Role of Extracellular Matrix in Development and Cancer Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
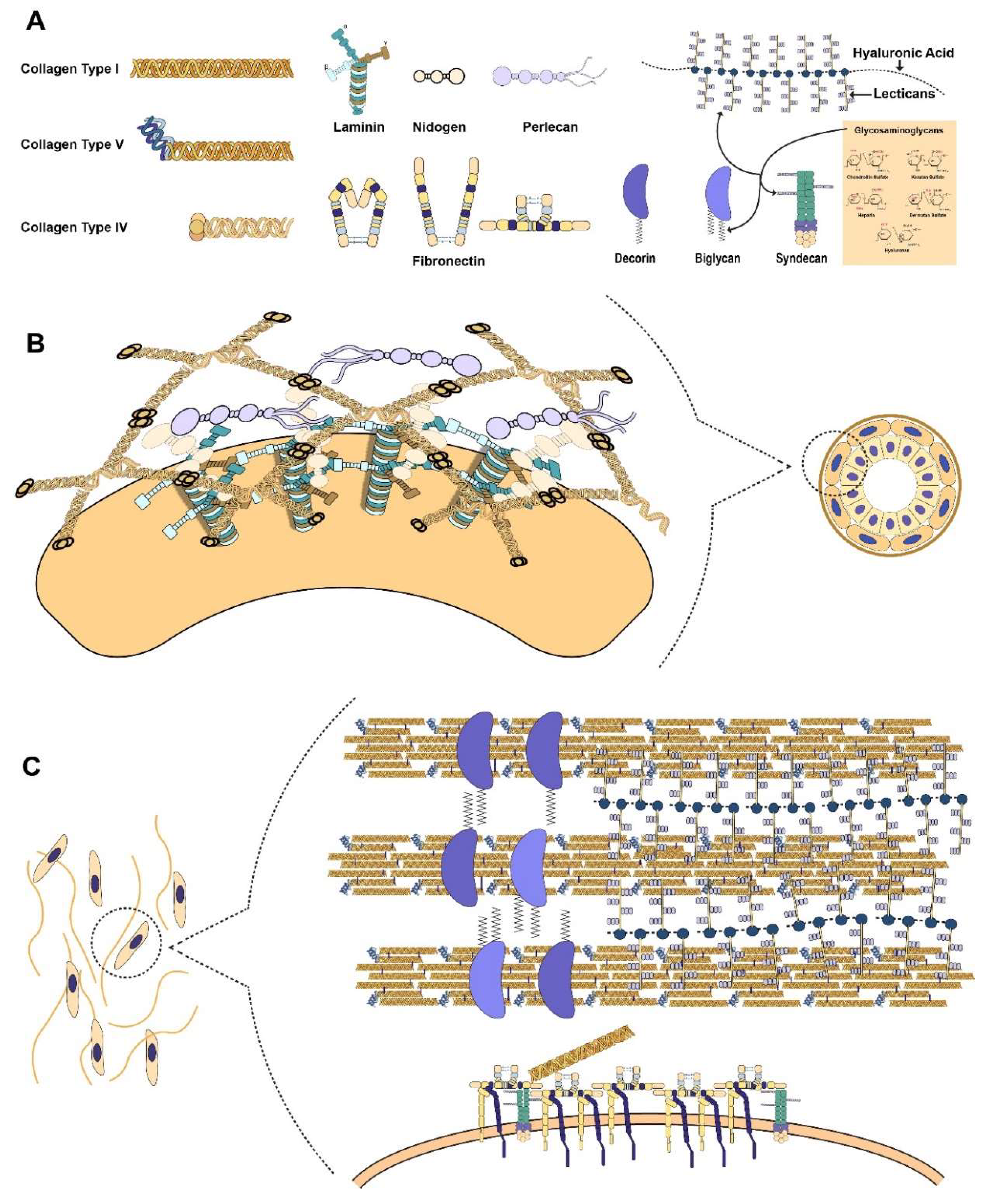
2. Primary Components of the Extracellular Matrix (ECM)
2.1. Collagen as the Basis of ECM Architecture
2.2. Proteoglycans as Functional Modifiers of the ECM
2.3. Connecting the Cell to the ECM through Laminin
2.4. Fibronectin as the Mechanosensitive Connection Between the Cell and ECM
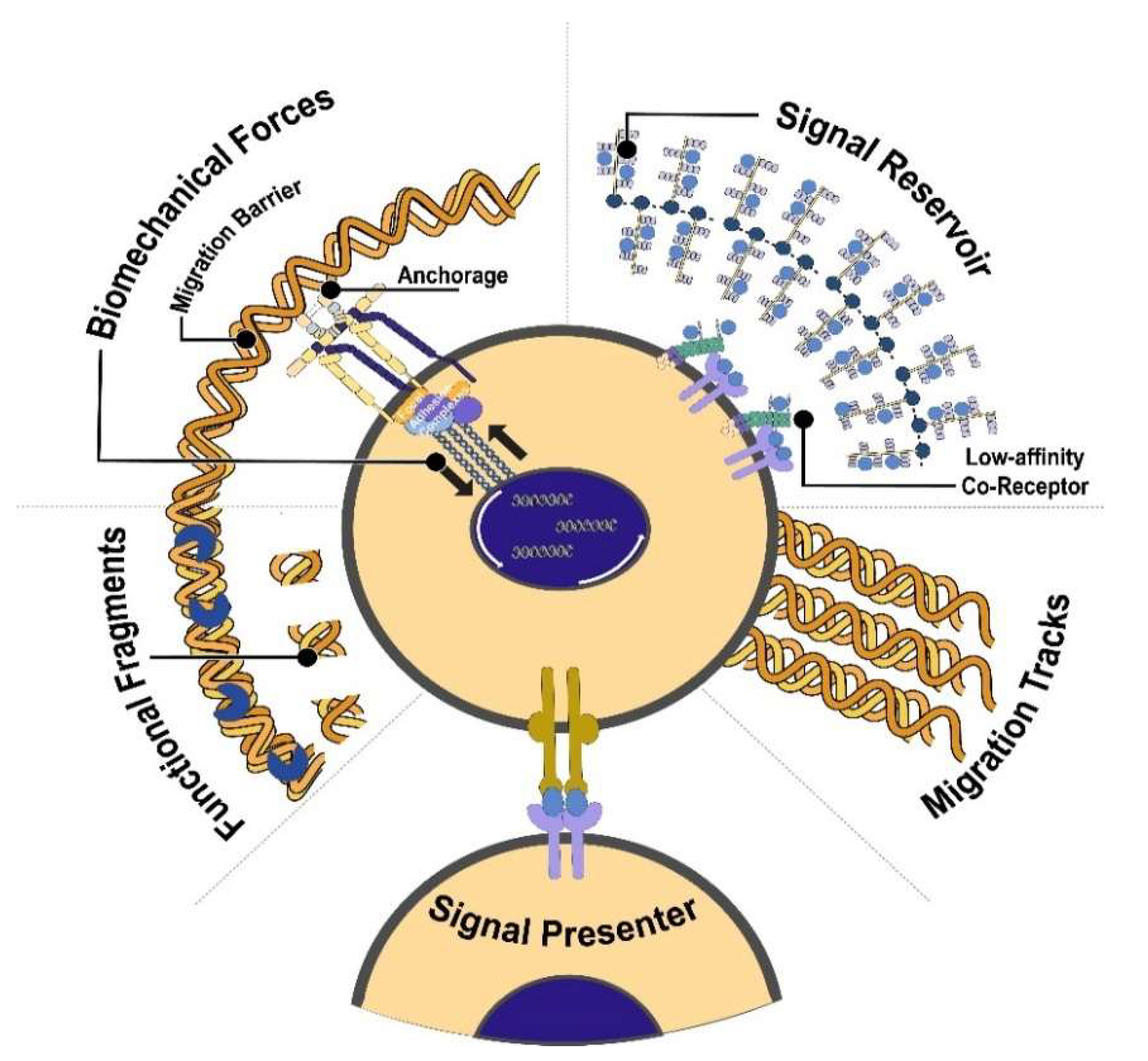
3. Function of ECM
3.1. ECM as Tracks for Migration and Proliferation
3.2. ECM as the Dynamic Blueprint for Development
3.3. ECM as the Driver for Cell Fate
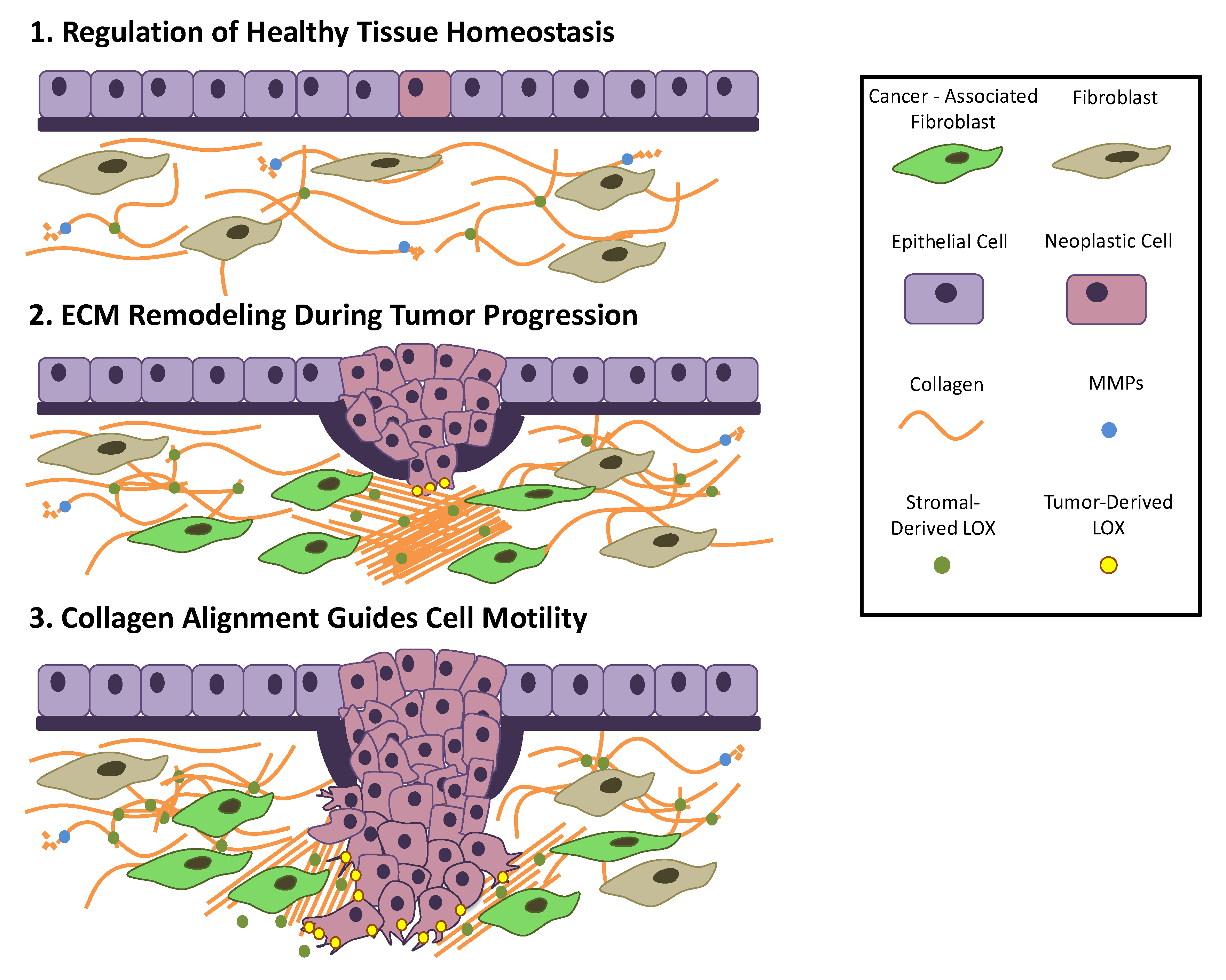
4. Tissue Homeostasis
5. ECM in Cancer
5.1. Dysregulation of ECM Molecules in Cancer Progression
5.2. Protein Unfolding Mediates Mechanotransduction
5.3. YAP & TAZ Mechanotransduction in Cancer Progression
5.4. ECM-Mediated Tumour Initiation and Migration
5.5. Metalloproteinases (MMPs) in Tumour Progression
5.6. Role of Mechanical Stress in Tumour Growth and Treatment
5.7. Quantification of Tumour Cell Mechanical Stress in vivo
5.8. Role of ECM Mechanics in Behaviour of Myofibroblastic Cells
- Tumour Rigidity
- ❖
- Caused by elevated ECM deposition
- ❖
- Mechanically activates pancreatic stellate cells (PSCs) to produce ECM
- ❖
- Induces EMT in epithelial cells
- ❖
- Amplifies growth-factor signalling
- Tumour Stress (Solid and Fluid)
- ❖
- Caused by increased cell proliferation and blood flow
- ❖
- Induces hypoxia in tumours
- ❖
- Augments cell proliferation
- ❖
- Increases chemo resistance
5.9. Seed and Soil
6. Challenges and Future Perspectives
Funding
Conflicts of Interest
References
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Laklai, H.; Weaver, V.M. Force matters: Biomechanical regulation of cell invasion and migration in disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (mmps), the main extracellular matrix (ecm) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M. Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell 1996, 84, 345–357. [Google Scholar] [CrossRef]
- Chen, K.D.; Li, Y.S.; Kim, M.; Li, S.; Yuan, S.; Chien, S.; Shyy, J.Y. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and shc. J. Biol. Chem. 1999, 274, 18393–18400. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, A.; Orr, A.W.; Tzima, E.; Schwartz, M.A. Integrins in mechanotransduction. J. Biol. Chem. 2004, 279, 12001–12004. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J.; Kivirikko, K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Bella, J.; Eaton, M.; Brodsky, B.; Berman, H. Crystal and molecular structure of a collagen-like peptide at 1.9 a resolution. Science 1994, 266, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Persikov, A.V.; Ramshaw, J.A.M.; Kirkpatrick, A.; Brodsky, B. Electrostatic interactions involving lysine make major contributions to collagen triple-helix stability. Biochemistry 2005, 44, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Muiznieks, L.D.; Keeley, F.W. Molecular assembly and mechanical properties of the extracellular matrix: A fibrous protein perspective. Biochim. Biophys. Acta 2013, 1832, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J. Intracellular post-translational modifications of collagens. In Collagen: Primer in Structure, Processing and Assembly; Brinckmann, J., Notbohm, H., Müller, P.K., Eds.; Springer-Verlag Berlin Heidelberg: Heidelberg, Germany, 2005; pp. 115–147. [Google Scholar]
- Birk, D.E.; Zycband, E.I.; Winkelmann, D.A.; Trelstad, R.L. Collagen fibrillogenesis in situ: Fibril segments are intermediates in matrix assembly. Proc. Natl. Acad. Sci. USA 1989, 86, 4549–4553. [Google Scholar] [CrossRef] [PubMed]
- Canty, E.G.; Lu, Y.; Meadows, R.S.; Shaw, M.K.; Holmes, D.F.; Kadler, K.E. Coalignment of plasma membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J. Cell Biol. 2004, 165, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Kalson, N.S.; Starborg, T.; Lu, Y.; Mironov, A.; Humphries, S.M.; Holmes, D.F.; Kadler, K.E. Nonmuscle myosin ii powered transport of newly formed collagen fibrils at the plasma membrane. Proc. Natl. Acad. Sci. USA 2013, 110, E4743–E4752. [Google Scholar] [CrossRef] [PubMed]
