Roles of Tristetraprolin in Tumorigenesis



Abstract
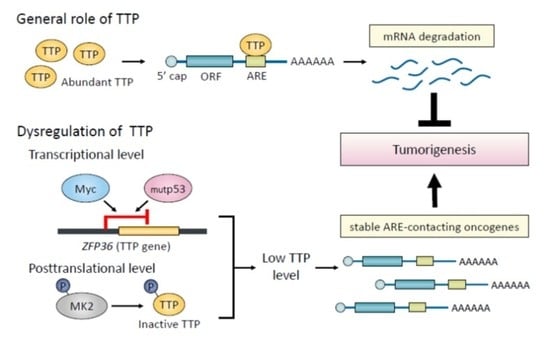
1. Introduction
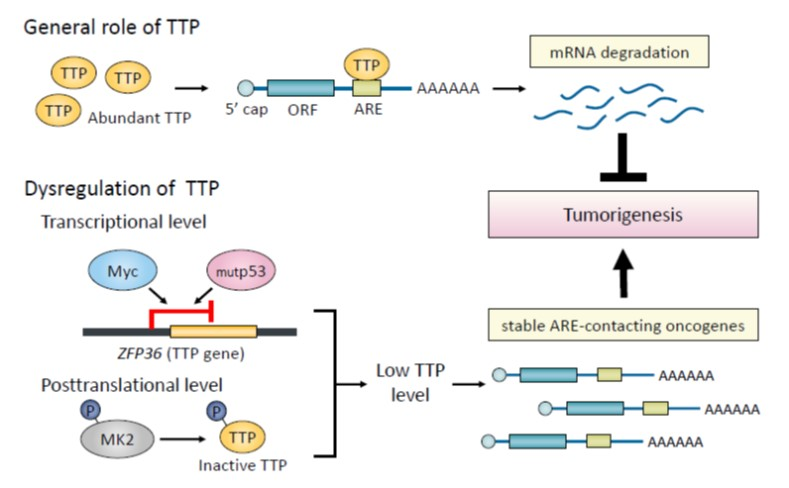
2. Oncogenes and Tumor Suppressor Genes Subjected to TTP-Mediated mRNA Decay
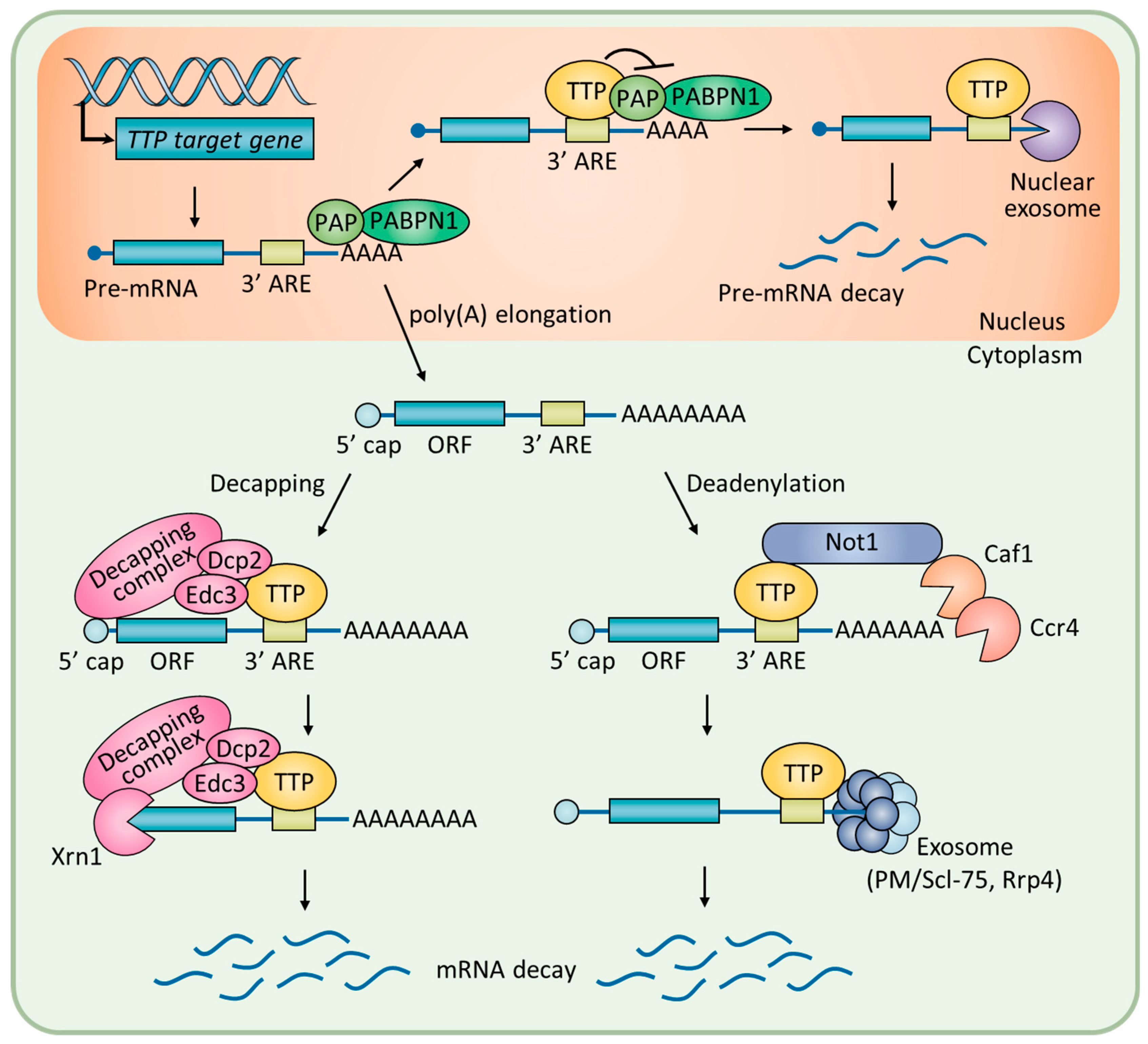
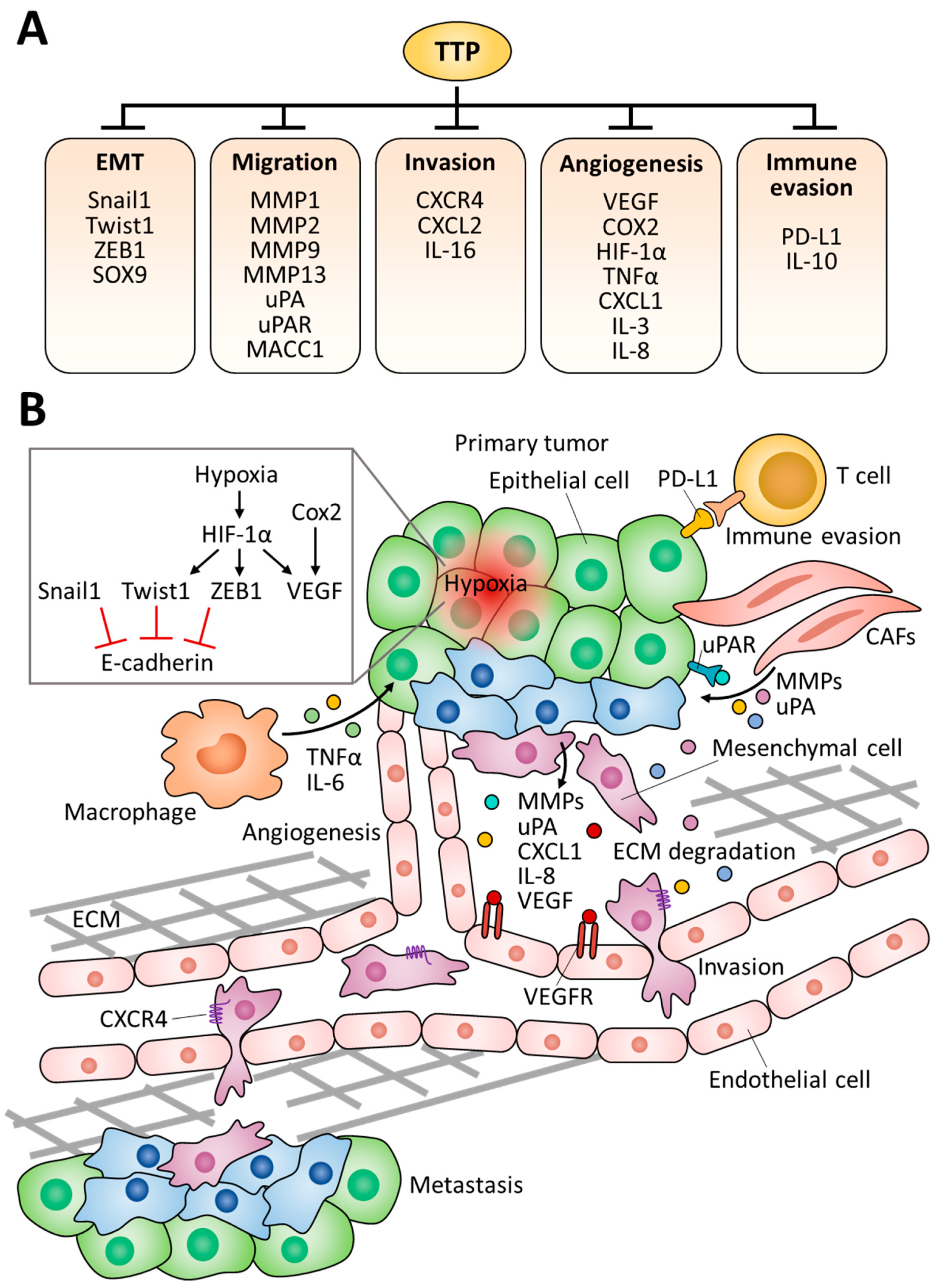
3. Roles of TTP in Tumor Progression
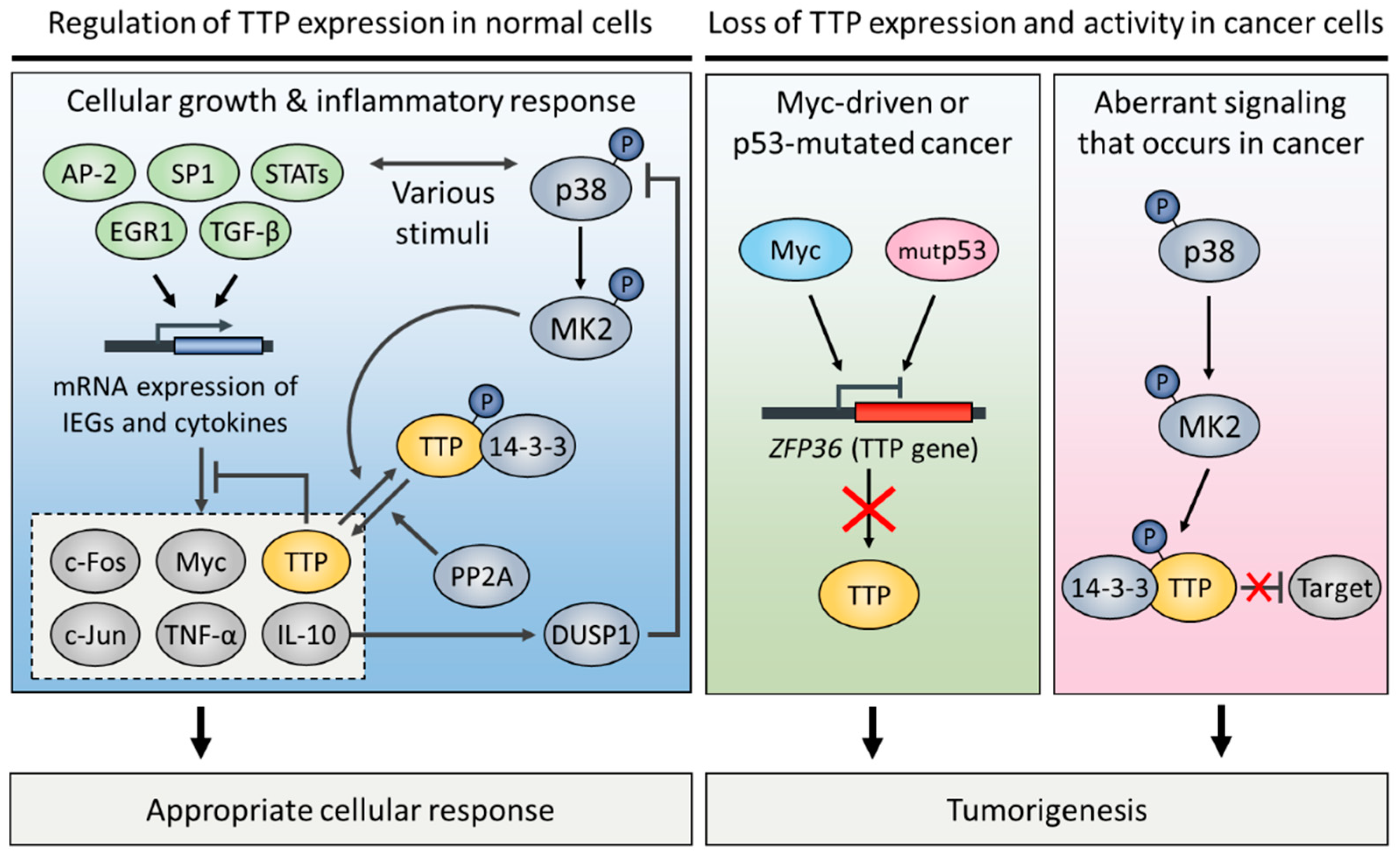
4. Regulation of TTP Expression in Normal and Cancer Cells
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| 3′ UTR | 3′ untranslated regions |
| AHRR | Aryl-hydrocarbon receptor repressor |
| AKT-1 | AKT serine/threonine kinase 1 |
| AP-1 | Activator protein 1 |
| AR | Androgen receptor |
| AREs | Adenosine and uridine-rich elements |
| BCL2 | B-cell CLL/Lymphoma 2 |
| BIRC3 | Baculoviral IAP repeat containing 3 |
| B-Raf | v-Raf murine sarcoma viral oncogene homolog B |
| CAFs | Carcinoma-associated fibroblasts |
| CCCH | Cysteine–cysteine-cysteine–histidine |
| CCNB1 | Cyclin B1 |
| CCND1 | Cyclin D1 |
| Ccr4 | Carbon catabolite repressor protein 4 |
| CDC25A | Cell division cycle 25 homolog A |
| CDK4 | Cyclin dependent kinase 4 |
| CDK6 | Cyclin dependent kinase 6 |
| CDKN1A | Cyclin dependent kinase inhibitor 1A |
| COX-2 | Cyclooxygenase 2 |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL2 | C-X-C motif chemokine ligand 2 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| Dcp2 | mRNA-decapping enzyme 2 |
| DUSP1 | Dual specificity phosphatase 1 |
| E2F1 | E2F transcription factor 1 |
| ECM | Extracellular matrix |
| EGR1 | Early growth response protein 1 |
| Edc3 | Enhancer of mRNA-decapping protein 3 |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinases |
| ERα | Estrogen receptor alpha |
| FOS | FBJ murine osteosarcoma viral oncogene homolog |
| GOS24 | G0/G1 switch regulatory protein 24 |
| GR | Glucocorticoid receptor |
| HDAC | Histone deacetylase |
| HIF-1α | Hypoxia-inducible factor 1α |
| HMGA2 | High mobility group A2 |
| IAP | Inhibitors of apoptosis proteins |
| IFN-γ | Interferon gamma |
| IL-6 | Interleukin-6 |
| iNOS | inducible nitric oxide synthase |
| Inr | Initiator element |
| JAK1 | Janus kinase 1 |
| JNK | c-Jun N-terminal kinases |
| JUN | v-jun avian sarcoma virus 17 oncogene homolog |
| KSRP | KH-type splicing regulatory protein |
| LATS2 | Large tumor suppressor kinase 2 |
| Lin28A | Lin-28 homolog A |
| MACC1 | Metastasis associated in colon cancer 1 |
| MAPK | Mitogen-activated protein kinase |
| miR-29a | MicroRNA-29a |
| MK2 | MAPK-activated protein kinase 2 |
| MMP-13 | Matrix metalloproteinase 13 |
| MMP-2 | Matrix metalloproteinase 2 |
| MMP-9 | Matrix metalloproteinase 9 |
| mRNA | Messenger RNAs |
| Myc | v-myc avian myelocytomatosis viral oncogene homolog |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Not1 | Negative on TATA 1 |
| NUP475 | Growth factor-inducible nuclear protein NUP475 |
| PABPN1 | Poly-A-binding protein nuclear 1 |
| PD-L1 | Programmed death-ligand 1 |
| PIM-1 | Proto-oncogene serine/threonine-protein kinase pim 1 |
| PM/Scl-75 | Polymyositis/systemic sclerosis 75 |
| PP2A | Protein phosphatase 2A |
| PR | Progesterone receptor |
| RB | Retinoblastoma 1 |
| Rrp4 | Ribosomal RNA processing 4 |
| SNAI1 | Zinc finger protein snail 1 |
| SOX9 | Sex-determining region Y box 9 |
| SP-1 | Specificity protein 1 |
| SRC-1 | Steroid receptor coactivator 1 |
| STAT | Signal transducer and activator of transcription |
| Tcf | T-cell factor |
| TGF-β1 | Transforming growth factor beta 1 |
| TIS | 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced sequence |
| TNF-α | Tumor necrosis factor alpha |
| TTP | Tristetraprolin |
| Twist1 | Twist-related protein 1 |
| uPA | Urokinase-type plasminogen activator |
| uPAR | Urokinase plasminogen activator receptor |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptors |
| XIAP | X-linked inhibitor of apoptosis |
| Xrn1 | 5′-3′ exoribonuclease |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
| ZFP36 | Zinc finger protein 36 |
References
- Guhaniyogi, J.; Brewer, G. Regulation of mRNA stability in mammalian cells. Gene 2001, 265, 11–23. [Google Scholar] [CrossRef]
- Park, J.M.; Kang, T.H. Transcriptional and posttranslational regulation of nucleotide excision repair: The guardian of the genome against ultraviolet radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef] [PubMed]
