Preamplification with dUTP and Cod UNG Enables Elimination of Contaminating Amplicons

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
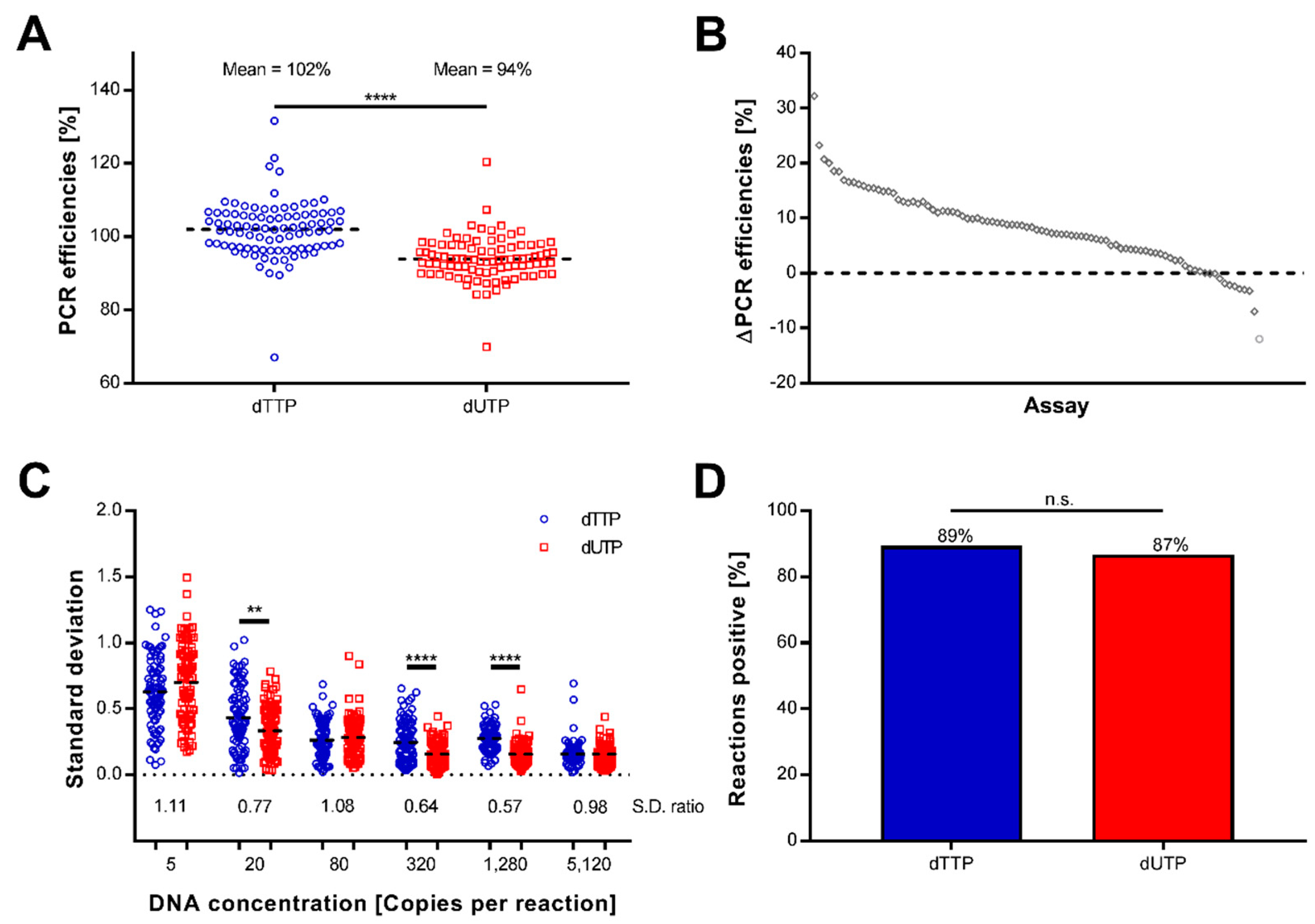
2.1. Preamplification Using Uracil Is Efficient, Reproducible, and Sensitive
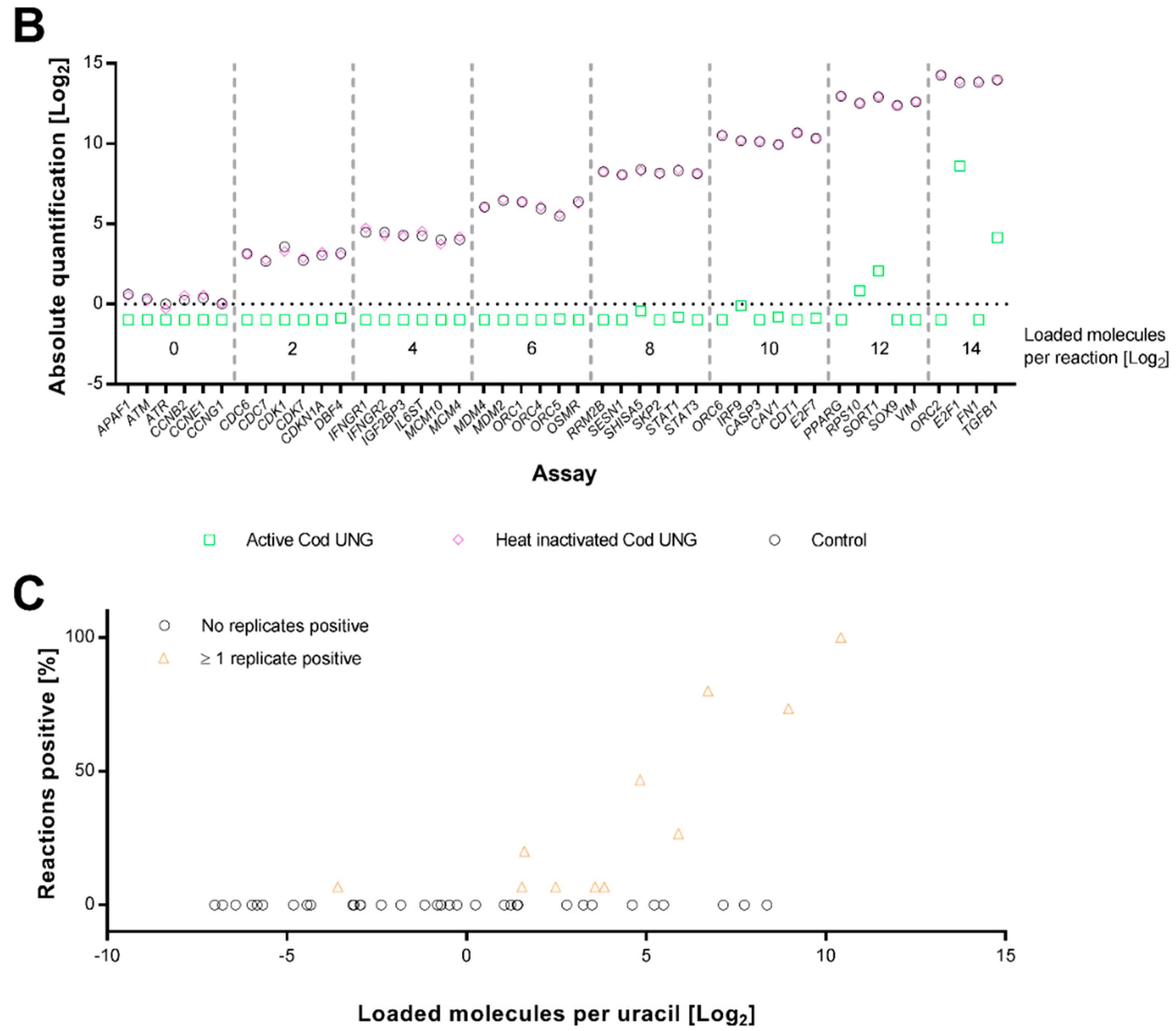
2.2. Cod UNG Is Compatible with Preamplification and Removes Carry-Over Contamination
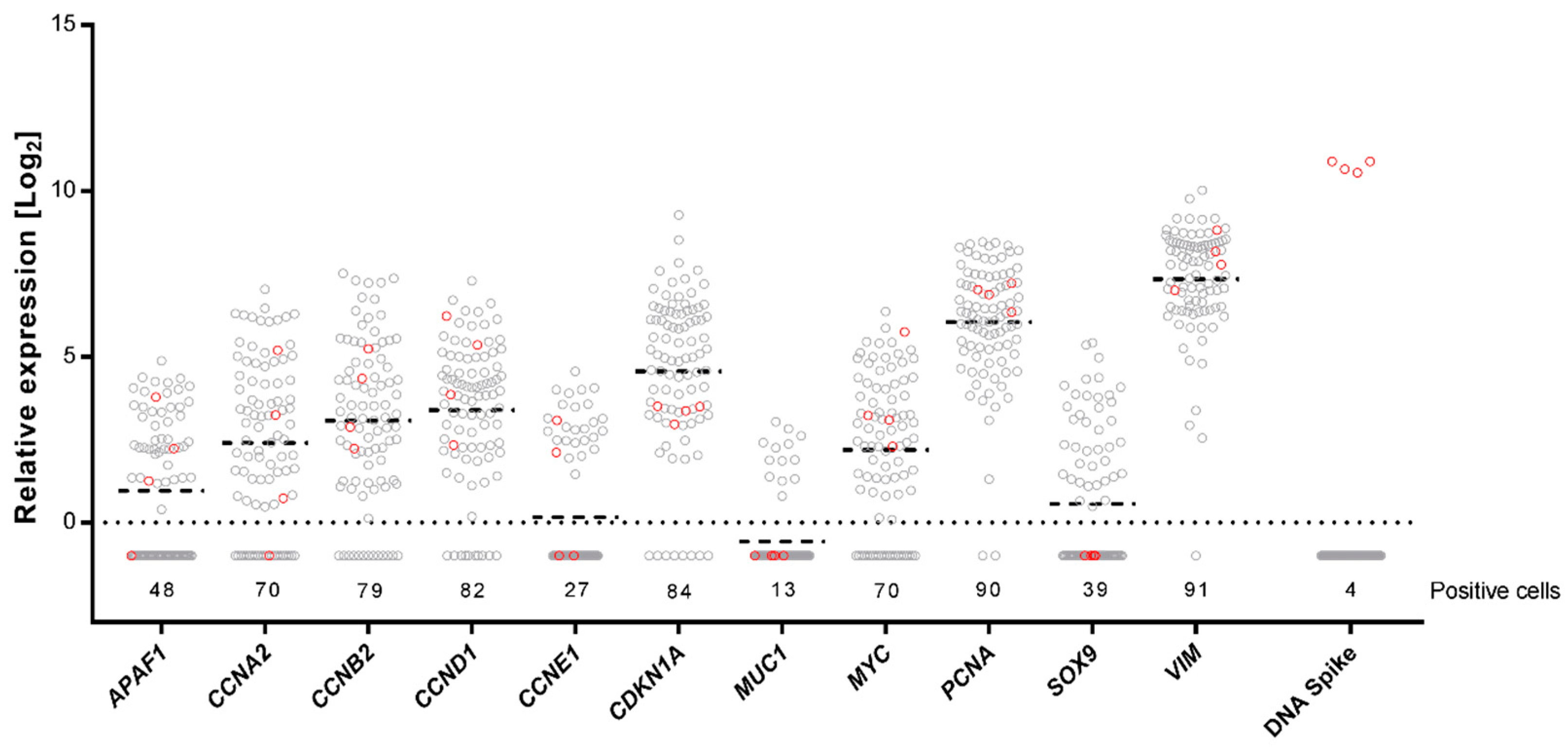
2.3. Single-Cell Gene Expression Profiling Using Preamplification with dUTP and Cod UNG
3. Discussion
4. Materials and Methods
4.1. DNA Standards
4.2. Target-Specific Preamplification
4.3. Cod UNG Treatment
4.4. High-Throughput Quantitative Real-Time PCR
4.5. Single-Cell Quantitative Real-Time PCR
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stahlberg, A.; Kubista, M. The workflow of single-cell expression profiling using quantitative real-time PCR. Expert Rev. Mol. Diagn. 2014, 14, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, A.; Kubista, M. Technical aspects and recommendations for single-cell qPCR. Mol. Asp. Med. 2018, 59, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Akrap, N.; Andersson, D.; Bom, E.; Gregersson, P.; Stahlberg, A.; Landberg, G. Identification of Distinct Breast Cancer Stem Cell Populations Based on Single-Cell Analyses of Functionally Enriched Stem and Progenitor Pools. Stem Cell Rep. 2016, 6, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Andersson, D.; Bom, E.; Walsh, C.; Stahlberg, A.; Landberg, G. Cellular organization and molecular differentiation model of breast cancer-associated fibroblasts. Mol. Cancer 2017, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Rusnakova, V.; Honsa, P.; Dzamba, D.; Stahlberg, A.; Kubista, M.; Anderova, M. Heterogeneity of astrocytes: From development to injury—Single cell gene expression. PLoS ONE 2013, 8, e69734. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Gallant, C.J.; Marinescu, V.D.; Niklasson, M.; Segerman, A.; Flamourakis, G.; Fredriksson, S.; Assarsson, E.; Lundberg, M.; Nelander, S.; et al. Simultaneous Multiplexed Measurement of RNA and Proteins in Single Cells. Cell Rep. 2016, 14, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Wills, Q.F.; Tipping, A.J.; Datta, K.; Mittal, R.; Goldson, A.J.; Sexton, D.W.; Holmes, C.C. Methods for qPCR gene expression profiling applied to 1440 lymphoblastoid single cells. Methods 2013, 59, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Smallwood, S.A.; Lee, H.J.; Krueger, F.; Reik, W.; Kelsey, G. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq). Nat. Protoc. 2017, 12, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Sugitani, A.; Karasaki, H.; Ogata, M.; Nozaki, R.; Sasajima, J.; Yokochi, T.; Asahara, S.; Koizumi, K.; Ando, K.; et al. An improved digital polymerase chain reaction protocol to capture low-copy KRAS mutations in plasma cell-free DNA by resolving ‘subsampling’ issues. Mol. Oncol. 2017, 11, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B.; Choi, D.S.; Luketich, J.D.; Pennathur, A.; Stahlberg, A.; Godfrey, T.E. Multiplex Preamplification of Serum DNA to Facilitate Reliable Detection of Extremely Rare Cancer Mutations in Circulating DNA by Digital PCR. J. Mol. Diagn. 2016, 18, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, A.; Krzyzanowski, P.M.; Jackson, J.B.; Egyud, M.; Stein, L.; Godfrey, T.E. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 2016, 44, e105. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.; Akrap, N.; Svec, D.; Godfrey, T.E.; Kubista, M.; Landberg, G.; Stahlberg, A. Properties of targeted preamplification in DNA and cDNA quantification. Expert Rev. Mol. Diagn. 2015, 15, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Kroneis, T.; Jonasson, E.; Andersson, D.; Dolatabadi, S.; Stahlberg, A. Global preamplification simplifies targeted mRNA quantification. Sci. Rep. 2017, 7, 45219. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.C.; Berninger, M.S.; Hartley, J.L. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene 1990, 93, 125–128. [Google Scholar] [CrossRef]
- Thornton, C.G.; Hartley, J.L.; Rashtchian, A. Utilizing uracil DNA glycosylase to control carryover contamination in PCR: Characterization of residual UDG activity following thermal cycling. Biotechniques 1992, 13, 180–184. [Google Scholar] [PubMed]
- ArcticZymes.com. Available online: https://arcticzymes.com/technology/cod-uracil-dna-glycosylase/ (accessed on 1 October 2018).
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006, 4, e309. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, M.; Stahlberg, A.; Rorsman, P.; Kubista, M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.L.; Rashtchian, A. Dealing with contamination: Enzymatic control of carryover contamination in PCR. PCR Methods Appl. 1993, 3, S10–S14. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.; Chan, K.C. Setting up a polymerase chain reaction laboratory. Methods Mol. Biol. 2006, 336, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Borst, A.; Box, A.T.; Fluit, A.C. False-positive results and contamination in nucleic acid amplification assays: Suggestions for a prevent and destroy strategy. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.E.; Wangh, L.J. Effectiveness and limitations of uracil-DNA glycosylases in sensitive real-time PCR assays. Biotechniques 2004, 36, 44–46. [Google Scholar] [CrossRef] [PubMed]