- Starborg, T.; Kalson, N.S.; Lu, Y.; Mironov, A.; Cootes, T.F.; Holmes, D.F.; Kadler, K.E. Using transmission electron microscopy and 3view to determine collagen fibril size and three-dimensional organization. Nat. Protoc. 2013, 8, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Hulmes, D.J. Building collagen molecules, fibrils, and suprafibrillar structures. J. Struct. Biol. 2002, 137, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Kadler, K.E.; Holmes, D.F.; Trotter, J.A.; Chapman, J.A. Collagen fibril formation. Biochem. J. 1996, 316, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hulmes, D.J.S. Collagen diversity, synthesis, and assembly. In Collagen: Structure and Mechanics, 1st ed.; Fratzl, P., Ed.; Springer: New York, NY, USA, 2008; pp. 15–47. [Google Scholar]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type v collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, P. Suprastructures of extracellular matrices: Paradigms of functions controlled by aggregates rather than molecules. Cell Tissue Res. 2009, 339, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Ameye, L.; Young, M.F. Mice deficient in small leucine-rich proteoglycans: Novel in vivo models for osteoporosis, osteoarthritis, ehlers-danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology 2002, 12, 107R–116R. [Google Scholar] [CrossRef] [PubMed]
- Molnar, J.; Fong, K.S.K.; He, Q.P.; Hayashi, K.; Kim, Y.; Fong, S.F.T.; Fogelgren, B.; Molnarne Szauter, K.; Mink, M.; Csiszar, K. Structural and functional diversity of lysyl oxidase and the lox-like proteins. Biochim. Biophys. Acta 2003, 1647, 220–224. [Google Scholar] [CrossRef]
- Fratzl, P.; Misof, K.; Zizak, I.; Rapp, G.; Amenitsch, H.; Bernstorff, S. Fibrillar structure and mechanical properties of collagen. J. Struct. Biol. 1997, 122, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, S.; Marinkovich, M.P. Molecular organization of the basement membrane zone. Clin. Dermatol. 2011, 29, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adh. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Leonova, E.I.; Galzitskaya, O.V. Structure and functions of syndecans in vertebrates. Biochem. (Mosc.) 2013, 78, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Miaczynska, M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Kashyap, R.; Zimmermann, P. Syntenin: Key player in cancer exosome biogenesis and uptake? CellCell Adh. Migr. 2017, 11, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Knox, S.M.; Whitelock, J.M. Perlecan: How does one molecule do so many things? Cell Mol. Life Sci. 2006, 63, 2435–2445. [Google Scholar] [CrossRef] [PubMed]
- Farach-Carson, M.C.; Carson, D.D. Perlecan—A multifunctional extracellular proteoglycan scaffold. Glycobiology 2007, 17, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.J.; La Pierre, D.P.; Wu, J.; Yee, A.J.; Yang, B.B. The interaction of versican with its binding partners. Cell Res. 2005, 15, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Yamada, K.M.; Yoneda, M.; Suzuki, S.; Kimata, K. Chondroitin sulfate proteoglycan (pg-m-like proteoglycan) is involved in the binding of hyaluronic acid to cellular fibronectin. J. Biol. Chem. 1986, 261, 13526–13535. [Google Scholar] [PubMed]
- Iozzo, R.V. The biology of the small leucine-rich proteoglycans. J. Biol. Chem. 1999, 274, 18843–18846. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Karamanos, N. Proteoglycans in health and disease: Emerging concepts and future directions. FEBS J. 2010, 277, 3863. [Google Scholar] [CrossRef] [PubMed]