- Sanduja, S.; Blanco, F.F.; Dixon, D.A. The roles of TTP and BRF proteins in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 2011, 2, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.G.; Blackshear, P.J. RNA-binding proteins in immune regulation: A focus on ccch zinc finger proteins. Nat. Rev. Immunol. 2017, 17, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.P.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the mRNA AU-rich element by the zinc finger domain of tis11d. Nat. Struct. Mol. Biol. 2004, 11, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.S.; Carballo, E.; Thorn, J.M.; Kennington, E.A.; Blackshear, P.J. Interactions of CCCH zinc finger proteins with mRNA—Binding of tristetraprolin-related zinc finger proteins to au-rich elements and destabilization of mRNA. J. Biol. Chem. 2000, 275, 17827–17837. [Google Scholar] [CrossRef] [PubMed]
- Sandler, H.; Kreth, J.; Timmers, H.T.M.; Stoecklin, G. Not1 mediates recruitment of the deadenylase CAF1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res. 2011, 39, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.L.; Wang, S.C.; Chiang, P.Y.; Lin, N.Y.; Shen, Y.F.; Chang, G.D.; Chang, C.J. Tristetraprolin inhibits poly(a)-tail synthesis in nuclear mRNA that contains AU-rich elements by interacting with poly(a)-binding protein nuclear 1. PLoS ONE 2012, 7, e41313. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Fenger-Gron, M.; Fillman, C.; Norrild, B.; Lykke-Andersen, J. Multiple processing body factors and the are binding protein ttp activate mRNA decapping. Mol. Cell 2005, 20, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.A.; Carballo, E.; Lee, D.M.; Lai, W.S.; Thompson, M.J.; Patel, D.D.; Schenkman, D.I.; Gilkeson, G.S.; Broxmeyer, H.E.; Haynes, B.F.; et al. A pathogenetic role for tnf alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996, 4, 445–454. [Google Scholar] [CrossRef]
- Probert, L.; Akassoglou, K.; Alexopoulou, L.; Douni, E.; Haralambous, S.; Hill, S.; Kassiotis, G.; Kontoyiannis, D.; Pasparakis, M.; Plows, D.; et al. Dissection of the pathologies induced by transmembrane and wild-type tumor necrosis factor in transgenic mice. J. Leukoc. Biol. 1996, 59, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of tnf biosynthesis in mice lacking tnf AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef]
- Lai, W.S.; Carballo, E.; Strum, J.R.; Kennington, E.A.; Phillips, R.S.; Blackshear, P.J. Evidence that tristetraprolin binds to au-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 1999, 19, 4311–4323. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Fallmann, J.; Kratochvill, F.; Kovarik, P.; Hofacker, I.L. Aresite: A database for the comprehensive investigation of au-rich elements. Nucleic Acids Res. 2011, 39, D66–D69. [Google Scholar] [CrossRef] [PubMed]
- Khabar, K.S.A. Hallmarks of cancer and AU-rich elements. Wiley Interdiscip. Rev. RNA 2017, 8, 1368. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, L.S.; Keene, J.D. RNA regulons in cancer and inflammation. Curr. Opin. Genet. Dev. 2018, 48, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Blackshear, P.J. Tristetraprolin (TTP): Interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Qu, H.H.; Chen, Y.; Xia, J.Z. The role of RNA-binding protein tristetraprolin in cancer and immunity. Med. Oncol. 2017, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, N.N.; Guo, J.; Xia, J.Z.; Ruan, Y.L. Dysregulation of TTP and hur plays an important role in cancers. Tumor Biol. 2016, 37, 14451–14461. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Vo, M.T.; Kim, H.J.; Lee, U.H.; Kim, C.W.; Kim, H.K.; Ko, M.S.; Lee, W.H.; Cha, S.J.; Min, Y.J.; et al. Stability of the lats2 tumor suppressor gene is regulated by tristetraprolin. J. Biol. Chem. 2010, 285, 17329–17337. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Kim, W.T.; Kim, D.H.; Park, J.W.; Kang, T.H.; Chung, J.W.; Leem, S.H. Tristetraprolin suppresses ahrr expression through mRNA destabilization. FEBS Lett. 2013, 587, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Al-Haj, L.; Blackshear, P.J.; Khabar, K.S.A. Regulation of P21/CIP1/WAF-1 mediated cell-cycle arrest by rnase l and tristetraprolin, and involvement of au-rich elements. Nucleic Acids Res. 2012, 40, 7739–7752. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Jacobs, N.C.; Hafner, M.; Kennington, E.A.; Nusbaum, J.D.; Tuschl, T.; Blackshear, P.J.; Ohler, U. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.D.H.; Koch, A.; Allister, A.; Saran, S.; Ewald, F.; Koch, M.; Nashan, B.; Tamura, T. Treatment with mapkap2 (MK2) inhibitor and DNA methylation inhibitor, 5-aza dc, synergistically triggers apoptosis in hepatocellular carcinoma (HCC) via tristetraprolin (TTP). Cell. Signal. 2016, 28, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Lee, J.H.; Jeong, W.W.; Kim, Y.H.; Cha, H.J.; Joe, Y.; Chung, H.T.; Cho, W.J.; Do, J.W.; Lee, B.J.; et al. TTP mediates cisplatin-induced apoptosis of head and neck cancer cells by down-regulating the expression of Bcl-2. J. Chemother. 