The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor

 , ,
, , 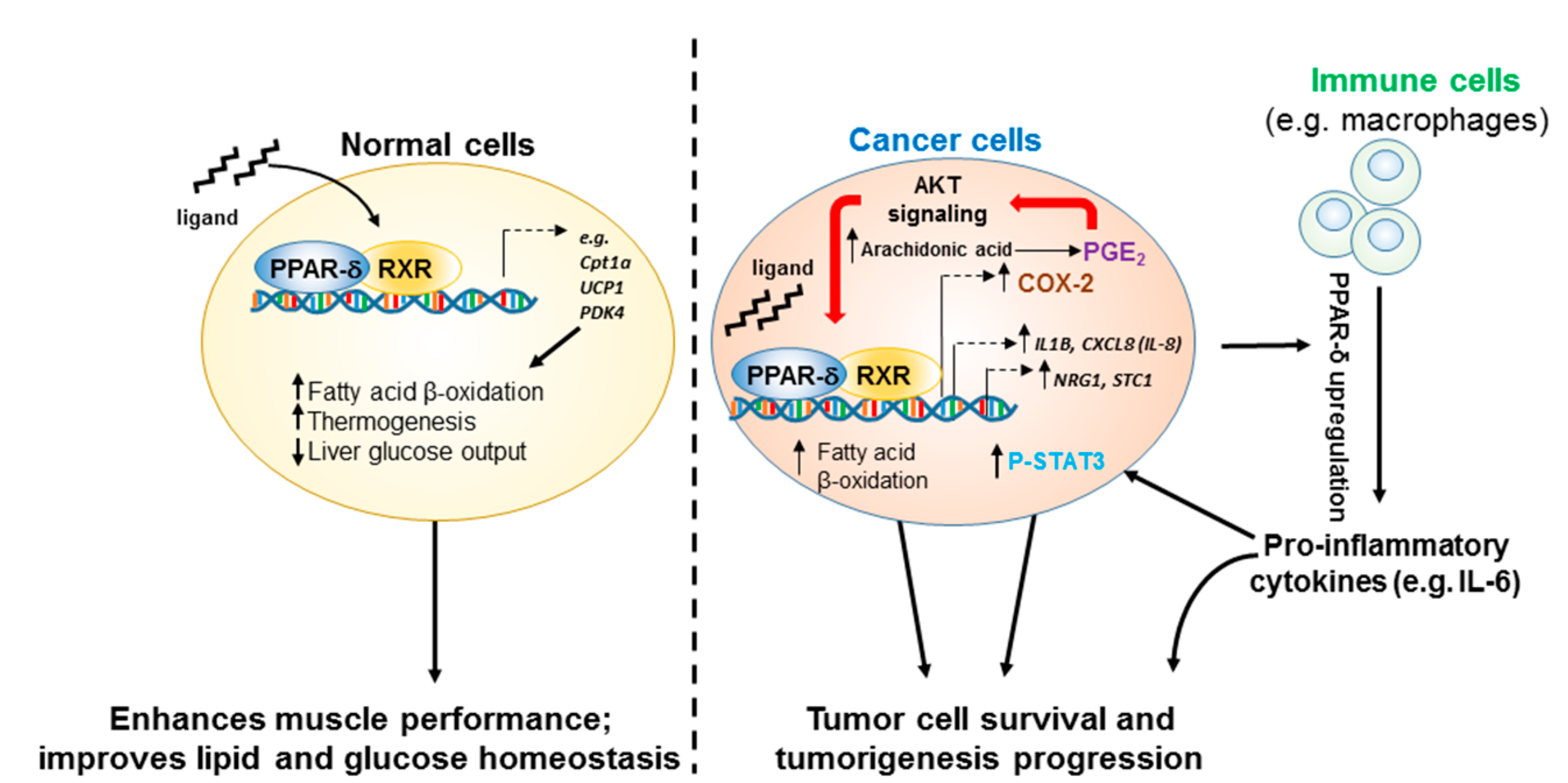{kind=link}
Abstract
1. Introduction
2. Metabolic Regulation by PPAR-δ
3. PPAR-δ in Inflammation-Related Diseases
4. Modulation of Inflammatory Actions in Immune Cells
5. The Role of PPAR-δ in Cancer
5.1. PPAR-δ Crosstalk between Inflammation and Cancer
5.2. PPAR-δ Promotion of Cancer
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | Cellular homolog of viral akt gene, also known as protein kinase B |
| AOM | azoxymethane |
| APC | Adenomatous polyposis coli |
| BCL-6 | B cell lymphoma 6 |
| CAC | colitis-associated cancer |
| CRC | Colorectal cancer |
| COX-2 | cyclooxygenase-2 |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 |
| CXCL2 | Chemokine (C-X-C motif) ligand 2 |
| CXCL4 | Chemokine (C-X-C motif) ligand 4 |
| DSS | dextran sodium sulfate |
| EAE | experimental autoimmune encepaholmyelitis |
| EMT | Epithelial mesenchymal transition |
| FABP | Fatty acid binding protein |
| FAT | Fatty acid translocase |
| FATP | Fatty acid transport protein |
| IDO-1 | indoleamine 2,3-dioxygenase 1 |
| IFN-γ | Interferon-γ |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| MCP | monocyte chemoattractive protein |
| MDM | monocyte-derived macrophage |
| MSC | mesenchymal stem cell |
| MMTV | Mouse mammary tumor virus |
| NRG1 | Neuregulin 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PGE2 | prostaglandin E2 |
| PI3K | phosphatidyl inositol 3 kinase |
| PPRE | PPAR response element |
| PUFA | polyunsaturated fatty acid |
| RXR | retinoid X receptor |
| STAT1/3 | Signal Transducer and Activator of Transcription 1/3 |
| STC1 | Stanniocalcin 1 |
| TAM | tumor-associated macrophage |
| TGF-β1 | transforming growth factor-β 1 |
| TNF-α | tumor necrosis factor α |
References
- Wagner, K.D.; Wagner, N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol. Ther. 2010, 125, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: No alteration in adipose tissue of obese and NIDDM patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zuo, X.; Shureiqi, I. Targeting peroxisome proliferator-activated receptor-beta/delta in colon cancer: How to aim? Biochem. Pharmacol. 2013, 85, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Nadra, K.; Anghel, S.I.; Joye, E.; Tan, N.S.; Basu-Modak, S.; Trono, D.; Wahli, W.; Desvergne, B. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor beta/delta. Mol. Cell. Boil. 2006, 26, 3266–3281. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Liao, D.; He, W.; Ong, E.S.; Nelson, M.C.; Olefsky, J.M.; Boland, R.; Evans, R.M. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Harmon, G.S.; Lam, M.T.; Glass, C.K. PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 2011, 111, 6321–6340. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular Recognition of Fatty Acids by Peroxisome Proliferator–Activated Receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Shureiqi, I.; Jiang, W.; Zuo, X.; Wu, Y.; Stimmel, J.B.; Leesnitzer, L.M.; Morris, J.S.; Fan, H.Z.; Fischer, S.M.; Lippman, S.M. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9968–9973. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Tan, J.; Krause, W.F.; Geraci, M.W.; Willson, T.M.; Dey, S.K.; DuBois, R.N. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 13275–13280. [Google Scholar] [CrossRef] [PubMed]