- Kalamajski, S.; Oldberg, A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010, 29, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as therapeutic targets: Successes and cancers. Cancers 2017, 9, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, A.; Naoe, T.; Matsushita, T.; Kaibuchi, K.; Schwartz, M.A. Integrin activation and matrix binding mediate cellular responses to mechanical stretch. J. Biol. Chem. 2005, 280, 16546–16549. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, T.; Katagiri, Y.; Faull, R.; Peter, K.; Tamura, R.; Quaranta, V.; Loftus, J.; Shattil, S.; Ginsberg, M. Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 1994, 124, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, M.; Renedo, A.S.; Huber, F.; Flück, M. How do fibroblasts translate mechanical signals into changes in extracellular matrix production? Matrix Biol. 2003, 22, 73–80. [Google Scholar] [CrossRef]
- Orr, A.W.; Helmke, B.P.; Blackman, B.R.; Schwartz, M.A. Mechanisms of mechanotransduction. Dev. Cell 2006, 10, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799–3804. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Wright, A.J.; Noble, M.; Mahoney, D.J.; Campbell, I.D.; Day, A.J.; Jackson, D.G. Structures of the cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat. Struct. Mol. Biol. 2007, 14, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between hyaluronan and its receptors (cd44, rhamm) regulate the activities of inflammation and cancer. Front. Immunol. 2015, 6, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.; McFerran, N.V.; Pivato, G.; Chambers, E.; Doherty, C.; Steele, D.; Timson, D.J. The 67 kda laminin receptor: Structure, function and role in disease. Biosci. Rep. 2008, 28, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Radziejewski, C.; Campbell, E.; Kovac, L.; McGlynn, M.; Ryan, T.E.; Davis, S.; Goldfarb, M.P.; Glass, D.J.; Lemke, G.; et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol. Cell 1997, 1, 25–34. [Google Scholar] [CrossRef]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, P.; Tomatis, D.; Rosas, M.; Grootjans, J.; Leenaerts, I.; Degeest, G.; Reekmans, G.; Coomans, C.; David, G. Characterization of syntenin, a syndecan-binding pdz protein, as a component of cell adhesion sites and microfilaments. Mol. Biol. Cell 2001, 13, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.K.; Tumova, S.; Couchman, J.R.; Woods, A. Syndecan-4 associates with alpha-actinin. J. Biol. Chem. 2003, 278, 7617–7623. [Google Scholar] [CrossRef] [PubMed]
- Okina, E.; Grossi, A.; Gopal, S.; Multhaupt, H.A.; Couchman, J.R. Alpha-actinin interactions with syndecan-4 are integral to fibroblast-matrix adhesion and regulate cytoskeletal architecture. Int. J. Biochem. Cell Biol. 2012, 44, 2161–2174. [Google Scholar] [CrossRef] [PubMed]
- 62. Bellin, R.M.; Kubicek, J.D.; Frigault, M.J.; Kamien, A.J.; Steward, R.L., Jr.; Barnes, H.M.; DiGlacomo, M.B.; Duncan, L.J.; Edgerly, C.K.; Morse, E.M.; et al. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc. Natl. Acad. Sci. USA 2009, 106, 22102–22107. [Google Scholar] [CrossRef] [PubMed]
- Domogatskaya, A.; Rodin, S.; Tryggvason, K. Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 2012, 28, 523–553. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.; Hunter, I.; Engel, J. Structure and function of laminin: Anatomy of a multidomain glycoprotein. FASEB J. 1990, 4, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Odermatt, E.; Engel, A.; Madri, J.A.; Furthmayr, H.; Rohde, H.; Timpl, R. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J. Mol. Biol. 1981, 150, 97–120. [Google Scholar] [CrossRef]
- Berrier, A.L.; Yamada, K.M. Cell-matrix adhesion. J. Cell Physiol. 2007, 213, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Poschl, E.; Schlotzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen iv is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Behrens, D.T.; Villone, D.; Koch, M.; Brunner, G.; Sorokin, L.; Robenek, H.; Bruckner-Tuderman, L.; Bruckner, P.; Hansen, U. The epidermal basement membrane is a composite of separate laminin- or collagen iv-containing networks connected by aggregated perlecan, but not by nidogens. J. Biol. Chem. 2012, 287, 18700–18709. [Google Scholar] [CrossRef] [PubMed]