2015, 27, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Kim, H.K.; Vo, M.T.; Lee, H.H.; Kim, H.J.; Min, Y.J.; Cho, W.J.; Park, J.W. Tristetraprolin controls the stability of CIAP2 mRNA through binding to the 3′ UTR of CIAP2 mRNA. Biochem. Biophys. Res. Commun. 2010, 400, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Jin, H.; Kim, W.T.; Kim, W.J.; Kim, S.Z.; Leem, S.H.; Kim, S.M. Tristetraprolin activation by resveratrol inhibits the proliferation and metastasis of colorectal cancer cells. Int. J. Oncol. 2018, 53, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.T.; Xiao, X.Q. Sodium butyrate down-regulates tristetraprolin-mediated cyclin b1 expression independent of the formation of processing bodies. Int. J. Biochem. Cell Biol. 2015, 69, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Marderosian, M.; Sharma, A.; Funk, A.P.; Vartanian, R.; Masri, J.; Jo, O.D.; Gera, J.F. Tristetraprolin regulates Cyclin D1 and c-Myc mRNA stability in response to rapamycin in an AKT-dependent manner via p38 MAPK signaling. Oncogene 2006, 25, 6277–6290. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Biswas, R.; Novotny, M.; Pavicic, P.G.; Herjan, T.; Mandal, P.; Hamilton, T.A. Tristetraprolin regulates cxcl1 (KC) mRNA stability. J. Immunol. 2008, 180, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Jalonen, U.; Nieminen, R.; Vuolteenaho, K.; Kankaanranta, H.; Moilanen, E. Down-regulation of tristetraprolin expression results in enhanced IL-12 and MIP-2 production and reduced MIP-3alpha synthesis in activated macrophages. Mediat. Inflamm. 2006, 2006, 40691. [Google Scholar] [CrossRef] [PubMed]
- Al-Souhibani, N.; Al-Ghamdi, M.; Al-Ahmadi, W.; Khabar, K.S.A. Posttranscriptional control of the chemokine receptor CXCR4 expression in cancer cells. Carcinogenesis 2014, 35, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Lee, S.R.; Leem, S.H. Tristetraprolin regulates prostate cancer cell growth through suppression of E2F1. J. Microbiol. Biotechnol. 2014, 24, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Citri, A.; Shay, T.; Lu, Y.L.; Katz, M.; Zhang, F.; Tarcic, G.; Siwak, D.; Lahad, J.; Jacob-Hirsch, J.; et al. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 2007, 39, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, F.Q.; Yang, Z.; Zhao, M.; Zhang, S.Q.; Gao, Y.; Feng, J.Y.; Yang, G.; Zhang, W.Y.; Ye, L.H.; et al. The fragment HMGA2-sh-3p20 from HMGA2 mRNA 3′UTR promotes the growth of hepatoma cells by upregulating HMGA2. Sci. Rep. 2017, 7, 2070. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, R.L.; SternJohn, J.R.; Rattenbacher, B.; Vlasova, I.A.; Williams, D.A.; Hau, H.H.; Blackshear, P.J.; Bohjanen, P.R. Tristetraprolin mediates interferon-gamma mRNA decay. J. Biol. Chem. 2009, 284, 11216–11223. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ning, H.; Gu, L.; Wang, Q.; Lu, W.; Peng, H.; Cui, W.; Ying, B.; Ross, C.R.; Wilson, G.M.; et al. Tristetraprolin induces cell cycle arrest in breast tumor cells through targeting AP-1/c-jun and nf-kappab pathway. Oncotarget 2015, 6, 41679–41691. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Vo, M.T.; Kim, H.K.; Lee, H.H.; Yoon, N.A.; Lee, B.J.; Min, Y.J.; Joo, W.D.; Cha, H.J.; Park, J.W.; et al. Ectopic over-expression of tristetraprolin in human cancer cells promotes biogenesis of let-7 by down-regulation of Lin28. Nucleic Acids Res. 2012, 40, 3856–3869. [Google Scholar] [CrossRef] [PubMed]
- Montorsi, L.; Guizzetti, F.; Alecci, C.; Caporali, A.; Martello, A.; Atene, C.G.; Parenti, S.; Pizzini, S.; Zanovello, P.; Bortoluzzi, S.; et al. Loss of zfp36 expression in colorectal cancer correlates to wnt/beta-catenin activity and enhances epithelial-to-mesenchymal transition through upregulation of zeb1, sox9 and macc1. Oncotarget 2016, 7, 59144–59157. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, C.W.; Vo, M.T.; Lee, H.H.; Lee, J.Y.; Yoon, N.A.; Lee, C.Y.; Moon, C.H.; Min, Y.J.; Park, J.W.; et al. Expression of proviral integration site for moloney murine leukemia virus 1 (pim-1) is post-transcriptionally regulated by tristetraprolin in cancer cells. J. Biol. Chem. 2012, 287, 28770–28778. [Google Scholar] [CrossRef] [PubMed]