- Naruhn, S.; Meissner, W.; Adhikary, T.; Kaddatz, K.; Klein, T.; Watzer, B.; Muller-Brusselbach, S.; Muller, R. 15-hydroxyeicosatetraenoic acid is a preferential peroxisome proliferator-activated receptor beta/delta agonist. Mol. Pharmacol. 2010, 77, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Baiga, T.J.; Downes, M.; La Clair, J.J.; Atkins, A.R.; Richard, S.B.; Fan, W.; Stockley-Noel, T.A.; Bowman, M.E.; Noel, J.P.; et al. Structural basis for specific ligation of the peroxisome proliferator-activated receptor delta. Proc. Natl. Acad. Sci. USA 2017, 114, E2563–E2570. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural Peroxisome Proliferator-Activated Receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Hotamisligil, G.S. Fatty acid binding proteins—The evolutionary crossroads of inflammatory and metabolic responses. J. Nutr. 2004, 134, 2464S–2468S. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Yousefnia, S.; Seyed Forootan, F.; Peymani, M.; Ghaedi, K.; Nasr Esfahani, M.H. Diverse roles of fatty acid binding proteins (FABPs) in development and pathogenesis of cancers. Gene 2018, 676, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Shaw, N.S.; Vinckenbosch, N.; Liu, P.; Yasmin, R.; Desvergne, B.; Wahli, W.; Noy, N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Boil. 2002, 22, 5114–5127. [Google Scholar] [CrossRef]
- Tan, N.S.; Vazquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARbeta/delta. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.H.; Goswami, D.; Griffin, P.R.; Noy, N.; Ortlund, E.A. Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor beta/delta (FABP5-PPARbeta/delta) signaling pathway. J. Biol. Chem. 2014, 289, 14941–14954. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.A.; Trivella, D.B.; Bernardes, A.; Gratieri, J.; Oliveira, P.S.; Figueira, A.C.; Webb, P.; Polikarpov, I. Structural insights into human peroxisome proliferator activated receptor delta (PPAR-delta) selective ligand binding. PLoS ONE 2012, 7, e33643. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Tudor, C.; Engelborghs, Y.; Wahli, W.; Desvergne, B. Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J. Biol. Chem. 2005, 280, 17880–17890. [Google Scholar] [CrossRef] [PubMed]
- Matsusue, K.; Miyoshi, A.; Yamano, S.; Gonzalez, F.J. Ligand-activated PPARbeta efficiently represses the induction of LXR-dependent promoter activity through competition with RXR. Mol. Cell. Endocrinol. 2006, 256, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-Regulated Gene Expression. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M.; Curtiss, L.K. Transcriptional repression of atherogenic inflammation: Modulation by PPARdelta. Science 2003, 302, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Staels, B. PPARbeta in macrophages and atherosclerosis. Biochimie 2017, 136, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Scholtysek, C.; Katzenbeisser, J.; Fu, H.; Uderhardt, S.; Ipseiz, N.; Stoll, C.; Zaiss, M.M.; Stock, M.; Donhauser, L.; Bohm, C.; et al. PPARbeta/delta governs Wnt signaling and bone turnover. Nat. Med. 2013, 19, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, T.; Wortmann, A.; Schumann, T.; Finkernagel, F.; Lieber, S.; Roth, K.; Toth, P.M.; Diederich, W.E.; Nist, A.; Stiewe, T.; et al. The transcriptional PPARbeta/delta network in human macrophages defines a unique agonist-induced activation state. Nucleic Acids Res. 2015, 43, 5033–5051. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARdelta Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta Mol. Cell Boil. Lipids 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor δ controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-Proliferator-Activated Receptor δ Activates Fat Metabolism to Prevent Obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Cheng, C.W.; Cao, A.Q.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 2018, 22, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Ravnskjaer, K.; Frigerio, F.; Boergesen, M.; Nielsen, T.; Maechler, P.; Mandrup, S. PPARdelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J. Lipid Res. 2010, 51, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Barg, S.; Vallois, D.; Lahiri, S.; Roger, C.; Yessoufou, A.; Pradevand, S.; McDonald, A.; Bonal, C.; Reimann, F.; et al. PPARbeta/delta affects pancreatic beta cell mass and insulin secretion in mice. J. Clin. Investig. 2012, 122, 4105–4117. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Abbott, M.J.; Ahmadian, M.; Lopes, A.B.; Wang, Y.; Sul, H.S. Desnutrin/ATGL activates PPARdelta to promote mitochondrial function for insulin secretion in islet beta cells. Cell Metab. 2013, 18, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, M.; Zwarts, I.; Zutphen, T.V.; Dijk, T.H.V.; Bloks, V.W.; Harkema, L.; Bruin, A.; Downes, M.; Evans, R.M.; Verkade, H.J.; et al. Intestinal PPARdelta protects against diet-induced obesity, insulin resistance and dyslipidemia. Sci. Rep. 2017, 7, 846. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Montagner, A.; Tan, N.; Wahli, W. Insights into the Role of PPARβ/δ in NAFLD. Int. J. Mol. Sci. 2018, 19, 1893. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Lee, C.H. PPAR delta as a therapeutic target in metabolic disease. FEBS Lett. 2008, 582, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sznaidman, M.L.; Haffner, C.D.; Maloney, P.R.; Fivush, A.; Chao, E.; Goreham, D.; Sierra, M.L.; LeGrumelec, C.; Xu, H.E.; Montana, V.G.; et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta)—Synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003, 13, 1517–1521. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Dunn, S.E.; Iorio, C.; Screaton, R.A.; Spaner, D.E. PPAR-delta promotes survival of chronic lymphocytic leukemia cells in energetically unfavorable conditions. Leukemia 2017, 31, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.L. Rationally designed PPARdelta-specific agonists and their therapeutic potential for metabolic syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 3284–3285. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Bothwell, A.L. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Mol. Cells 2012, 33, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kanakasabai, S.; Chearwae, W.; Walline, C.C.; Iams, W.; Adams, S.M.; Bright, J.J. Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology 2010, 130, 572–588. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.E.; Bhat, R.; Straus, D.S.; Sobel, R.A.; Axtell, R.; Johnson, A.; Nguyen, K.; Mukundan, L.; Moshkova, M.; Dugas, J.C.; et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J. Exp. Med. 2010, 207, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Choi, R.; Kim, H.M.; Cho, E.J.; Kim, B.H.; Choi, Y.S.; Naowaboot, J.; Lee, E.Y.; Yang, Y.C.; Shin, J.Y.; et al. Peroxisome proliferator-activated receptor delta agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp. Mol. Med. 2012, 44, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Ogawa, D.; Wada, J.; Yamamoto, N.; Shikata, K.; Sato, C.; Tachibana, H.; Toyota, N.; Makino, H. Activation of peroxisome proliferator-activated receptor delta inhibits streptozotocin-induced diabetic nephropathy through anti-inflammatory mechanisms in mice. Diabetes 2011, 60, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Wong, W.T.; Zhao, L.; Xu, J.; Wang, L.; Lau, C.W.; Chen, Z.Y.; Ma, R.C.; Xu, A.; Wang, N.; et al. PPARdelta Is Required for Exercise to Attenuate Endoplasmic Reticulum Stress and Endothelial Dysfunction in Diabetic Mice. Diabetes 2017, 66, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Romanowska, M.; Reilly, L.; Palmer, C.N.; Gustafsson, M.C.; Foerster, J. Activation of PPARbeta/delta causes a psoriasis-like skin disease in vivo. PLoS ONE 2010, 5, e9701. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Ipseiz, N.; Espinosa-Carrasco, G.; Caicedo, A.; Tejedor, G.; Toupet, K.; Loriau, J.; Scholtysek, C.; Stoll, C.; Khoury, M.; et al. PPARbeta/delta directs the therapeutic potential of mesenchymal stem cells in arthritis. Ann. Rheum. Dis. 2016, 75, 2166–2174. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, L.; Odegaard, J.I.; Morel, C.R.; Heredia, J.E.; Mwangi, J.W.; Ricardo-Gonzalez, R.R.; Goh, Y.P.; Eagle, A.R.; Dunn, S.E.; Awakuni, J.U.; et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 2009, 15, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Muller, R. PPARbeta/delta in human cancer. Biochimie 2017, 136, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 2012, 10, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. PPARdelta and PGE2 signaling pathways communicate and connect inflammation to colorectal cancer. Inflamm. Cell Signal. 2014, 1. [Google Scholar] [CrossRef]
- Backlund, M.G.; Mann, J.R.; Dubois, R.N. Mechanisms for the prevention of gastrointestinal cancer: The role of prostaglandin E2. Oncology 2005, 69, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H.; Shi, Q.; Katkuri, S.; Walhi, W.; Desvergne, B.; Das, S.K.; Dey, S.K.; DuBois, R.N. Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell 2004, 6, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Xu, M.; Zuo, X.; Yu, J.; Xu, W.; Moussalli, M.J.; Elias, E.; Li, H.S.; Watowich, S.S.; Shureiqi, I. 15-Lipoxygenase-1 suppression of colitis-associated colon cancer through inhibition of the IL-6/STAT3 signaling pathway. FASEB J. 2015, 29, 2359–2370. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, L.; Ning, W.; Guo, L.; Sun, X.; Dey, S.K.; Chaturvedi, R.; Wilson, K.T.; DuBois, R.N. Peroxisome proliferator-activated receptor delta promotes colonic inflammation and tumor growth. Proc. Natl. Acad. Sci. USA 2014, 111, 7084–7089. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol. Cell. Boil. 2000, 20, 5119–5128. [Google Scholar] [CrossRef]