- Oberbaumer, I.; Wiedemann, H.; Timpl, R.; Kuhn, K. Shape and assembly of type iv procollagen obtained from cell culture. EMBO J. 1982, 1, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Tsilibary, E.C.; Koliakos, G.G.; Charonis, A.S.; Vogel, A.M.; Raeger, L.A.; Furcht, L.T. Heparin type iv collagen interactions: Equilibrium binding and inhibition of type iv collagen self-assembly. J. Biol. Chem. 1988, 263, 19112–19116. [Google Scholar] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbauer, J.E. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 1991, 113, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fogetry, F.J.; Akiyama, S.K.; Yamada, K.M.; Mosher, D.F. Inhibition of binding of fibronectin to matrix assembly sites by anti-integrin (α5β1) antibodies. J. Cell Biol. 1990, 111, 699–708. [Google Scholar]
- McDonald, J.A.; Quade, B.J.; Broekelmann, T.J.; LaChance, R.; Forsman, K.; Hasegawa, E.; Akiyama, S. Fibronectin’s cell-adhesive domain and an amino-terminal matrix assembly domain participate in its assembly into fibroblast pericellular matrix. J. Biol. Chem. 1987, 262, 2957–2967. [Google Scholar] [PubMed]
- Chung, C.Y.; Erickson, H.P. Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-c. J. Cell Biol. 1997, 110, 1413–1419. [Google Scholar]
- Galante, L.L.; Schwarzbauer, J.E. Requirements for sulfate transport and the diastrophic dysplasia sulfate transporter in fibronectin matrix assembly. J. Cell Biol. 2007, 179, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Morla, A.; Ruoslahti, E. A fibronectin self-assembly site involved in fibronectin matrix assembly: Reconstruction in a synthetic peptide. J. Cell Biol. 1992, 118, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Klass, C.M.; Couchman, J.R.; Woods, A. Control of extracellular matrix assembly by syndecan-2 proteoglycan. J. Cell Sci. 2000, 113, 493–506. [Google Scholar] [PubMed]
- Woods, A. Syndecans: Transmembrane modulators of adhesion and matrix assembly. J. Clin. Invest. 2001, 107, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin regulates latent transforming growth factor-beta (tgf beta) by controlling matrix assembly of latent tgf beta-binding protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef] [PubMed]
- Sottile, J.; Hocking, D.C. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol. Biol. Cell 2002, 13, 3546–3559. [Google Scholar] [CrossRef] [PubMed]
- Dzamba, B.J.; Wu, H.; Jaenisch, R.; Peters, D.M. Fibronectin binding site in type i collagen regulates fibronectin fibril formation. J. Cell Biol. 1993, 121, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Zoppi, N.; De Petro, G.; Marchina, E.; Gardella, R.; Tavian, D.; Ferraboli, S.; Barlati, S. Matrix assembly induction and cell migration and invasion inhibition by a 13-amino acid fibronectin peptide. J. Biol. Chem. 2003, 278, 14346–14355. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.H. Extracellular matrix in development: Insights from mechanisms conserved between invertebrates and vertebrates. Cold Spring Harb. Perspect Biol. 2011, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Entchev, E.V.; Gonzalez-Gaitan, M.A. Morphogen gradient formation and vesicular trafficking. Traffic 2002, 3, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Uhler, C.; Shivashankar, G.V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Knecht, A.K.; Bronner-Fraser, M. Induction of the neural crest: A multigene process. Nat. Rev. Genet. 2002, 3, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Ng, C.; Jana, A.; Padhi, A.; Szymanski, P.; Lee, J.S.H.; Behkam, B.; Nain, A.S. Aligned fibers direct collective cell migration to engineer closing and nonclosing wound gaps. Mol. Biol. Cell 2017, 28, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Motalleb, R.; Berns, E.J.; Patel, P.; Gold, J.; Stupp, S.I.; Georg Kuhn, H. In vivo migration of endogenous brain progenitor cells guided by an injectable peptide amphiphile biomaterial. J. Tissue Eng. Regen. Med. 2018, 12, e2123–e2133. [Google Scholar] [CrossRef] [PubMed]
