PWCDA: Path Weighted Method for Predicting circRNA-Disease Associations

Abstract
1. Introduction
2. Results and Discussion
2.1. Effect of Parameter
2.2. LOOCV
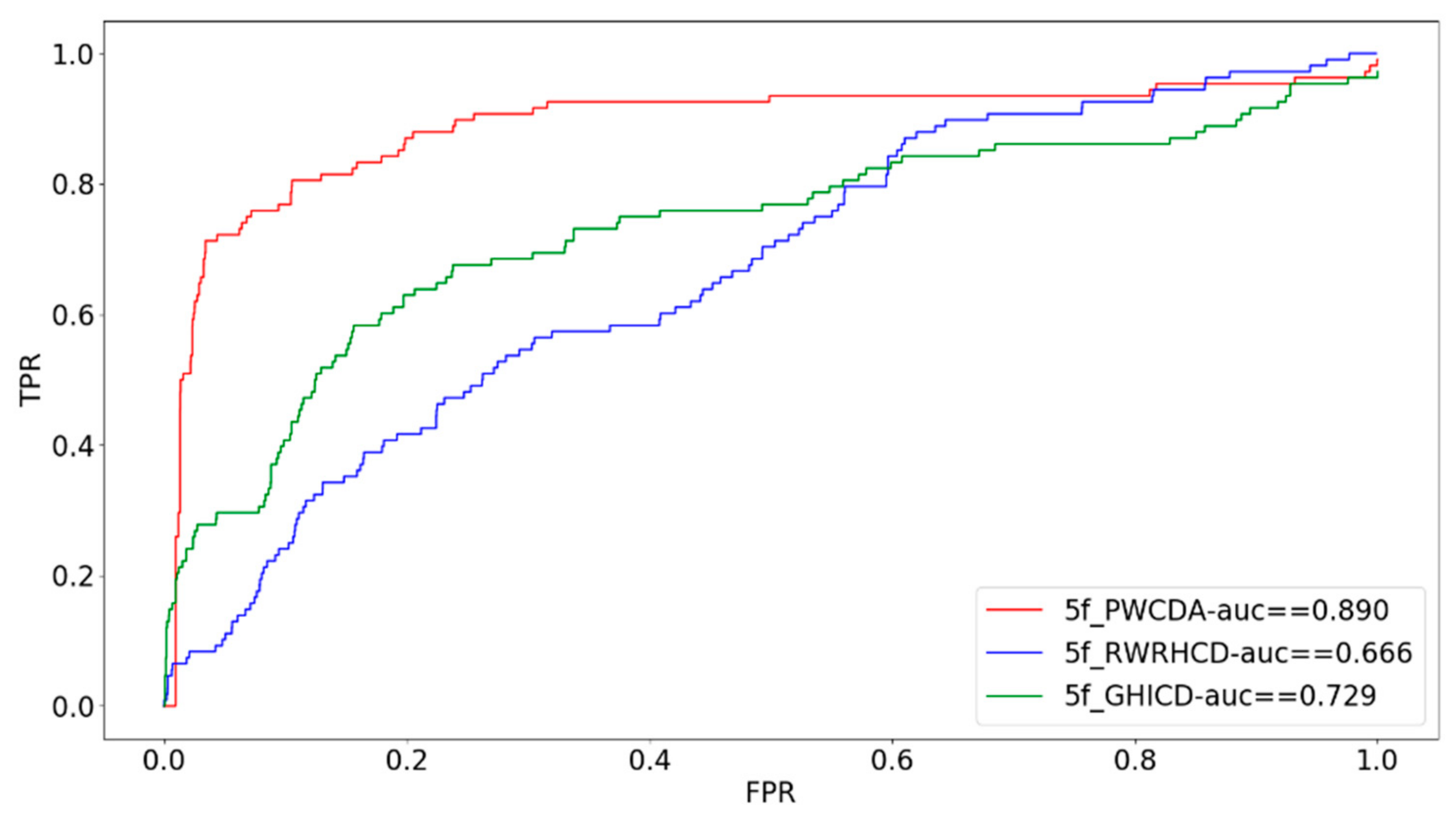
2.3. Five-Fold Cross Validation
2.4. Case Studies
3. Materials and Methods
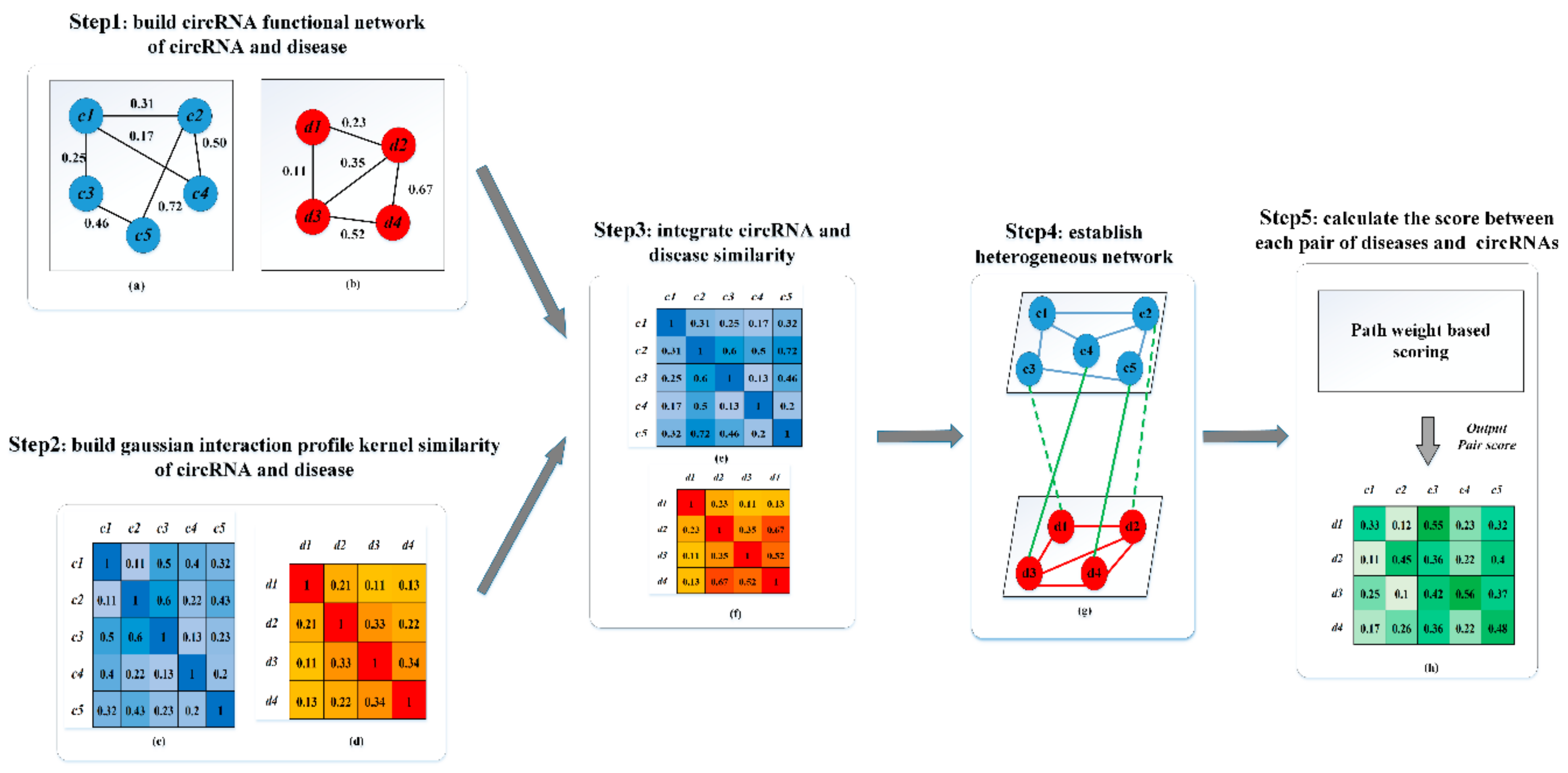
3.1. Human circRNA-Disease Associations Network
3.2. CircRNA Semantic Similarity
3.3. Disease Functional Similarity
3.4. CircRNA GIP Kernel Similarity
3.5. Disease GIP Kernel Similarity
3.6. Combine Multiple Similarity (circRNA and Disease)
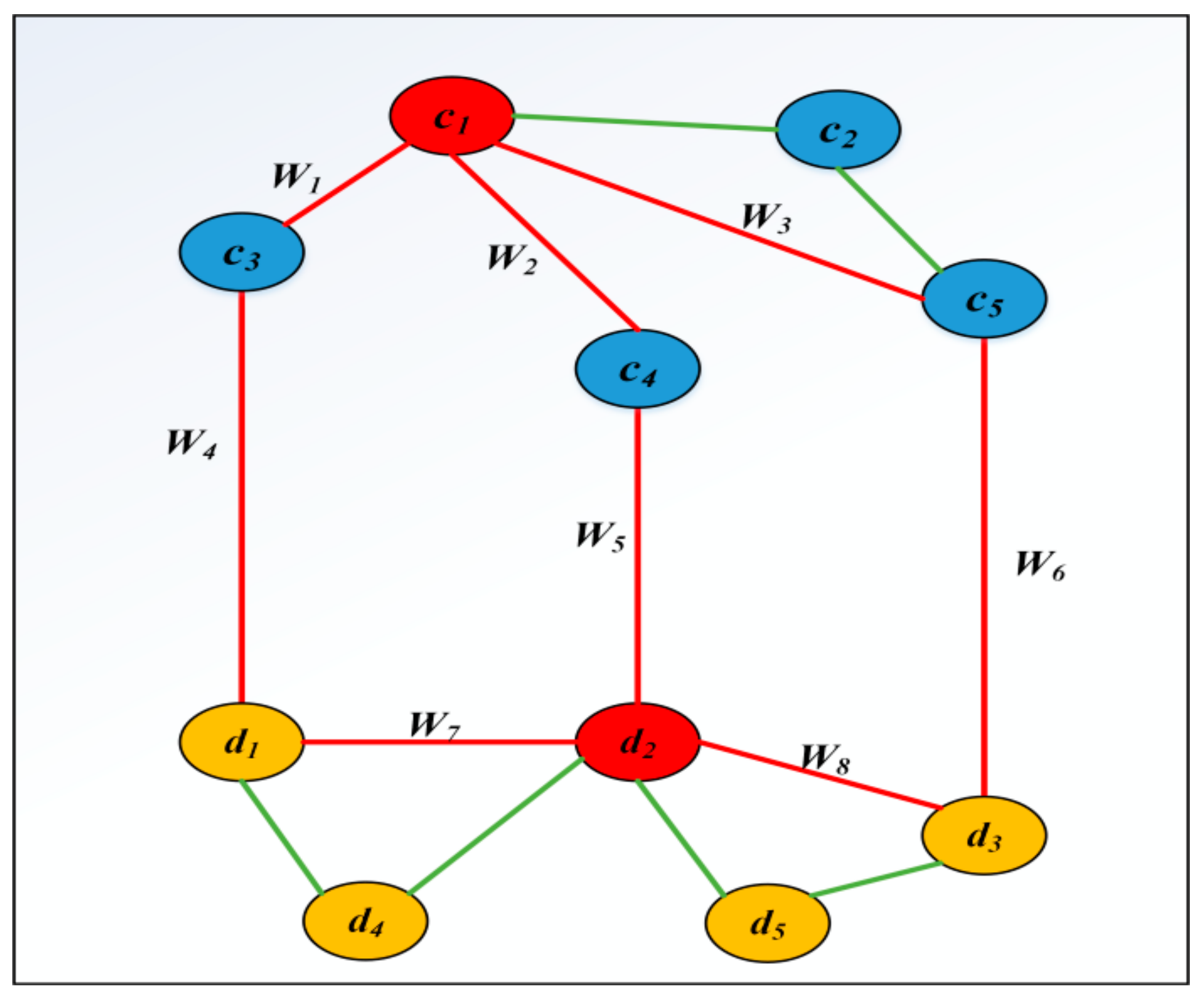
3.7. Constructing Heterogeneous Network
3.8. Perfomance Metrics
3.9. PWCDA
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| PWCDA | Path Weighed method, for predicting CircRNA-Disease Associations |
| LOOCV | leave one out cross validation |
| GIP | Gaussian Interaction Profile |
| GO | Gene Ontology |
| AUC | Area Under roc Curve |
| ROC | Receiver Operating Characteristic Curve |
| TPR | True Positive Rate |
| FPR | False Positive Rate |
References
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular Intronic Long Noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, P.J.; Zaug, A.J.; Cech, T.R. The intervening sequence of the ribosomal RNA precursor is converted to a circular RNA in isolated nuclei of Tetrahymena. Cell 1981, 23, 467–476. [Google Scholar] [CrossRef]
- Meng, S.; Zhou, H.; Feng, Z.; Xu, Z.; Tang, Y.; Li, P.; Wu, M. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 2017, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nazarali, A.J.; Ji, S. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am. J. Cancer Res. 2016, 6, 1167–1176. [Google Scholar] [PubMed]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 426. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; Bao, Y.; Yee, M.-C.; Barrett, S.P.; Hogan, G.J.; Olsen, M.N.; Dinneny, J.R.; Brown, P.O.; Salzman, J. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 2014, 9, e90859. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Fang, L.; Yang, W.; Wu, N.; Awan, F.M.; Yang, Z.; Yang, B.B. Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ. 