- Hollingshead, H.E.; Morimura, K.; Adachi, M.; Kennett, M.J.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peters, J.M. PPARbeta/delta protects against experimental colitis through a ligand-independent mechanism. Dig. Dis. Sci. 2007, 52, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Martín-Martín, N.; Zabala-Letona, A.; Fernández-Ruiz, S.; Arreal, L.; Camacho, L.; Castillo-Martin, M.; Cortazar, A.R.; Torrano, V.; Astobiza, I.; Zúñiga-García, P.; et al. PPARδ Elicits Ligand-Independent Repression of Trefoil Factor Family to Limit Prostate Cancer Growth. Cancer Res. 2018, 78, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Reinartz, S.; Finkernagel, F.; Adhikary, T.; Rohnalter, V.; Schumann, T.; Schober, Y.; Nockher, W.A.; Nist, A.; Stiewe, T.; Jansen, J.M.; et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Colvin, E.K. Tumor-Associated Macrophages Contribute to Tumor Progression in Ovarian Cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Lu, J.; Xiao, J.; Upadhyay, G.; Umans, R.; Kallakury, B.; Yin, Y.; Fant, M.E.; Kopelovich, L.; Glazer, R.I. PPARdelta induces estrogen receptor-positive mammary neoplasia through an inflammatory and metabolic phenotype linked to mTOR activation. Cancer Res. 2013, 73, 4349–4361. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Han, C.; Lim, K.; Wu, T. Cross-talk between Peroxisome Proliferator-Activated Receptor δ and Cytosolic Phospholipase A2α/Cyclooxygenase-2/Prostaglandin E2 Signaling Pathways in Human Hepatocellular Carcinoma Cells. Cancer Res. 2006, 66, 11859–11868. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Gonzalez, F.J.; Muller, R. Establishing the Role of PPARbeta/delta in Carcinogenesis. Trends Endocrinol. Metab. 2015, 26, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Takayama, O.; Yamamoto, H.; Damdinsuren, B.; Sugita, Y.; Ngan, C.Y.; Xu, X.; Tsujino, T.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; et al. Expression of PPAR[delta] in multistage carcinogenesis of the colorectum: Implications of malignant cancer morphology. Br. J. Cancer 2006, 95, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, M.; Taki, K.; Somada, S.; Sakiyama, Y.; Kubo, N.; Kaku, T.; Tsuruta, S.; Kusumoto, T.; Sakai, H.; Nakamura, K.; et al. The expression of both peroxisome proliferator-activated receptor delta and cyclooxygenase-2 in tissues is associated with poor prognosis in colorectal cancer patients. Dig. Dis. Sci. 2011, 56, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Xu, W.; Xu, M.; Tian, R.; Moussalli, M.J.; Mao, F.; Zheng, X.; Wang, J.; Morris, J.S.; Gagea, M.; et al. Metastasis regulation by PPARD expression in cancer cells. JCI Insight 2017, 2, e91419. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, A.; Schwager, C.; Kleeff, J.; Esposito, I.; Domhan, S.; Peschke, P.; Hauser, K.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12890–12895. [Google Scholar] [CrossRef] [PubMed]
