Chem. 2016, 291, 22757–22768. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Fahlman, R.P. The anti-apoptotic form of tyrosine kinase lyn that is generated by proteolysis is degraded by the N-end rule pathway. Oncotarget 2014, 5, 2714–2722. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Yamanouchi, D.; Esbona, K.; Kamiya, K.; Zhang, F.; Kent, K.C.; Liu, B. Caspase-mediated protein kinase c-δ cleavage is necessary for apoptosis of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2253–H2261. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.-G. Cleavage of the death domain kinase rip by caspase-8 prompts tnf-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Kojima, H.; Yoshida, K.; Kufe, D. Caspase-3-mediated cleavage of protein kinase c θ in induction of apoptosis. J. Boil. Chem. 1997, 272, 20317–20320. [Google Scholar] [CrossRef]
- Wickliffe, K.E.; Leppla, S.H.; Moayeri, M. Killing of macrophages by anthrax lethal toxin: Involvement of the n-end rule pathway. Cell. Microbiol. 2008, 10, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Birnbaum, M.D.; Patel, D.M.; Morgan, W.M.; Singh, J.; Barrientos, A.; Zhang, F. Posttranslational arginylation enzyme ate1 affects DNA mutagenesis by regulating stress response. Cell Death Dis. 2016, 7, e2378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhu, W.; Zhang, J.; Guo, L. Tyrosine kinase etk/bmx protects nasopharyngeal carcinoma cells from apoptosis induced by radiation. Cancer Boil. Ther. 2011, 11, 690–698. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Huang, C.-L.; Kung, H.-J.; Huang, C.-Y.F. Proteolytic activation of etk/bmx tyrosine kinase by caspases. J. Boil. Chem. 2001, 276, 17672–17678. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Kumagai, A.; Dunphy, W.G.; Varshavsky, A. Dissection of c-mos degron. Embo J. 2002, 21, 6061–6071. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Payoe, R.; Fahlman, R.P. The c-terminal proteolytic fragment of the breast cancer susceptibility type 1 protein (brca1) is degraded by the n-end rule pathway. J. Boil. Chem. 2012, 287, 7495–7502. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Pharmacological manipulation of cell death: Clinical applications in sight? J. Clin. Investig. 2005, 115, 2610–2617. [Google Scholar] [CrossRef] [PubMed]
- Schwerk, C.; Schulze-Osthoff, K. Non-apoptotic functions of caspases in cellular proliferation and differentiation. Biochem. Pharmacol. 2003, 66, 1453–1458. [Google Scholar] [CrossRef]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Kuranaga, E.; Miura, M. Nonapoptotic functions of caspases: Caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Boil. 2007, 17, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Arama, E.; Agapite, J.; Steller, H. Caspase activity and a specific cytochrome c are required for sperm differentiation in drosophila. Dev. Cell 2003, 4, 687–697. [Google Scholar] [CrossRef]
- Dahm, R. Lens fibre cell differentiation—A link with apoptosis? Ophthalmic Res. 1999, 31, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; He, Z.; Shen, J.; Huang, Q.; Li, W.; Liu, X.; He, Y.; Wolf, F.; Li, C.-Y. Apoptotic caspases regulate induction of ipscs from human fibroblasts. Cell Stem Cell 2010, 7, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Rébé, C.; Plenchette, S.; Zermati, Y.; Hermine, O.; Vainchenker, W.; Garrido, C.; Solary, E.; Dubrez-Daloz, L. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 2002, 100, 4446–4453. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Kondo, S.; Krzyzanowska, A.; Hiromi, Y.; Truman, J.W. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 2006, 9, 1234–1236. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.T.; Zhu, S.; Younger, S.; Jan, L.Y.; Jan, Y.N. Identification of e2/e3 ubiquitinating enzymes and caspase activity regulating drosophila sensory neuron dendrite pruning. Neuron 2006, 51, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Helfer, B.; Boswell, B.C.; Finlay, D.; Cipres, A.; Vuori, K.; Kang, T.B.; Wallach, D.; Dorfleutner, A.; Lahti, J.M.; Flynn, D.C. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006, 66, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Oshima, K.; Takeda, M.; Kuranaga, E.; Ueda, R.; Aigaki, T.; Miura, M.; Hayashi, S. Ikkε regulates f actin assembly and interacts with drosophila iap1 in cellular morphogenesis. Curr. Boil. 2006, 16, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.-Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; So, C.; Lam, H.-M.; Fung, M.-C.; Tsang, S.-Y. Apoptosis Reversal Promotes Cancer Stem Cell-Like Cell Formation. Neoplasia 2018, 20, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Zhang, F.; Colavita, K.; Leu, N.A.; Kurosaka, S.; Kumar, A.; Birnbaum, M.D.; Győrffy, B.; Dong, D.W.; Shtutman, M.; et al. Arginyltransferase suppresses cell tumorigenic potential and inversely correlates with metastases in human cancers. Oncogene 2016, 35, 4058–4068. [Google Scholar] [CrossRef] [PubMed]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Denault, J.B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. Xiap inhibits caspase-3 and-7 using two binding sites: Evolutionarily conserved mechanism of iaps. Embo J. 2005, 24, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.C.; Mahrus, S.; Wells, J.A. Activation of specific apoptotic caspases with an engineered small-molecule-activated protease. Cell 2010, 142, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-M.; Butterworth, M.; MacFarlane, M.; Dubiel, W.; Ciechanover, A.; Cohen, G.M. Caspase activation inhibits proteasome function during apoptosis. Mol. Cell 2004, 14, 81–93. [Google Scholar] [CrossRef]
- Weaver, B.P.; Weaver, Y.M.; Mitani, S.; Han, M. Coupled Caspase and N-End Rule Ligase Activities Allow Recognition and Degradation of Pluripotency Factor LIN-28 during Non-Apoptotic Development. Dev. Cell 2017, 41, 665–673.e6. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.-I.; Kuranaga, E. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017, 24, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Leitao, L.C.; Fahlman, R.P. Emerging branches of the n-end rule pathways are revealing the sequence complexities of n-termini dependent protein degradation. Biochem. Cell Boil. 2017, 96, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Ragheb, M.A. Post-translational N-terminal arginylation of protein fragments: A pivotal portal to proteolysis. Curr. Protein Pept. Sci. 2018, 19, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Jönsson, J.; Larsson, C. The regulatory domain of protein kinase cθ localises to the golgi complex and induces apoptosis in neuroblastoma and jurkat cells. Cell Death Differ. 2003, 10, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Eldeeb, M.; Wang, X. Beyond deubiquitylation: Usp30-mediated regulation of mitochondrial homeostasis. In Mitochondrial DNA and Diseases; Sun, H., Wang, X., Eds.; Springer: Singapore, 2017; pp. 133–148. [Google Scholar]
- Wang, Z.; Xia, X.; Yang, X.; Zhang, X.; Liu, Y.; Wu, D.; Fang, Y.; Liu, Y.; Xu, J.; Qiu, Y.; et al. A picorna-like virus suppresses the N-end rule pathway to inhibit apoptosis. eLife 2017, 6, e30590. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eldeeb, M.A.; Fahlman, R.P.; Esmaili, M.; Ragheb, M.A. Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells. Int. J. Mol. Sci. 2018, 19, 3414. https://doi.org/10.3390/ijms19113414
Eldeeb MA, Fahlman RP, Esmaili M, Ragheb MA. Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells. International Journal of Molecular Sciences. 2018; 19(11):3414. https://doi.org/10.3390/ijms19113414
Chicago/Turabian StyleEldeeb, Mohamed A., Richard P. Fahlman, Mansoore Esmaili, and Mohamed A. Ragheb. 2018. "Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells" International Journal of Molecular Sciences 19, no. 11: 3414. https://doi.org/10.3390/ijms19113414
APA StyleEldeeb, M. A., Fahlman, R. P., Esmaili, M., & Ragheb, M. A. (2018). Regulating Apoptosis by Degradation: The N-End Rule-Mediated Regulation of Apoptotic Proteolytic Fragments in Mammalian Cells. International Journal of Molecular Sciences, 19(11), 3414. https://doi.org/10.3390/ijms19113414