- Palecek, S.P.; Loftus, J.C.; Ginsberg, M.H.; Lauffenburger, D.A.; Horwitz, A.F. Integrin-ligand binding properties govern cell migration speed through cell substratum adhesiveness. Nature 1997, 385, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Hartman, C.D.; Isenberg, B.C.; Chua, S.G.; Wong, J.Y. Extracellular matrix type modulates cell migration on mechanical gradients. Exp. Cell Res. 2017, 359, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hamilla, S.; Cam, M.; Aranda-Espinoza, H.; Mili, S. Extracellular matrix stiffness and cell contractility control rna localization to promote cell migration. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fata, J.E.; Werb, Z.; Bissell, M.J. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res. 2004, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, G.; Strickland, P.; Coleman, S.; Daniel, C.W. Epithelium-dependent extracellular matrix synthesis in transforming-growth-factor β1-growth-inhibited mouse mammary gland. J. Cell Biol. 1990, 110, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Hinck, L.; Silberstein, G.B. Key stages in mammary gland development: The mammary end bud as a motile organ. Breast Cancer Res. 2005, 7, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Alford, D.; Baeckstrom, D.; Geyp, M.; Pitha, P.; Taylor-Papadimitriou, J. Integrin-matrix interactions affect the form of the structures developing from human mammary epithelial cells in collagen or fibrin gel. J. Cell Sci. 1998, 111, 521–532. [Google Scholar] [PubMed]
- Vogel, W.F.; Aszodi, A.; Alves, F.; Pawson, T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol. Cell Biol. 2001, 21, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.M.; VanDujin, M.M.; Inman, J.L.; Fletcher, D.A.; Bissell, M.J. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science 2006, 314, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Nelson, C.M. Endogenous patterns of mechanical stress are required for branching morphogenesis. Integr. Biol. 2010, 2, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.K.; Cortes, E.; Rice, A.J.; Sarper, M.; Del Rio Hernandez, A. Quantitative analysis of 3d extracellular matrix remodelling by pancreatic stellate cells. Biol Open 2016, 5, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Ortega, N. New functional roles for non-collagenous domains of basement membrane collagens. J. Cell Sci. 2002, 115, 4201–4214. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Kouros-Mehr, H.; Lu, P.; Werb, Z. Hormonal and local control of mammary branching morphogenesis. Differentiation 2006, 74, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Streuli, C.H.; Schmidhauser, C.; Bailey, N.; Yurchenco, P.D.; Skubitz, A.P.N.; Roskelley, C.; Bissell, M.J. Laminin mediates tissue-specific gene expression in mammary epithelia. J. Cell Biol. 1995, 129, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of yap and taz in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of yap/taz in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by yap and taz. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Schroeder, M.C.; Halder, G. Regulation of the hippo pathway by cell architecture and mechanical signals. Semin. Cell Dev. Biol. 2012, 23, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Diaz, M.F.; Price, K.M.; Ozuna, J.A.; Zhang, S.; Sevick-Muraca, E.M.; Hagan, J.P.; Wenzel, P.L. Fluid shear stress activates yap1 to promote cancer cell motility. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Yamamoto, K.; Agarwala, S.; Terai, K.; Fukui, H.; Fukuhara, S.; Ando, K.; Miyazaki, T.; Yokota, Y.; Schmelzer, E.; et al. Flow-dependent endothelial yap regulation contributes to vessel maintenance. Dev. Cell 2017, 40, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, J.Y.; Li, B.; Tian, X.Y.; Chen, L.J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-yap/taz-jnk cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.J.; Chien, S. Flow-dependent yap/taz activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [PubMed]