- SciencePrimer.com. Available online: http://scienceprimer.com/copy-number-calculator-for-realtime-pcr (accessed on 12 September 2018).
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, A.; Rusnakova, V.; Forootan, A.; Anderova, M.; Kubista, M. RT-qPCR work-flow for single-cell data analysis. Methods 2013, 59, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Svec, D.; Andersson, D.; Pekny, M.; Sjoback, R.; Kubista, M.; Stahlberg, A. Direct cell lysis for single-cell gene expression profiling. Front. Oncol. 2013, 3, 274. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, D.; Svec, D.; Pedersen, C.; Henriksen, J.R.; Ståhlberg, A. Preamplification with dUTP and Cod UNG Enables Elimination of Contaminating Amplicons. Int. J. Mol. Sci. 2018, 19, 3185. https://doi.org/10.3390/ijms19103185
Andersson D, Svec D, Pedersen C, Henriksen JR, Ståhlberg A. Preamplification with dUTP and Cod UNG Enables Elimination of Contaminating Amplicons. International Journal of Molecular Sciences. 2018; 19(10):3185. https://doi.org/10.3390/ijms19103185
Chicago/Turabian StyleAndersson, Daniel, David Svec, Cathrine Pedersen, Jørn Remi Henriksen, and Anders Ståhlberg. 2018. "Preamplification with dUTP and Cod UNG Enables Elimination of Contaminating Amplicons" International Journal of Molecular Sciences 19, no. 10: 3185. https://doi.org/10.3390/ijms19103185
APA StyleAndersson, D., Svec, D., Pedersen, C., Henriksen, J. R., & Ståhlberg, A. (2018). Preamplification with dUTP and Cod UNG Enables Elimination of Contaminating Amplicons. International Journal of Molecular Sciences, 19(10), 3185. https://doi.org/10.3390/ijms19103185