2017, 24, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Armakola, M.; Higgins, M.J.; Figley, M.D.; Barmada, S.J.; Scarborough, E.A.; Diaz, Z.; Fang, X.; Shorter, J.; Krogan, N.J.; Finkbeiner, S.; et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 2012, 44, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, A. Circular RNAs as miRNA sponges. Nat. Rev. Mol. Cell Boil. 2013, 14, 195. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Zheng, Q.; Bao, C.; He, J.; Chen, B.; Lyu, D.; Zheng, B.; Xu, Y.; Long, Z.; et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017, 388, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Sève, P.; Reiman, T.; Dumontet, C. The role of betaIII tubulin in predicting chemoresistance in non-small cell lung cancer. Lung Cancer 2010, 67, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xu, X.; Ouyang, Y.; Wang, Y.; Yang, J.; Yin, L.; Ge, J.; Wang, H. Microarray expression profile analysis of circular RNAs in pancreatic cancer. Mol. Med. Rep. 2018, 17, 7661–7671. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Lei, X.; Fang, Z.; Jiang, Q.; Wu, F.-X. CircR2Disease: A manually curated database for experimentally supported circular RNAs associated with various diseases. Database 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Zhang, L.; Zheng, M.; Sun, X.; Lu, Y.; Liu, P. Circ2Disease: A manually curated database of experimentally validated circRNAs in human disease. Sci. Rep. 2018, 8, 11018. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Liu, B.; Yan, C. SACMDA: MiRNA-Disease Association Prediction with Short Acyclic Connections in Heterogeneous Graph. Neuroinformatics 2018, 16, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.-Y.; Huang, L. NDAMDA: Network distance analysis for MiRNA-disease association prediction. J. Cell. Mol. Med. 2018, 22, 2884–2895. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, X.; He, Z.; Zou, Q. Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Chen, Z.; Deng, L. Integrating Multiple Heterogeneous Networks for Novel LncRNA-disease Association Inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Wang, J.; Domeniconi, C.; Yu, G. Matrix factorization-based data fusion for the prediction of lncRNA-disease associations. Bioinformatics 2018, 34, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, N.; Chen, P.; Zhang, J.; Wang, B. DrugECs: An Ensemble System with Feature Subspaces for Accurate Drug-Target Interaction Prediction. Biomed. Res. Int. 2017, 2017, 6340316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, Y.; Li, D. Drug-Target Interaction Prediction through Label Propagation with Linear Neighborhood Information. Molecules 2017, 22, 2056. [Google Scholar] [CrossRef] [PubMed]
