In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses




Abstract

1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Identifiers
4.2. In Silico Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beutler, E.; Grabowski, G.A. Gaucher disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3635–3668. [Google Scholar]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. α-Galactosidase A deficiency: Fabry disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Lieberman, R.L.; D’Aquino, J.A.; Ringe, D.; Petsko, G.A. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry 2009, 48, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Amberg, A. In Silico Methods. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays; Vogel, H.G., Maas, J., Hock, F.J., Mayer, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Tchernitchko, D.; Goossens, M.; Wajcman, H. In silico prediction of the deleterious effect of a mutation: Proceed with caution in clinical genetics. Clin. Chem. 2004, 50, 1974–1978. [Google Scholar] [CrossRef] [PubMed]
- Rigsby, R.E.; Parker, A.B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. 2016, 44, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Bromberg, Y.; Rost, B. News from the protein mutability landscape. J. Mol. Biol. 2013, 425, 3937–3948. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. 8), S1. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Amaral, O.; Marcao, A.; Sa Miranda, M.; Desnick, R.J.; Grace, M.E. Gaucher disease: Expression and characterization of mild and severe acid beta-glucosidase mutations in Portuguese type 1 patients. Eur. J. Hum. Genet. 2000, 8, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Montfort, M.; Chabas, A.; Vilageliu, L.; Grinberg, D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum. Mutat. 2004, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Ron, I.; Dagan, A.; Gatt, S.; Pasmanik-Chor, M.; Horowitz, M. Use of fluorescent substrates for characterization of Gaucher disease mutations. Blood Cells Mol. Dis. 2005, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Chabas, A.; Gort, L.; Diaz-Font, A.; Montfort, M.; Santamaria, R.; Cidras, M.; Grinberg, D.; Vilageliu, L. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: Effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol. Dis. 2005, 35, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.E.; Berg, A.; He, G.S.; Goldberg, L.; Horowitz, M.; Grabowski, G.A. Gaucher disease: Heterologous expression of two alleles associated with neuronopathic phenotypes. Am. J. Hum. Genet. 1991, 49, 646–655. [Google Scholar] [PubMed]