- Selmi, T.; Martello, A.; Vignudelli, T.; Ferrari, E.; Grande, A.; Gemelli, C.; Salomoni, P.; Ferrari, S.; Zanocco-Marani, T. zfp36 Expression impairs glioblastoma cell lines viability and invasiveness by targeting multiple signal transduction pathways. Cell Cycle 2012, 11, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.A.; Jo, H.G.; Lee, U.H.; Park, J.H.; Yoon, J.E.; Ryu, J.; Kang, S.S.; Min, Y.J.; Ju, S.A.; Seo, E.H.; et al. Tristetraprolin suppresses the EMT through the down-regulation of twist1 and snail1 in cancer cells. Oncotarget 2016, 7, 8931–8943. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kramer, A.; Matthess, Y.; Yan, R.; Spankuch, B.; Gatje, R.; Knecht, R.; Kaufmann, M.; Strebhardt, K. Stable gene silencing of Cyclin b1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene 2006, 25, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, H.; Koizumi, H.; Uchikoshi, T. Expression of the G2-M checkpoint regulators Cyclin b1 and cdc2 in nonmalignant and malignant human breast lesions: Immunocytochemical and quantitative image analyses. Am. J. Pathol. 1997, 150, 15–23. [Google Scholar] [PubMed]
- Wang, A.; Yoshimi, N.; Ino, N.; Tanaka, T.; Mori, H. Overexpression of Cyclin b1 in human colorectal cancers. J. Cancer Res. Clin. Oncol. 1997, 123, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Jang, S.J.; Khuri, F.R.; Hassan, K.; Liu, D.; Hong, W.K.; Mao, L. Overexpression of Cyclin b1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 2000, 60, 4000–4004. [Google Scholar] [PubMed]
- Diehl, J.A. Cycling to cancer with Cyclin d1. Cancer Biol. Ther. 2002, 1, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin d as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Nath, N.; Fusaro, G.; Chellappan, S. Rb and prohibitin target distinct regions of e2f1 for repression and respond to different upstream signals. Mol. Cell. Biol. 1999, 19, 7447–7460. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Roh, Y.G.; Kim, S.K.; Lee, J.S.; Seol, S.Y.; Lee, H.H.; Kim, W.T.; Kim, W.J.; Heo, J.; Cha, H.J.; et al. Activation of ezh2 and suz12 regulated by E2F1 predicts the disease progression and aggressive characteristics of bladder cancer. Clin. Cancer Res. 2015, 21, 5391–5403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Magnuson, N.S. Pim-1 kinase-dependent phosphorylation of p21Cip1/WAF1 regulates its stability and cellular localization in H1299 cells. Mol. Cancer Res. 2007, 5, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.R.; Liang, C.; Feng, D.; Cheng, Y.J.; Wang, W.M.; Yang, D.J.; Wang, Y.X.; Cai, Q.P. Low tristetraprolin expression promotes cell proliferation and predicts poor patients outcome in pancreatic cancer. Oncotarget 2016, 7, 17737–17750. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, H.J.; Yoon, N.A.; Lee, W.H.; Min, Y.J.; Ko, B.K.; Lee, B.J.; Lee, A.; Cha, H.J.; Cho, W.J.; et al. Tumor suppressor p53 plays a key role in induction of both tristetraprolin and let-7 in human cancer cells. Nucleic Acids Res. 2013, 41, 5614–5625. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Zhang, S.Z.; Shan, J.L.; Hu, Z.J.; Liu, X.Y.; Chen, L.R.; Ren, X.C.; Yao, L.F.; Sheng, H.Q.; Li, L.; et al. Elevated HMGA2 expression is associated with cancer aggressiveness and predicts poor outcome in breast cancer. Cancer Lett. 2016, 376, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.P.; Youle, R.J. Balancing cell growth and death. Curr. Opin. Cell Biol. 2012, 24, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Otake, Y.; Soundararajan, S.; Sengupta, T.K.; Kio, E.A.; Smith, J.C.; Pineda-Roman, M.; Stuart, R.K.; Spicer, E.K.; Fernandes, D.J. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood 2007, 109, 3069–3075. [Google Scholar] [PubMed]
- Rounbehler, R.J.; Fallahi, M.; Yang, C.; Steeves, M.A.; Li, W.; Doherty, J.R.; Schaub, F.X.; Sanduja, S.; Dixon, D.A.; Blackshear, P.J.; et al. Tristetraprolin impairs Myc-induced lymphoma and abolishes the malignant state. Cell 2012, 150, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Ning, H.; Qian, X.; Huang, Q.; Hou, R.; Almourani, R.; Fu, M.; Blackshear, P.J.; Liu, J. Suppression of il-12 production by tristetraprolin through blocking nf-kcyb nuclear translocation. J. Immunol. 2013, 191, 3922–3930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Jiang, Y.W.; Su, Y.L.; Lee, S.C.; Chang, M.S.; Chang, C.J. Transcriptional regulation of tristetraprolin by NF-kappaB signaling in lps-stimulated macrophages. Mol. Biol. Rep. 2013, 40, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Schichl, Y.M.; Resch, U.; Hofer-Warbinek, R.; de Martin, R. Tristetraprolin impairs NF-kappaB/p65 nuclear translocation. J. Biol. Chem. 2009, 284, 29571–29581. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Mohle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Van Tubergen, E.A.; Banerjee, R.; Liu, M.; Broek, R.V.; Light, E.; Kuo, S.; Feinberg, S.E.; Willis, A.L.; Wolf, G.; Carey, T.; et al. Inactivation or loss of TTP promotes invasion in head and neck cancer via transcript stabilization and secretion of MMP9, MMP2, and IL-6. Clin. Cancer Res. 2013, 19, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Roussos, E.T.; Keckesova, Z.; Haley, J.D.; Epstein, D.M.; Weinberg, R.A.; Condeelis, J.S. AACR special conference on epithelial-mesenchymal transition and cancer progression and treatment. Cancer Res. 2010, 70, 7360–7364. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmadi, W.; Al-Ghamdi, M.; Al-Souhibani, N.; Khabar, K.S.A. miR-29a inhibition normalizes hur over-expression and aberrant AU-rich mRNA stability in invasive cancer. J. Pathol. 2013, 230, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Ueno, Y.; Hayashi, S.; Fukushima, T. The role of proteolysis in tumor invasiveness in glioblastoma and metastatic brain tumors. Anticancer Res. 2002, 22, 4265–4268. [Google Scholar] [PubMed]