- Ba-Alawi, W.; Soufan, O.; Essack, M.; Kalnis, P.; Bajic, V.B. DASPfind: New efficient method to predict drug-target interactions. J. Cheminform. 2016, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Patra, J.C. Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics (Oxf. Engl.) 2010, 26, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H.; Huang, Y.-A.; Yan, G.-Y. HGIMDA: Heterogeneous graph inference for miRNA-disease association prediction. Oncotarget 2016, 7, 65257–65269. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Y.; Xu, J.; Bai, W.; Ren, X.; Wu, H. CircRNAs as biomarkers of cancer: A meta-analysis. BMC Cancer 2018, 18, 303. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Grammatikakis, I.; Kim, K.M.; De, S.; Martindale, J.L.; Munk, R.; Yang, X.; Abdelmohsen, K.; Gorospe, M. Identification of senescence-associated circular RNAs (SAC-RNAs) reveals senescence suppressor CircPVT1. Nucleic Acids Res. 2017, 45, 4021–4035. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, L.; Su, Y.; Zhang, X. Screening potential biomarkers for colorectal cancer based on circular RNA chips. Oncol. Rep. 2018, 39, 2499–2512. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, K.; Wu, F.; Wang, W.; Zhang, K.; Hu, H.; Liu, Y.; Jiang, T. circRNA disease: A manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis. 2018, 9, 475. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Baehner, F.; Dabbs, D.J.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Breast cancer prognostic classification in the molecular era: The role of histological grade. Breast Cancer Res. 2010, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Lan, F.; Ouyang, X.; Wang, K.; Lin, Y.; Yu, Y.; Wang, L.; Wang, Y.; Huang, Q. Expression and clinical significance of long non-coding RNA HNF1A-AS1 in human gastric cancer. World J. Surg. Oncol. 2015, 13, 302. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Ouyang, X.; Zhang, F.; Wang, K.; Lin, Y.; Sun, B.; Wang, Y.; Wang, L.; Huang, Q. Circular RNAs expression profiles in human gastric cancer. Sci. Rep. 2017, 7, 9060. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, J.; Zhao, J.; Wang, X. Silencing of hsa_circ_0007534 suppresses proliferation and induces apoptosis in colorectal cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 118–126. [Google Scholar] [PubMed]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Price, T.; Peña, F.I.; Cho, Y.-R. Survey: Enhancing protein complex prediction in PPI networks with GO similarity weighting. Interdiscip. Sci. 2013, 5, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, T.; Pakhomov, S.V.S.; Patwardhan, S.; Chute, C.G. Measures of semantic similarity and relatedness in the biomedical domain. J. Biomed. Inf. 2007, 40, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Mina, M.; Guerra, C.; Cannataro, M. Semantic similarity analysis of protein data: Assessment with biological features and issues. Brief. Bioinform. 2012, 13, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-A.; Chan, K.C.C.; You, Z.-H. Constructing prediction models from expression profiles for large scale lncRNA-miRNA interaction profiling. Bioinformatics 2018, 34, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Queralt-Rosinach, N.; Bravo, A.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Oyston, J. Online Mendelian Inheritance in Man. Anesthesiology 1998, 89, 811–812. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.-X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Gonçalves, J.P.; Larminie, C.; Przulj, N. Predicting disease associations via biological network analysis. BMC Bioinform. 2014, 15, 304. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, M.; Shi, H.; Ju, H.; Jiang, Q.; Cheng, L. Measuring disease similarity and predicting disease-related ncRNAs by a novel method. BMC Med. Genom. 2017, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Jiang, Y.; Wang, Z.; Shi, H.; Sun, J.; Yang, H.; Zhang, S.; Hu, Y.; Zhou, M. DisSim: An online system for exploring significant similar diseases and exhibiting potential therapeutic drugs. Sci. Rep. 2016, 6, 30024. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhao, L.; Liu, Z.; Ju, H.; Shi, H.; Xu, P.; Wang, Y.; Cheng, L. DisSetSim: An online system for calculating similarity between disease sets. J. Biomed. Semant. 2017, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.-Y. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, A.; Feng, H.; Wang, M. NTSMDA: Prediction of miRNA-disease associations by integrating network topological similarity. Mol. Biosyst. 2016, 12, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 2015, 5, 16840. [Google Scholar] [CrossRef] [PubMed]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zeng, T.; Liu, X.; Chen, L. Diagnosing phenotypes of single-sample individuals by edge biomarkers. J. Mol. Cell Biol. 2015, 7, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, J.; Sun, S.; Zhou, X.; Zeng, T.; Chen, L. Individual-specific edge-network analysis for disease prediction. Nucleic Acids Res. 2017, 45, e170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhou, Y.; Zhang, X.J.; Chen, L. Part mutual information for quantifying direct associations in networks. Proc. Natl. Acad. Sci. USA 2016, 113, 5130–5135. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, R.; Liu, Z.P.; Li, M.; Aihara, K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2012, 2, 342. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, M.; Tang, W.; Liu, W.; Zhang, S.; Chen, L.; Xia, J. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat. Commun. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.; Liu, W.; Liu, C.; Cui, J.; Li, Q.; Ni, H.; Yang, Y.; Wu, C.; Chen, C.; et al. Dysfunction of PLA2G6 and CYP2C44 associated network signals imminent carcinogenesis from chronic inflammation to hepatocellular carcinoma. J. Mol. Cell Biol. 2018, 9, 489–503. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α | 0.5 | 1 | 1.5 | 2 | 3 | 3.5 | 4 | 4.5 | 5 |
| AUC | 0.97100 | 0.97209 | 0.97206 | 0.97208 | 0.97202 | 0.97010 | 0.97010 | 0.97010 | 0.96879 |
| γ | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 | 0.6 |
| AUC | 0.96483 | 0.96483 | 0.96483 | 0.96500 | 0.97209 | 0.97205 |
| Breast Cancer | |||||
|---|---|---|---|---|---|
| Rank | circRNA Name/id | Evidences | Rank | circRNA Name/id | Evidences |
| 1 | circpvt1/hsa_circ_0001821 | PMID:279280058 | 16 | hsa_circ_0001667 | circRNAdisease |
| 2 | circ-foxo3 | circRNAdisease | 17 | hsa_circ_0085495 | circRNAdisease |
| 3 | hsa_circ_0001313/circccdc66 | PMID:28249903 | 18 | hsa_circ_0086241 | circRNAdisease |
| 4 | hsa_circ_0007534 | PMID:29593432 | 19 | hsa_circ_0092276 | circRNAdisease |
| 5 | hsa_circ_0000284/circhipk3 | PMID:27050392 | 20 | hsa_circ_0003838 | circRNAdisease |