- Pedchenko, T.V.; Gonzalez, A.L.; Wang, D.; DuBois, R.N.; Massion, P.P. Peroxisome proliferator-activated receptor beta/delta expression and activation in lung cancer. Am. J. Respir. Cell Mol. Boil. 2008, 39, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Harman, F.S.; Nicol, C.J.; Marin, H.E.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat. Med. 2004, 10, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H.; Guo, Y.; Ning, W.; Katkuri, S.; Wahli, W.; Desvergne, B.; Dey, S.K.; DuBois, R.N. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. USA 2006, 103, 19069–19074. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Peng, Z.; Moussalli, M.J.; Morris, J.S.; Broaddus, R.R.; Fischer, S.M.; Shureiqi, I. Targeted Genetic Disruption of Peroxisome Proliferator-Activated Receptor-{delta} and Colonic Tumorigenesis. J. Natl. Cancer Inst. 2009, 101, 762–767. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar] [CrossRef]
- Zuo, X.; Xu, M.; Yu, J.; Wu, Y.; Moussalli, M.J.; Manyam, G.C.; Lee, S.I.; Liang, S.; Gagea, M.; Morris, J.S.; et al. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR-delta/beta overexpression. J. Natl. Cancer Inst. 2014, 106, dju052. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Ai, Y.; Narko, K.; Wang, Z.; Peters, J.M.; Hla, T. PPARδ is pro-tumorigenic in a mouse model of COX-2-induced mammary cancer. Prostaglandins Other Lipid Mediat. 2009, 88, 97–100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Muller-Brusselbach, S.; Komhoff, M.; Rieck, M.; Meissner, W.; Kaddatz, K.; Adamkiewicz, J.; Keil, B.; Klose, K.J.; Moll, R.; Burdick, A.D.; et al. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARbeta-deficient mice. EMBO J. 2007, 26, 3686–3698. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.A.; Yoo, T.; Hwang, J.S.; Kang, E.S.; Lee, W.J.; Paek, K.S.; Park, C.; Kim, J.H.; Do, J.T.; Lim, D.S.; et al. Ligand-activated PPARdelta modulates the migration and invasion of melanoma cells by regulating Snail expression. Am. J. Cancer Res. 2014, 4, 674–682. [Google Scholar] [PubMed]
- Her, N.G.; Jeong, S.I.; Cho, K.; Ha, T.K.; Han, J.; Ko, K.P.; Park, S.K.; Lee, J.H.; Lee, M.G.; Ryu, B.K.; et al. PPARdelta promotes oncogenic redirection of TGF-beta1 signaling through the activation of the ABCA1-Cav1 pathway. Cell Cycle 2013, 12, 1521–1535. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Colby, J.K.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int. J. Mol. Sci. 2018, 19, 3339. https://doi.org/10.3390/ijms19113339
Liu Y, Colby JK, Zuo X, Jaoude J, Wei D, Shureiqi I. The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. International Journal of Molecular Sciences. 2018; 19(11):3339. https://doi.org/10.3390/ijms19113339
Chicago/Turabian StyleLiu, Yi, Jennifer K. Colby, Xiangsheng Zuo, Jonathan Jaoude, Daoyan Wei, and Imad Shureiqi. 2018. "The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor" International Journal of Molecular Sciences 19, no. 11: 3339. https://doi.org/10.3390/ijms19113339
APA StyleLiu, Y., Colby, J. K., Zuo, X., Jaoude, J., Wei, D., & Shureiqi, I. (2018). The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. International Journal of Molecular Sciences, 19(11), 3339. https://doi.org/10.3390/ijms19113339