- Ryu, J.; Yoon, N.A.; Seong, H.; Jeong, J.Y.; Kang, S.; Park, N.; Choi, J.; Lee, D.H.; Roh, G.S.; Kim, H.J.; et al. Resveratrol induces glioma cell apoptosis through activation of tristetraprolin. Mol. Cells 2015, 38, 991–997. [Google Scholar] [PubMed]
- Ryu, J.; Yoon, N.A.; Lee, Y.K.; Jeong, J.Y.; Kang, S.; Seong, H.; Choi, J.; Park, N.; Kim, N.; Cho, W.J.; et al. Tristetraprolin inhibits the growth of human glioma cells through downregulation of urokinase plasminogen activator/urokinase plasminogen activator receptor mRNAs. Mol. Cells 2015, 38, 156–162. [Google Scholar] [PubMed]
- Xiong, T.; Liu, X.W.; Huang, X.L.; Xu, X.F.; Xie, W.Q.; Zhang, S.J.; Tu, J. Tristetraprolin: A novel target of diallyl disulfide that inhibits the progression of breast cancer. Oncol. Lett. 2018, 15, 7817–7827. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Qu, H.H.; Shan, T.; Chen, Y.G.; Chen, Y.; Xia, J.Z. Tristetraprolin overexpression in gastric cancer cells suppresses PD-L1 expression and inhibits tumor progression by enhancing antitumor immunity. Mol. Cells 2018, 41, 653–664. [Google Scholar] [PubMed]
- Coelho, M.A.; Trecesson, S.D.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic Ras signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Milke, L.; Schulz, K.; Weigert, A.; Sha, W.X.; Schmid, T.; Brune, B. Depletion of tristetraprolin in breast cancer cells increases interleukin-16 expression and promotes tumor infiltration with monocytes/macrophages. Carcinogenesis 2013, 34, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Essafi-Benkhadir, K.; Onesto, C.; Stebe, E.; Moroni, C.; Pages, G. Tristetraprolin inhibits ras-dependent tumor vascularization by inducing vascular endothelial growth factor mRNA degradation. Mol. Biol. Cell 2007, 18, 4648–4658. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Son, Y.J.; Lee, W.H.; Park, Y.W.; Chae, S.W.; Cho, W.J.; Kim, Y.M.; Choi, H.J.; Choi, D.H.; Jung, S.W.; et al. Tristetraprolin regulates expression of VEGF and tumorigenesis in human colon cancer. Int. J. Cancer 2010, 126, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Gately, S.; Li, W.W. Multiple roles of cox-2 in tumor angiogenesis: A target for antiangiogenic therapy. Semin. Oncol. 2004, 31, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Sawaoka, H.; Dixon, D.A.; Oates, J.A.; Boutaud, O. Tristetraprolin binds to the 3′-untranslated region of cyclooxygenase-2 mRNA. A polyadenylation variant in a cancer cell line lacks the binding site. J. Biol. Chem. 2003, 278, 13928–13935. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.J.; Lee, H.H.; Chae, S.W.; Cho, W.J.; Kim, Y.M.; Choi, H.J.; Choi, D.H.; Jung, S.W.; Min, Y.J.; Lee, B.J.; et al. Tristetraprolin downregulates the expression of both VEGF and COX-2 in human colon cancer. Hepatogastroenterology 2011, 58, 790–795. [Google Scholar] [PubMed]
- Hau, H.H.; Walsh, R.J.; Ogilvie, R.L.; Williams, D.A.; Reilly, C.S.; Bohjanen, P.R. Tristetraprolin recruits functional mRNA decay complexes to are sequences. J. Cell. Biochem. 2007, 100, 1477–1492. [Google Scholar] [CrossRef] [PubMed]
- Winzen, R.; Thakur, B.K.; Dittrich-Breiholz, O.; Shah, M.; Redich, N.; Dhamija, S.; Kracht, M.; Holtmann, H. Functional analysis of ksrp interaction with the au-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol. Cell. Biol. 2007, 27, 8388–8400. [Google Scholar] [CrossRef] [PubMed]
- Fechir, M.; Linker, K.; Pautz, A.; Hubrich, T.; Forstermann, U.; Rodriguez-Pascual, F.; Kleinert, H. Tristetraprolin regulates the expression of the human inducible nitric-oxide synthase gene. Mol. Pharmacol. 2005, 67, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.C.; Han, X.D.; Peng, J.Y.; Qin, H.L.; Wang, Y. The role of CXC chemokines and their receptors in the progression and treatment of tumors. J. Mol. Histol. 2012, 43, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Zeelenberg, I.S.; Ruuls-Van Stalle, L.; Roos, E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003, 63, 3833–3839. [Google Scholar] [PubMed]