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of variant acid beta-glucosidases: Effects of Gaucher disease mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, G.H.; Kim, S.S.; Ko, J.M.; Lee, J.J.; Yoo, H.W. Effects of a chemical chaperone on genetic mutations in alpha-galactosidase A in Korean patients with Fabry disease. Exp. Mol. Med. 2009, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Ortiz, A.; Germain, D.P.; Viana-Baptista, M.; Caldeira-Gomes, A.; Camprecios, M.; Fenollar-Cortes, M.; Gallegos-Villalobos, A.; Garcia, D.; Garcia-Robles, J.A.; et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metab. 2015, 114, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Froissart, R.; Guffon, N.; Vanier, M.T.; Desnick, R.J.; Maire, I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Yasuda, M.; Shabbeer, J.; Benson, S.D.; Maire, I.; Burnett, R.M.; Desnick, R.J. Fabry disease: Characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum. Mutat. 2003, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, U.; Zlotogora, J.; Kafert, S.; Gieselmann, V. Multiple mutations are responsible for the high frequency of metachromatic leukodystrophy in a small geographic area. Am. J. Hum. Genet. 1995, 56, 51–57. [Google Scholar] [PubMed]
- Berná, L.; Gieselmann, V.; Poupetová, H.; Hrebícek, M.; Elleder, M.; Ledvinová, J. Novel mutations associated with metachromatic leukodystrophy: Phenotype and expression studies in nine Czech and Slovak patients. Am. J. Med. Genet. A 2004, 129A, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kafert, S.; Heinisch, U.; Wenger, D.A.; Zlotogora, J.; Gieselmann, V. Characterization of two arylsulfatase A missense mutations D335V and T274M causing late infantile metachromatic leukodystrophy. Hum. Mutat. 1996, 7, 311–317. [Google Scholar] [CrossRef]
- Marcão, A.; Amaral, O.; Pinto, E.; Pinto, R.; Sá Miranda, M.C. Metachromatic leucodystrophy in Portugal-finding of four new molecular lesions: C300F, P425T, g.1190-1191insC, and g.2408delC. Mutations in brief no. 232. Online. Hum. Mutat. 1999, 13, 337–338. [Google Scholar] [CrossRef]