- Starr, D.A.; Fridolfsson, H.N. Interactions between nuclei and the cytoskeleton are mediated by sun-kash nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 2010, 26, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef] [PubMed]
- Attwood, S.J.; Cortes, E.; Haining, A.W.; Robinson, B.; Li, D.; Gautrot, J.; Del Rio Hernandez, A. Adhesive ligand tether length affects the size and length of focal adhesions and influences cell spreading and attachment. Sci. Rep. 2016, 6, 34334. [Google Scholar] [CrossRef] [PubMed]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.G.; Parks, W.C. Matrix remodeling by mmps during wound repair. Matrix Biol. 2015, 44–46, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tzima, E. Pecam-1 is necessary for flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Osborne, L.D.; Guilluy, C.; Chen, Z.; O’Brien, E.T., 3rd; Reader, J.S.; Burridge, K.; Superfine, R.; Tzima, E. Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.K.; Mauad, T.; Tijin, G.; Karlsson, J.C.; Westergren-Thorsson, G. The extracellular matrix - the under-recognized element in lung disease? J. Pathol. 2016, 240, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Gauldie, J.; Bellaye, P.S. Editorial: Extracellular matrix: The common thread of disease progression in fibrosis? Arthritis Rheumatol. 2016, 68, 1053–1056. [Google Scholar] [PubMed]
- Friedl, P.; Wolf, K. Tube travel: The role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008, 68, 7247–7249. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.A.; Rivera, L.B.; Miller, A.F.; Carbon, J.G.; Dineen, S.P.; Xie, Y.; Castrillon, D.H.; Sage, E.H.; Puolakkainen, P.; Bradshaw, A.D.; et al. Lack of host sparc enhances vascular function and tumor spread in an orthotopic murine model of pancreatic carcinoma. Dis. Model. Mech. 2010, 3, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Ge, G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenviron 2012, 5, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Chen, S.; Yuan, W.; Fan, Q.; Tian, J.; Wang, X.; Chen, L.; Zhang, X.; Wei, W.; Liu, R.; et al. Oriented collagen fibers direct tumor cell intravasation. Proc. Natl. Acad. Sci. USA 2016, 113, 11208–11213. [Google Scholar] [CrossRef] [PubMed]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Riching, K.M.; Cox, B.L.; Salick, M.R.; Pehlke, C.; Riching, A.S.; Ponik, S.M.; Bass, B.R.; Crone, W.C.; Jiang, Y.; Weaver, A.M.; et al. 3d collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys. J. 2014, 107, 2546–2558. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikstrom, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A., Jr.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Invest. 2000, 106, 349–360. [Google Scholar] [CrossRef] [PubMed]
- McAtee, C.O.; Barycki, J.J.; Simpson, M.A. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv. Cancer Res. 2014, 123, 1–34. [Google Scholar] [PubMed]
- Huveneers, S.; Danen, E.H. Adhesion signaling - crosstalk between integrins, src and rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth factors, matrices, and forces combine and control stem cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and yap-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The hippo pathway effectors taz and yap in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S. Role of yap/taz in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Haining, A.W.; von Essen, M.; Attwood, S.J.; Hytonen, V.P.; Del Rio Hernandez, A. All subdomains of the talin rod are mechanically vulnerable and may contribute to cellular mechanosensing. ACS Nano. 2016, 10, 6648–6658. [Google Scholar] [CrossRef] [PubMed]