- Bourcier, C.; Griseri, P.; Grepin, R.; Bertolotto, C.; Mazure, N.; Pages, G. Constitutive erk activity induces downregulation of tristetraprolin, a major protein controlling interleukin8/CXCL8 mRNA stability in melanoma cells. Am. J. Physiol. Cell Physiol. 2011, 301, C609–C618. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Harris, A.L. Tumour hypoxia, hypoxia signaling pathways and hypoxia inducible factor expression in human cancer. Anticancer Res. 2001, 21, 4317–4324. [Google Scholar] [PubMed]
- Kim, T.W.; Yim, S.; Choi, B.J.; Jang, Y.; Lee, J.J.; Sohn, B.H.; Yoo, H.S.; Il Yeom, Y.; Park, K.C. Tristetraprolin regulates the stability of hif-1 alpha mRNA during prolonged hypoxia. Biochem. Biophys. Res. Commun. 2010, 391, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Suswam, E.; Li, Y.; Zhang, X.; Gillespie, G.Y.; Li, X.; Shacka, J.J.; Lu, L.; Zheng, L.; King, P.H. Tristetraprolin down-regulates interleukin-8 and vascular endothelial growth factor in malignant glioma cells. Cancer Res 2008, 68, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.E.; Kuwano, Y.; Alkharouf, N.; Blackshear, P.J.; Gorospe, M.; Wilson, G.M. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res. 2009, 69, 5168–5176. [Google Scholar] [CrossRef] [PubMed]
- Gebeshuber, C.A.; Zatloukal, K.; Martinez, J. Mir-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 2009, 10, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Carrick, D.M.; Blackshear, P.J. Comparative expression of tristetraprolin (TTP) family member transcripts in normal human tissues and cancer cell lines. Arch. Biochem. Biophys. 2007, 462, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Sanduja, S.; Kaza, V.; Dixon, D.A. The mRNA decay factor tristetraprolin (ttp) induces senescence in human papillomavirus-transformed cervical cancer cells by targeting E6-AP ubiquitin ligase. Aging 2009, 1, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Sanduja, S.; Bemis-Standoli, K.; Pena, E.A.; Price, R.L.; Dixon, D.A. The mRNA binding proteins hur and tristetraprolin regulate cyclooxygenase 2 expression during colon carcinogenesis. Gastroenterology 2009, 136, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Sohn, B.H.; Park, I.Y.; Lee, J.J.; Yang, S.J.; Jang, Y.J.; Park, K.C.; Kim, D.J.; Lee, D.C.; Sohn, H.A.; Kim, T.W.; et al. Functional switching of TGF-beta1 signaling in liver cancer via epigenetic modulation of a single CPG site in ttp promoter. Gastroenterology 2010, 138, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.S.; Stumpo, D.J.; Blackshear, P.J. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J. Biol. Chem. 1990, 265, 16556–16563. [Google Scholar] [PubMed]
- DuBois, R.N.; McLane, M.W.; Ryder, K.; Lau, L.F.; Nathans, D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 1990, 265, 19185–19191. [Google Scholar] [PubMed]
- Lim, R.W.; Varnum, B.C.; Herschman, H.R. Cloning of tetradecanoyl phorbol ester-induced ‘primary response’ sequences and their expression in density-arrested swiss 3T3 cells and a TPA non-proliferative variant. Oncogene 1987, 1, 263–270. [Google Scholar] [PubMed]
- Gomperts, M.; Pascall, J.C.; Brown, K.D. The nucleotide sequence of a cdna encoding an EGF-inducible gene indicates the existence of a new family of mitogen-induced genes. Oncogene 1990, 5, 1081–1083. [Google Scholar] [PubMed]
- Lai, W.S.; Thompson, M.J.; Taylor, G.A.; Liu, Y.; Blackshear, P.J. Promoter analysis of zfp-36, the mitogen-inducible gene encoding the zinc finger protein tristetraprolin. J. Biol. Chem. 1995, 270, 25266–25272. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Chen, F.; Kim, Y.J.; Chen, Y. Transcriptional regulation of tristetraprolin by transforming growth factor-beta in human T cells. J. Biol. Chem. 2003, 278, 30373–30381. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.F.; Sanduja, S.; Deane, N.G.; Blackshear, P.J.; Dixon, D.A. Transforming growth factor beta regulates p-body formation through induction of the mRNA decay factor tristetraprolin. Mol. Cell. Biol. 2014, 34, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Sauer, I.; Schaljo, B.; Vogl, C.; Gattermeier, I.; Kolbe, T.; Muller, M.; Blackshear, P.J.; Kovarik, P. Interferons limit inflammatory responses by induction of tristetraprolin. Blood 2006, 107, 4790–4797. [Google Scholar] [CrossRef] [PubMed]