- Lukatela, G.; Krauss, N.; Theis, K.; Selmer, T.; Gieselmann, V.; von Figura, K.; Saenger, W. Crystal structure of human arylsulfatase A: The aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry 1998, 37, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Kawame, H.; Ida, H.; Ohashi, T.; Eto, Y. Single exon mutation in arylsulfatase A gene has two effects: Loss of enzyme activity and aberrant splicing. Hum. Genet. 1994, 93, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; Kukita, Y.; Nagano, S.; Sakai, Y.; Yamashita, Y.; Fukuyama, H.; Inatomi, Y.; Saito, Y.; Koike, R.; Tsuji, S.; et al. Adult onset globoid cell leukodystrophy (Krabbe disease): Analysis of galactosylceramidase cDNA from four Japanese patients. Hum. Genet. 1997, 100, 450–456. [Google Scholar] [CrossRef] [PubMed]
- De Gasperi, R.; Gama Sosa, M.A.; Sartorato, E.; Battistini, S.; Raghavan, S.; Kolodny, E.H. Molecular basis of late-life globoid cell leukodystrophy. Hum. Mutat. 1999, 14, 256–262. [Google Scholar] [CrossRef]
- Fu, L.; Inui, K.; Nishigaki, T.; Tatsumi, N.; Tsukamoto, H.; Kokubu, C.; Muramatsu, T.; Okada, S. Molecular heterogeneity of Krabbe disease. J. Inherit. Metab. Dis. 1999, 22, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Duarte, A.J.; Ribeiro, D.; Chaves, J.; Amaral, O.; Alves, S. Correction of a Splicing Mutation Affecting an Unverricht-Lundborg Disease Patient by Antisense Therapy. Genes 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E. Identification of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM1). Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar] [PubMed]
- Auerswald, E.A.; Nägler, D.K.; Assfalg-Machleidt, I.; Stubbs, M.T.; Machleidt, W.; Fritz, H. Hairpin loop mutations of chicken cystatin have different effects on the inhibition of cathepsin B, cathepsin L and papain. FEBS Lett. 1995, 361, 179–184. [Google Scholar] [CrossRef]
- Joensuu, T.; Kuronen, M.; Alakurtti, K.; Tegelberg, S.; Hakala, P.; Aalto, A.; Huopaniemi, L.; Aula, N.; Michellucci, R.; Eriksson, K.; et al. Cystatin B: Mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients. Eur. J. Hum. Genet. 2007, 15, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, K.; Weber, E.; Rinne, R.; Theil, G.; de Haan, G.J.; Lindhout, D.; Salmikangas, P.; Saukko, P.; Lahtinen, U.; Lehesjoki, A.E. Loss of lysosomal association of cystatin B proteins representing progressive myoclonus epilepsy, EPM1, mutations. Eur. J. Hum. Genet. 2005, 13, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Pasmanik-Chor, M.; Ron, I.; Kolodny, E.H. The enigma of the E326K mutation in acid β-glucocerebrosidase. Mol. Genet. Metab. 