| 6 | hsa_circ_0011946 | PMID:29593432 | 21 | circvrk1 | PMID:29221160 |
| 7 | hsa_circ_0093869 | PMID: 29593432 | 22 | circbrip | PMID: 29221160 |
| 8 | hsa_circ_0001982 | circRNAdisease | 23 | circola | PMID: 29221160 |
| 9 | hsa_circ_0001785 | circRNAdisease | 24 | circetfa | PMID: 29221160 |
| 10 | hsa_circ_0108942 | circRNAdisease | 25 | circmed13 | PMID: 29221160 |
| 11 | hsa_circ_0068033 | circRNAdisease | 26 | circbc111b | PMID:28739726 |
| 12 | circamot11/hsa_circ_0004214 | circRNAdisease | 27 | circdennd4c | circRNAdisease |
| 13 | hsa_circ_0006528 | circRNAdisease | 28 | hsa_circ_103110/hsa_circ_0004771 | circRNAdisease |
| 14 | hsa_circ_0002113 | circRNAdisease | 29 | hsa_circ_104689/hsa_circ_0001824 | unconfirmed |
| 15 | hsa_circ_0002874 | circRNAdisease | 30 | hsa_circ_104821/hsa_circ_0001875 | circRNAdisease |
| Gastric Cancer | |||||
|---|---|---|---|---|---|
| Rank | circRNA Name/id | Evidences | Rank | circRNA Name/id | Evidences |
| 1 | hsa_circ_0076305 | circRNAdisease | 16 | circma0138960/hsa-circma7690-15 | circRNAdisease |
| 2 | hsa_circ_0076304 | circRNAdisease | 17 | hsa_circ_0000181 | circRNAdisease |
| 3 | circpvt1/hsa_circ_0001821 | circRNAdisease | 18 | hsa_circ_0000745 | circRNAdisease |
| 4 | hsa_circ_0001649 | unconfirmed | 19 | hsa_circ_0085616 | circRNAdisease |
| 5 | hsa_circ_0000284/circhipk3 | unconfirmed | 20 | hsa_circ_0006127 | circRNAdisease |
| 6 | hsa_circ_0014717 | circRNAdisease | 21 | hsa_circ_0000026 | circRNAdisease |
| 7 | cdr1as/cirs-7/hsa_circ_0001946 | unconfirmed | 22 | hsa_circ_0000144 | circRNAdisease |
| 8 | hsa_circ_0003195 | circRNAdisease | 23 | hsa_circ_0032821 | circRNAdisease |
| 9 | hsa_circ_0000520 | circRNAdisease | 24 | hsa_circ_0005529 | circRNAdisease |
| 10 | hsa_circ_0074362 | circRNAdisease | 25 | hsa_circ_0061274 | circRNAdisease |
| 11 | hsa_circ_0001017 | circRNAdisease | 26 | hsa_circ_0005927 | circRNAdisease |
| 12 | hsa_circ_0061276 | circRNAdisease | 27 | hsa_circ_0092341 | circRNAdisease |
| 13 | circ-zfr | unconfirmed | 28 | hsa_circ_0001561 | unconfirmed |
| 14 | circma0047905/hsa_circ_0047905 | circRNAdisease | 29 | circlarp4 | circRNAdisease |
| 15 | circma0138960/hsa_circ_0138960 | circRNAdisease | 30 | hsa_circ_0035431 | circRNAdisease |
| Colorectal Cancer | |||||
|---|---|---|---|---|---|