- Haining, A.W.M.; Rahikainen, R.; Cortes, E.; Lachowski, D.; Rice, A.; von Essen, M.; Hytonen, V.P.; Del Rio Hernandez, A. Mechanotransduction in talin through the interaction of the r8 domain with dlc1. PLoS Biol. 2018, 16, e2005599. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a twist1-g3bp2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of yap/taz: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Guan, K.L. The yap and taz transcription co-activators: Key downstream effectors of the mammalian hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. Yap/taz as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Lakshmanan, M.; Swa, H.L.; Chen, J.; Zhang, X.; Ong, Y.S.; Loo, L.S.; Akincilar, S.C.; Gunaratne, J.; Tergaonkar, V.; et al. An oncogenic role of agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat. Commun. 2015, 6, 6184. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Njah, K.; Pobbati, A.V.; Lim, Y.B.; Raju, A.; Lakshmanan, M.; Tergaonkar, V.; Lim, C.T.; Hong, W. Agrin as a mechanotransduction signal regulating yap through the hippo pathway. Cell Rep. 2017, 18, 2464–2479. [Google Scholar] [CrossRef] [PubMed]
- Tatrai, P.; Dudas, J.; Batmunkh, E.; Mathe, M.; Zalatnai, A.; Schaff, Z.; Ramadori, G.; Kovalszky, I. Agrin, a novel basement membrane component in human and rat liver, accumulates in cirrhosis and hepatocellular carcinoma. Lab. Invest. 2006, 86, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Hong, W. Linking extracellular matrix agrin to the hippo pathway in liver cancer and beyond. Cancers (Basel) 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.C.; Lu, S.; Pan, M.; Hong, A.W.; et al. Rap2 mediates mechanoresponses of the hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Taira, K.; Umikawa, M.; Takei, K.; Myagmar, B.E.; Shinzato, M.; Machida, N.; Uezato, H.; Nonaka, S.; Kariya, K. The traf2- and nck-interacting kinase as a putative effector of rap2 to regulate actin cytoskeleton. J. Biol. Chem. 2004, 279, 49488–49496. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Hagedorn, E.J.; Sherwood, D.R. Cell invasion through basement membrane: The netrin receptor dcc guides the way. Worm 2013, 2, e26169. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.C.; Lohmer, L.L.; Hagedorn, E.J.; Sherwood, D.R. Traversing the basement membrane in vivo: A diversity of strategies. J. Cell Biol. 2014, 204, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, R.; Matsuoka, T.; Kimura-Yoshida, C.; Han, S.W.; Mochida, K.; Adachi, T.; Takayama, S.; Matsuo, I. External mechanical cues trigger the establishment of the anterior-posterior axis in early mouse embryos. Dev. Cell 2013, 27, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, E.J.; Ziel, J.W.; Morrissey, M.A.; Linden, L.M.; Wang, Z.; Chi, Q.; Johnson, S.A.; Sherwood, D.R. The netrin receptor dcc focuses invadopodia-driven basement membrane transmigration in vivo. J. Cell Biol. 2013, 201, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.; Wiesner, C.; Himmel, M. Degrading devices: Invadosomes in proteolytic cell invasion. Annu. Rev. Cell Dev. Biol. 2011, 27, 185–211. [Google Scholar] [CrossRef] [PubMed]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Ihara, S.; Hagedorn, E.J.; Morrissey, M.A.; Chi, Q.; Motegi, F.; Kramer, J.M.; Sherwood, D.R. Basement membrane sliding and targeted adhesion remodels tissue boundaries during uterine-vulval attachment in caenorhabditis elegans. Nat. Cell Biol. 2011, 13, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-β1 release. Biochem. J. 1997, 322, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Wang, C.Y.; Werb, Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015, 44–46, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. Tgf-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Moberg, P.E.; Miles, L.A.; Wagner, S.; Soloway, P.; Gardner, H.A. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc. Natl. Acad. Sci. USA 2000, 97, 2202–2207. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papatsoris, A.G.; Papavassiliou, A.G. Matrix metalloproteinase inhibitors as anticancer agents. Int. J. Biochem. Cell Biol. 2008, 40, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Dutton, C.M.; Qi, W.-N.; Block, J.A.; Brodt, P.; Durko, M.; Scully, S.P. Inhibition of mmp-1 expression by antisense rna decreases invasiveness of human chondrosarcoma. J. Orthop. Res. 2003, 21, 1063–1070. [Google Scholar] [CrossRef]