2011, 104, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Freitas, J.; Duarte, A.J.; Ribeiro, I.; Ribeiro, D.; Lima, J.L.; Chaves, J.; Amaral, O. Unverricht-Lundborg disease: Homozygosity for a new splicing mutation in the cystatin B gene. Epilepsy Res. 2012, 99, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.J.; Ribeiro, D.; Chaves, J.; Amaral, O. Characterization of a rare Unverricht-Lundborg disease mutation. Mol. Genet. Metab. Rep. 2015, 4, 68–71. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.; de Souza, R.M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Parkinsons Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139 (Suppl. 1), 77–90. [Google Scholar] [CrossRef] [PubMed]
- Berge-Seidl, V.; Pihlstrøm, L.; Maple-Grødem, J.; Forsgren, L.; Linder, J.; Larsen, J.P.; Tysnes, O.B.; Toft, M. The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci. Lett. 2017, 658, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Thirumal Kumar, D.; Eldous, H.G.; Mahgoub, Z.A.; George Priya Doss, C.; Zayed, H. Computational modelling approaches as a potential platform to understand the molecular genetics association between Parkinson’s and Gaucher diseases. Metab. Brain Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouze, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Orlando, FL, USA, 7–10 October 2012; pp. 414–417. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. 1), S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gene Mutants | PROVEAN | PolyPhen-2 | SNAP2 | ExPASy | Protein Function and Structure |
|---|---|---|---|---|---|
| Prediction Expected Accuracy | |||||
| GBA1 Mutants | |||||
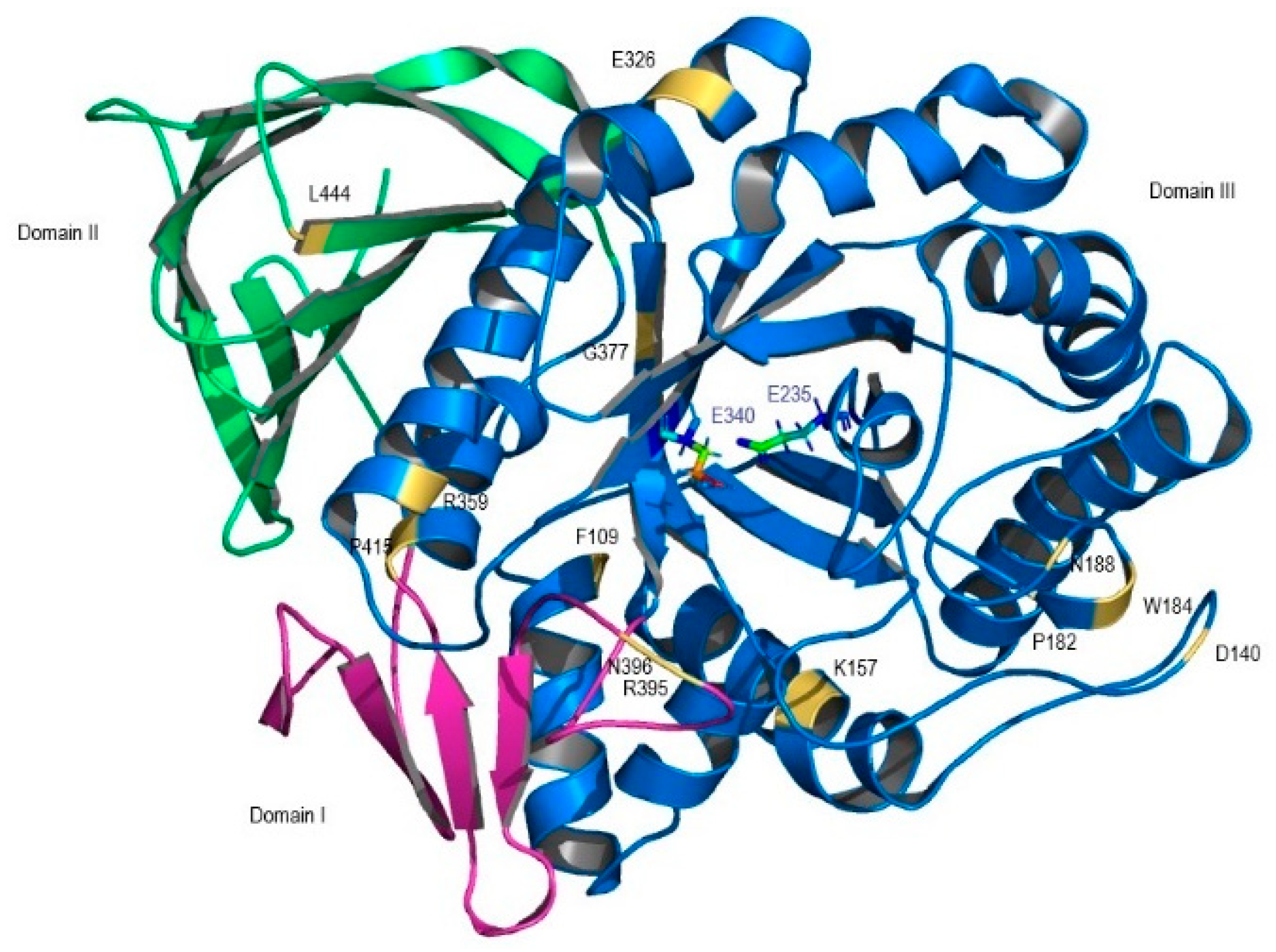
| F109V CM005404 | Deleterious | Probably damaging | Effect 75% | NA | 15% of wt activity; weakly conserved; domain III; stable protein [12] |
| P182L rs80205046 | Deleterious | Probably damaging | Effect 85% | NA | Near null activity; buried site; domain III; unstable protein [13] |
| D140H rs147138516 | Neutral | Benign | Neutral 57% | Disease | 73% of wt activity; domain III [14] |
| K157Q rs121908297 | Deleterious | Probably damaging | Neutral 57% | Disease | 9.7% of wt activity; domain III; conserved region; unstable protein [14] |
| W184R rs61748906 | Deleterious | Probably Damaging | Effect 66% | Disease | Inactive enzyme; domain III periphery; alteration of enzyme geometry; unstable protein [12] |
| N188S rs364897 | Deleterious | Benign | Effect 66% | Disease | 66.6% of wt activity; domain III periphery; stable protein [13] |
| E326K rs2230288 | Neutral | Benign | Neutral 56% | Disease | 42.7–25% of wt activity; domain III; stable protein [15] |