| Rank | circRNA Name/id | Evidences | Rank | circRNA Name/id | Evidences |
| 1 | hsa_circ_0001649 | PMID:29421663 | 16 | has-circ_0006174 | circRNAdisease |
| 2 | hsa_circ_0007534 | PMID:29364478 | 17 | hsa_circ_0008509 | circRNAdisease |
| 3 | cdr1as/cirs-7/hsa_circ_0001946 | circRNAdisease | 18 | hsa_circ_0084021 | circRNAdisease |
| 4 | hsa_circ_0000284/circhipk3 | PMID:27050392 | 19 | circ_banp | circRNAdisease |
| 5 | hsa_circ_0001313/circccdc66 | circRNAdisease | 20 | hsa_circrna_103809 | circRNAdisease |
| 6 | ciritch/hsa_circ_0001141/hsa_circ_001763 | unconfirmed | 21 | hsa_circrna_104700 | circRNAdisease |
| 7 | hsa_circ_0014717 | PMID:29571246 | 22 | hsa_circ_0000069 | circRNAdisease |
| 8 | hsa_circ_0000567 | PMID:29333615 | 23 | hsa_circ_001988/hsa_circ_0001451 | circRNAdisease |
| 9 | hsa_circ_000984/hsa_circ_0001724 | circRNAdisease | 24 | hsa_circ_0000677/hsa_circ_001569/circabcc | circRNAdisease |
| 10 | hsa_circ_0020397 | circRNAdisease | 25 | circ_kldhc10/hsa_circ_0082333 | PMID:26138677 |
| 11 | hsa_circ_0007031 | circRNAdisease | 26 | circ_stxbp51 | unconfirmed |
| 12 | hsa_circ_0000504 | circRNAdisease | 27 | circ-shkbp1 | unconfirmed |
| 13 | hsa_circ_0007006 | circRNAdisease | 28 | circ-fbxw7 | unconfirmed |
| 14 | hsa_circ_0074930 | circRNAdisease | 29 | hsa_circ_0046701 | unconfirmed |
| 15 | hsa_circ_0048232 | circRNAdisease | 30 | circttbk2/hsa_circ_0000594 | unconfirmed |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.; Fang, Z.; Chen, L.; Wu, F.-X. PWCDA: Path Weighted Method for Predicting circRNA-Disease Associations. Int. J. Mol. Sci. 2018, 19, 3410. https://doi.org/10.3390/ijms19113410
Lei X, Fang Z, Chen L, Wu F-X. PWCDA: Path Weighted Method for Predicting circRNA-Disease Associations. International Journal of Molecular Sciences. 2018; 19(11):3410. https://doi.org/10.3390/ijms19113410
Chicago/Turabian StyleLei, Xiujuan, Zengqiang Fang, Luonan Chen, and Fang-Xiang Wu. 2018. "PWCDA: Path Weighted Method for Predicting circRNA-Disease Associations" International Journal of Molecular Sciences 19, no. 11: 3410. https://doi.org/10.3390/ijms19113410
APA StyleLei, X., Fang, Z., Chen, L., & Wu, F.-X. (2018). PWCDA: Path Weighted Method for Predicting circRNA-Disease Associations. International Journal of Molecular Sciences, 19(11), 3410. https://doi.org/10.3390/ijms19113410