- Gaba, A.; Grivennikov, S.I.; Do, M.V.; Stumpo, D.J.; Blackshear, P.J.; Karin, M. Cutting edge: IL-10-mediated tristetraprolin induction is part of a feedback loop that controls macrophage STAT3 activation and cytokine production. J. Immunol. 2012, 189, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nakajima, H.; Ikeda, K.; Maezawa, Y.; Suto, A.; Takatori, H.; Saito, Y.; Iwamoto, I. IL-4-STAT6 signaling induces tristetraprolin expression and inhibits TNF-alpha production in mast cells. J. Exp. Med. 2003, 198, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Tchen, C.R.; Brook, M.; Saklatvala, J.; Clark, A.R. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem. 2004, 279, 32393–32400. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Connolly, J.E.; Rigby, W.F. The role of mRNA turnover in the regulation of tristetraprolin expression: Evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol. 2004, 172, 7263–7271. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bhat, A.A.; Krishnan, M.; Singh, A.B.; Dhawan, P. Trichostatin-a modulates claudin-1 mRNA stability through the modulation of hu antigen r and tristetraprolin in colon cancer cells. Carcinogenesis 2013, 34, 2610–2621. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Sanduja, S.; Blanco, F.F.; Hu, L.; Dixon, D.A. Histone deacetylase inhibitors activate tristetraprolin expression through induction of early growth response protein 1 (EGR1) in colorectal cancer cells. Biomolecules 2015, 5, 2035–2055. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Liu, B.Y.; Yan, S.; Jiang, T.H.; Cheng, H.Q.; Jiang, H.S.; Cao, Y.; Mao, A.W. MicroRNA-29a promotes pancreatic cancer growth by inhibiting tristetraprolin. Cell. Physiol. Biochem. 2015, 37, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Dzineku, F.; Blackshear, P.J. Expression and purification of recombinant tristetraprolin that can bind to tumor necrosis factor-alpha mRNA and serve as a substrate for mitogen-activated protein kinases. Arch. Biochem. Biophys. 2003, 412, 106–120. [Google Scholar] [CrossRef]
- Carballo, E.; Cao, H.; Lai, W.S.; Kennington, E.A.; Campbell, D.; Blackshear, P.J. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J. Biol. Chem. 2001, 276, 42580–42587. [Google Scholar] [CrossRef] [PubMed]
- Rigby, W.F.; Roy, K.; Collins, J.; Rigby, S.; Connolly, J.E.; Bloch, D.B.; Brooks, S.A. Structure/function analysis of tristetraprolin (TTP): P38 stress-activated protein kinase and lipopolysaccharide stimulation do not alter ttp function. J. Immunol. 2005, 174, 7883–7893. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Aubareda, A.; Tudor, C.; Saklatvala, J.; Clark, A.R.; Dean, J.L. Mapkap kinase 2 blocks tristetraprolin-directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J. Biol. Chem. 2010, 285, 27590–27600. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.L.; Scheckel, C.; Stoecklin, G.; Lykke-Andersen, J. Phosphorylation of tristetraprolin by MK2 impairs au-rich element mRNA decay by preventing deadenylase recruitment. Mol. Cell. Biol. 2011, 31, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Stehn, J.R.; Yaffe, M.B.; Blackwell, T.K. Cytoplasmic localization of tristetraprolin involves 14-3-3-dependent and -independent mechanisms. J. Biol. Chem. 2002, 277, 18029–18036. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Lei, T.; Song, Y.; Yanes, N.; Qi, Y.; Fu, M. RNA-destabilizing factor tristetraprolin negatively regulates NF-kappaB signaling. J. Biol. Chem. 2009, 284, 29383–29390. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Garcia, T.; Gomez-Romero, V.; Tecalco-Cruz, A.; Valadez-Graham, V.; Leon-Del-Rio, A. Nuclear tristetraprolin acts as a corepressor of multiple steroid nuclear receptors in breast cancer cells. Mol. Genet. Metab. Rep. 2016, 7, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Garcia, T.; Tecalco-Cruz, A.; Gomez-Romero, V.; Reyes-Carmona, S.; Meneses-Morales, I.; Leon-Del-Rio, A. Tristetraprolin represses estrogen receptor alpha transactivation in breast cancer cells. J. Biol. Chem. 2014, 289, 15554–15565. [Google Scholar] [CrossRef] [PubMed]
- Hitti, E.; Iakovleva, T.; Brook, M.; Deppenmeier, S.; Gruber, A.D.; Radzioch, D.; Clark, A.R.; Blackshear, P.J.; Kotlyarov, A.; Gaestel, M. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol. Cell. Biol. 2006, 26, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, I.; Torrejon, B.; Madoz-Gurpide, J.; Rojo, F.; Garcia-Foncillas, J. PP2A plays a key role in inflammation and cancer through tristetraprolin activation. Ann. Rheum. Dis. 2017, 76. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.D.; Ammit, A.J.; Clark, A.R. Mapk p38 regulates inflammatory gene expression via tristetraprolin: Doing good by stealth. Int. J. Biochem. Cell. Biol. 2018, 94, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Rumzhum, N.N.; Hansbro, P.M.; Morris, J.C.; Clark, A.R.; Verrills, N.M.; Ammit, A.J. Activating protein phosphatase 2a (PP2A) enhances tristetraprolin (TTP) anti-inflammatory function in a549 lung epithelial cells. Cell Signal. 2016, 28, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Stoecklin, G.; Van Way, S.; Hinkovska-Galcheva, V.; Guo, R.F.; Anderson, P.; Shanley, T.P. Tristetraprolin (TTP)-14-3-3 complex formation protects ttp from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J. Biol. Chem. 2007, 282, 3766–3777. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.R.; Dean, J.L. The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: A tale of two phosphatases. Biochem. Soc. Trans. 2016, 44, 1321–1337. [Google Scholar] [CrossRef] [PubMed]