| R359Q rs74979486 | Deleterious | Probably Damaging | Effect 75% | Disease | 4.5% of wt activity; domain III stable protein; highly conserved region [12] |
| G377S rs121908311 | Deleterious | Probably Damaging | Effect 91% | Disease | 17% of wt activity; domain III; stable protein [12] |
| R395P | Deleterious | Benign | Effect 75% | NA | 4.5% of wt activity; domain I, loop 2; stable protein [12] |
| N396T rs75385858 | Deleterious | Probably Damaging | Effect 85% | NA | 14% of wt activity; domain I; stable protein [12] |
| P415R rs121908295 | Deleterious | Probably damaging | Effect 59% | Disease | Near null activity; conserved region; unstable protein [16] |
| L444P rs421016 | Deleterious | Possibly damaging | Effect 91% | NA | 5.7–9% of wt activity; unstable protein [12,17]. |
| GLA Mutants | |||||
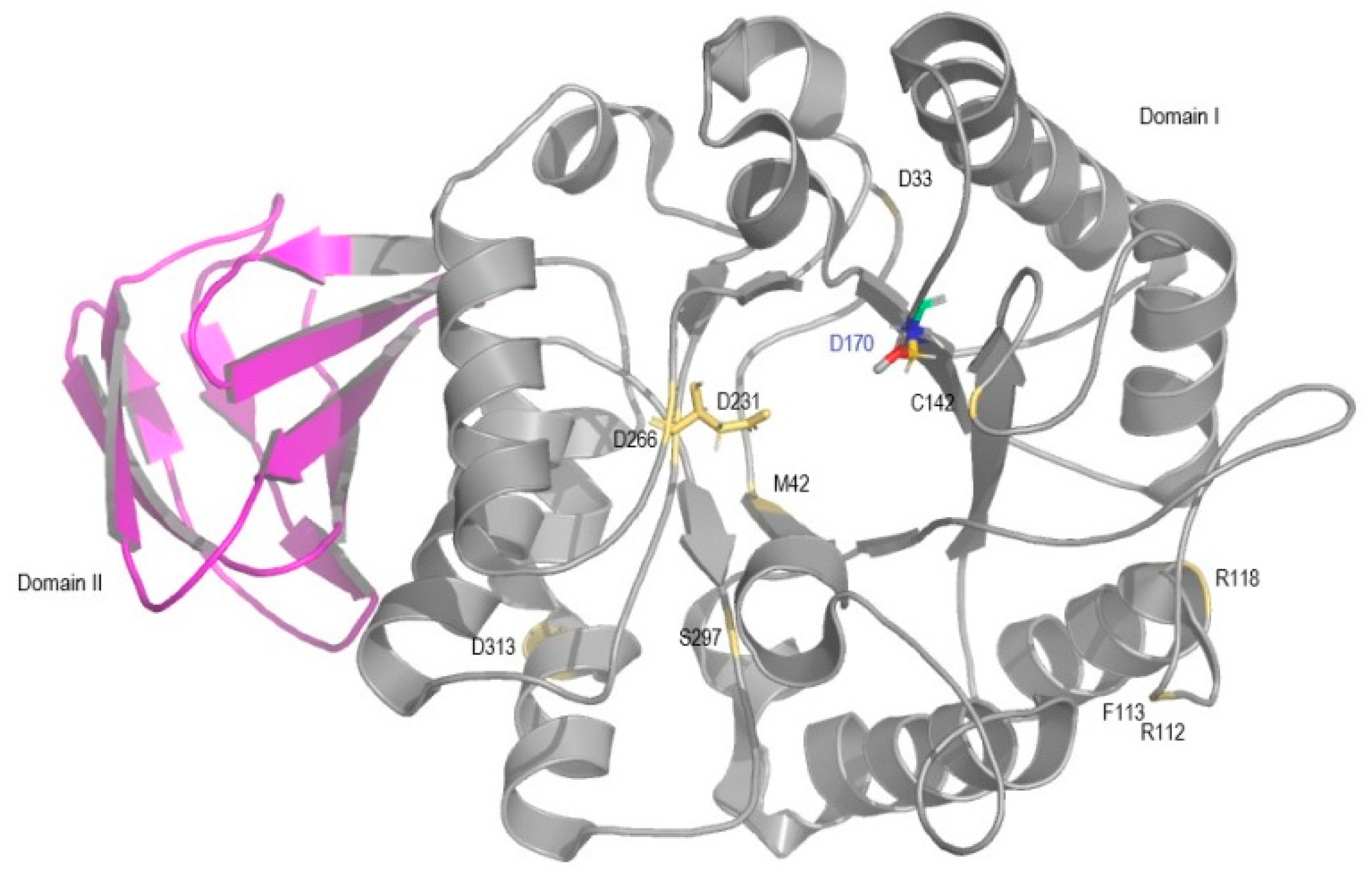
| D33G rs869312136 | Deleterious | Possibly damaging | Effect 75% | Unclassified | 37% of wt activity; periphery of domain I [18] |
| M42V | Deleterious | Probably damaging | Effect 85% | Disease | 7% of wt activity; domain I; unstable protein [19] |
| R112C rs104894834 | Deleterious | Probably damaging | Effect 91% | Disease | 5% of wt activity; periphery of domain I; unstable protein [19] |
| F113L rs869312142 | Deleterious | Probably damaging | Effect 91% | Disease | 20% of wt activity; periphery of domain I; altered alpha-GAL surface; unstable protein [19] |
| R118C rs148158093 | Deleterious | Probably Damaging | Effect 53% | NA | 29–32% of wt activity; periphery of domain I; unstable protein [20,21]. |
| C142W | Deleterious | Probably damaging | Effect 95% | NA | 5% of wt activity; domain I; near active site pocket; unstable protein [19]. |
| D231G | Deleterious | Probably damaging | Effect 95% | NA | 4% of wt activity; domain I, active site pocket; stable protein [19] |
| D266N rs869312407 | Deleterious | Probably damaging | Effect 95% | Disease | 5% of wt activity; domain I, near the active site pocket; buried; unstable protein [19] |
| S297F rs28935489 | Deleterious | Probably damaging | Effect 95% | Disease | 5% of wt activity; unstable protein [19] |
| D313Y rs28935490 | Deleterious | Probably damaging | Effect 95% | Disease | 76% of wt activity; in domain I periphery; stable protein [22,23] |
| Gene Mutants | PROVEAN | PolyPhen-2 | SNAP2 | ExPASy | Protein Function and Structure |
|---|---|---|---|---|---|
| Prediction Expected Accuracy | |||||
| ARSA mutants | |||||
| G86D rs74315460 | Deleterious | Probably damaging | Effect 95% | Disease | Null activity; unstable protein [24] |
| C156R rs199476348 | Deleterious | Probably Damaging | Effect 59% | Disease | 50% of wt activity [25] |
| T274M rs74315472 | Deleterious | Probably Damaging | Effect 95% | Disease | 35% of wt activity [26] |
| C300F rs74315484 | Deleterious | Probably Damaging | Effect 95% | Disease | Null activity; disruption of disulfide bond linking major and minor β-sheets [27,28] |
| T409I rs74315481 | Neutral | Possibly damaging | Effect 75% | Disease | 60% of wt activity [29] |
| GALC Mutants | |||||
| I82M without reference SNP (rs) | Deleterious | Probably Damaging | Neutral 57% | Disease | Normal activity [30] |
| G286D rs199847983 | Deleterious | Probably Damaging | Effect 71% | Disease | 17.5% of wt activity [31] |
| Y335C rs757407613 | Deleterious | Probably Damaging | Effect 75% | Disease | 10% of wt activity [32] |
| G553R rs748573754 | Deleterious | Probably Damaging | Effect 91% | Disease | 1.8% of wt activity [31] |
| L634S rs138577661 | Deleterious | Probably Damaging | Effect 95% | Disease | 12% of wt activity [30] |
| CSTB mutants | |||||
| Q22Q rs386833443 | Neutral | NA | Neutral 82% | NA | Expected abnormal peptide with premature truncation [33] |
| G4R rs74315443 | Deleterious | Probably Damaging | Effect 85% | Disease | Binding pocket modification; interaction properties compromised [34] |
| G50E rs312262708 | Deleterious | Possibly Damaging | Effect 95% | NA | Altered stability and interaction with target proteins [35,36] |
| Q71P rs796052392 | Deleterious | Possibly Damaging | Effect 75% | NA | Changes in second binding loop; altered binding affinities [37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, A.J.; Ribeiro, D.; Moreira, L.; Amaral, O. In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses. Int. J. Mol. Sci. 2018, 19, 3409. https://doi.org/10.3390/ijms19113409
Duarte AJ, Ribeiro D, Moreira L, Amaral O. In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses. International Journal of Molecular Sciences. 2018; 19(11):3409. https://doi.org/10.3390/ijms19113409
Chicago/Turabian StyleDuarte, Ana Joana, Diogo Ribeiro, Luciana Moreira, and Olga Amaral. 2018. "In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses" International Journal of Molecular Sciences 19, no. 11: 3409. https://doi.org/10.3390/ijms19113409
APA StyleDuarte, A. J., Ribeiro, D., Moreira, L., & Amaral, O. (2018). In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses. International Journal of Molecular Sciences, 19(11), 3409. https://doi.org/10.3390/ijms19113409