- Suswam, E.A.; Shacka, J.J.; Walker, K.; Lu, L.; Li, X.; Si, Y.; Zhang, X.; Zheng, L.; Nabors, L.B.; Cao, H.; et al. Mutant tristetraprolin: A potent inhibitor of malignant glioma cell growth. J. Neurooncol. 2013, 113, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Medicherla, S.; Reddy, M.; Ying, J.; Navas, T.A.; Li, L.; Nguyen, A.N.; Kerr, I.; Hanjarappa, N.; Protter, A.A.; Higgins, L.S. P38alpha-selective map kinase inhibitor reduces tumor growth in mouse xenograft models of multiple myeloma. Anticancer Res. 2008, 28, 3827–3833. [Google Scholar] [PubMed]
- Banerjee, R.; Van Tubergen, E.A.; Scanlon, C.S.; Vander Broek, R.; Lints, J.P.; Liu, M.; Russo, N.; Inglehart, R.C.; Wang, Y.; Polverini, P.J.; et al. The G protein-coupled receptor GALR2 promotes angiogenesis in head and neck cancer. Mol. Cancer Ther. 2014, 13, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Dufies, M.; Giuliano, S.; Ambrosetti, D.; Claren, A.; Ndiaye, P.D.; Mastri, M.; Moghrabi, W.; Cooley, L.S.; Ettaiche, M.; Chamorey, E.; et al. Sunitinib stimulates expression of vegfc by tumor cells and promotes lymphangiogenesis in clear cell renal cell carcinomas. Cancer Res. 2017, 77, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhang, T.; Yang, Z.; Wei, J.C.; Shen, H.F.; Xiao, D.; Wang, Q.; Yang, P.; Chen, H.C.; Hu, H.; et al. Gambogic acid efficiently kills stem-like colorectal cancer cells by upregulating zfp36 expression. Cell. Physiol. Biochem. 2018, 46, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, C.; He, G. Tristetraprolin: A novel mediator of the anticancer properties of resveratrol. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | ARE Sequences | Regulation by TTP | |||
|---|---|---|---|---|---|
| ARE Binding 1 | 3′ UTR Binding 2 | mRNA Decay 3 | Ref. | ||
| AHRR | TTCTGGCCTCTGGGCATTTATGGATTTAAGACCAGGATGGTATTTCAGAAGCTT | O | O | O | [23] |
| AKT-1 | TTTTTTTACAACATTCAACTTTAGT | O | ND | O | [25,26] |
| BCL2 | ATTTATTTATTTA | ND | O | O | [27] |
| BIRC3 (cIAP2) | TTTGGTTTCCTTAAAATTTTTATTTATTTACAACTCAAAAAACATTGTTTTG | O | O | O | [28,29] |
| CCNB1 (cyclin B1) | TTATTTACTTTTACCACTATTTAAG | O | O | O | [25,30] |
| CCND1 (cyclin D1) | TTATTATATTCCGTAGGTAGATGTG, ACATAATATATTCTATTTTTATACTCT | O | O | O | [25,31] |
| CDKN1A (p21) | TAGTCTCAGTTTGTGTGTCTTAATTATTATTTGTGTTTTAATTTAAACACCTCCT | O | O | O | [24] |
| CXCL1 | TCTTCTATTTATTTATTTATTCATTAGTT | O | O | O | [25,32] |
| CXCL2 (MIP-2) | CACACTCTCCCATTATATTTATTG | O | ND | O | [25,33] |
| CXCR4 | ACTTATTTATATAAATTTTTTTTG | O | O | O | [25,34] |
| E2F1 | CTTTAATGGAGCGTTATTTATTTATCGAGGCCTCTTTG | O | O | O | [29,35] |
| FOS (c-Fos) | TAATTTATTTATT | ND | O | O | [36] |
| HMGA2 | TGTAATTTAATGA | ND | O | O | [37] |
| IFN-γ | CTATTTATTAATATTTAA | O | O | O | [38] |
| JUN (c-Jun) | TTCTCTATTAGACTTTAGAAA, AGCACTCTGAGTTTACCATTTG | O | ND | ND | [25,39] |
| LATS2 | TTCAAATTAGTATGATTCCTATTTAAAGTGATTTATATTTGAGTAAAAAGTTCAA | O | O | O | [22] |
| Lin28A | TTTTATTTATTTG | O | O | O | [29,40] |
| MACC1 | TATAATTTAATAT | ND | O | O | [41] |
| MYC (Myc) | AATTTCAATCCTAGTATATAGTACCTAGTATTATAGGTACTATAAACCCTAATTTTTTTTATTTAA | O | O | O | [25,31] |
| PIM-1 | CCTGGAGGTCAATGTTATGTATTTATTTATTTATTTATTTGGTTCCCTTCCTATTCC | O | O | O | [42] |
| PIM-3 | TTTAATTTATTTG | ND | O | O | [43] |
| SNAI1 (Snail1) | GTTATATGTACAGTTTATTGATATTCAATAAAGCAGTTAATTTATATATTAAAAA | O | O | O | [44] |
| XIAP | CAAATTTATTTTATTTATTTAATT | O | O | O | [25,43] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-M.; Lee, T.-H.; Kang, T.-H. Roles of Tristetraprolin in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 3384. https://doi.org/10.3390/ijms19113384
Park J-M, Lee T-H, Kang T-H. Roles of Tristetraprolin in Tumorigenesis. International Journal of Molecular Sciences. 2018; 19(11):3384. https://doi.org/10.3390/ijms19113384
Chicago/Turabian StylePark, Jeong-Min, Tae-Hee Lee, and Tae-Hong Kang. 2018. "Roles of Tristetraprolin in Tumorigenesis" International Journal of Molecular Sciences 19, no. 11: 3384. https://doi.org/10.3390/ijms19113384
APA StylePark, J.-M., Lee, T.-H., & Kang, T.-H. (2018). Roles of Tristetraprolin in Tumorigenesis. International Journal of Molecular Sciences, 19(11), 3384. https://doi.org/10.3390/ijms19113384