- Fingleton, B. Mmps as therapeutic targets—still a viable option? Semin. Cell Dev. Biol. 2008, 19, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, O.; Koshy, S.T.; Branco da Cunha, C.; Shin, J.W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, R.W.; Cowan, C.R.; Mih, J.D.; Koryakina, Y.; Gioeli, D.; Slack-Davis, J.K.; Blackman, B.R.; Tschumperlin, D.J.; Parsons, J.T. Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS One 2010, 5, e12905. [Google Scholar] [CrossRef] [PubMed]
- Mpekris, F.; Angeli, S.; Pirentis, A.P.; Stylianopoulos, T. Stress-mediated progression of solid tumors: Effect of mechanical stress on tissue oxygenation, cancer cell proliferation, and drug delivery. Biomech. Model. Mechanobiol. 2015, 14, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. An indirect way to tame cancer. Sci. Am. 2014, 310, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T. The solid mechanics of cancer and strategies for improved therapy. J. Biomech. Eng. 2017, 139. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Boucher, Y.; Ferrone, C.R.; Roberge, S.; Martin, J.D.; Stylianopoulos, T.; Bardeesy, N.; DePinho, R.A.; Padera, T.P.; Munn, L.L.; et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 2014, 26, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Tse, J.; Jain, R.K.; Munn, L.L. Micro-environmental mechanical stress controls tumor spheroid size and morphology by suppressing proliferation and inducing apoptosis in cancer cells. PLoS One 2009, 4, e4632. [Google Scholar] [CrossRef] [PubMed]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling stress: The mechanics of cancer progression and aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed]
- Koumoutsakos, P.; Pivkin, I.; Milde, F. The fluid mechanics of cancer and its therapy. Annu. Rev. Fluid Mech. 2013, 45, 325–355. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Zanetti, M. Tumor stress inside out: Cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment. J. Immunol. 2011, 187, 4403–4409. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed]
- Sarntinoranont, M.; Rooney, F.; Ferrari, M. Interstitial stress and fluid pressure within a growing tumor. Ann. Biomed. Eng. 2003, 31, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Gaustad, J.V.; Egeland, T.A.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Jain, R.K. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 18632–18637. [Google Scholar] [CrossRef] [PubMed]
- Nia, H.T.; Liu, H.; Seano, G.; Datta, M.; Jones, D.; Rahbari, N.; Incio, J.; Chauhan, V.P.; Jung, K.; Martin, J.D.; et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat.Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Campas, O.; Mammoto, T.; Hasso, S.; Sperling, R.A.; O’Connell, D.; Bischof, A.G.; Maas, R.; Weitz, D.A.; Mahadevan, L.; Ingber, D.E. Quantifying cell-generated mechanical forces within living embryonic tissues. Nat. Methods 2014, 11, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Lucio, A.A.; Mongera, A.; Shelton, E.; Chen, R.; Doyle, A.M.; Campas, O. Spatiotemporal variation of endogenous cell-generated stresses within 3d multicellular spheroids. Sci. Rep. 2017, 7, 12022. [Google Scholar] [CrossRef] [PubMed]
- Lucio, A.A.; Ingber, D.E.; Campas, O. Generation of biocompatible droplets for in vivo and in vitro measurement of cell-generated mechanical stresses. Methods Cell Biol. 2015, 125, 373–390. [Google Scholar] [PubMed]
- Rowghanian, P.; Meinhart, C.D.; Campàs, O. Dynamics of ferrofluid drop deformations under spatially uniform magnetic fields. J. Fluid Mech. 2016, 802, 245–262. [Google Scholar] [CrossRef]
- Serwane, F.; Mongera, A.; Rowghanian, P.; Kealhofer, D.A.; Lucio, A.A.; Hockenbery, Z.M.; Campas, O. In vivo quantification of spatially varying mechanical properties in developing tissues. Nat. Methods 2017, 14, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
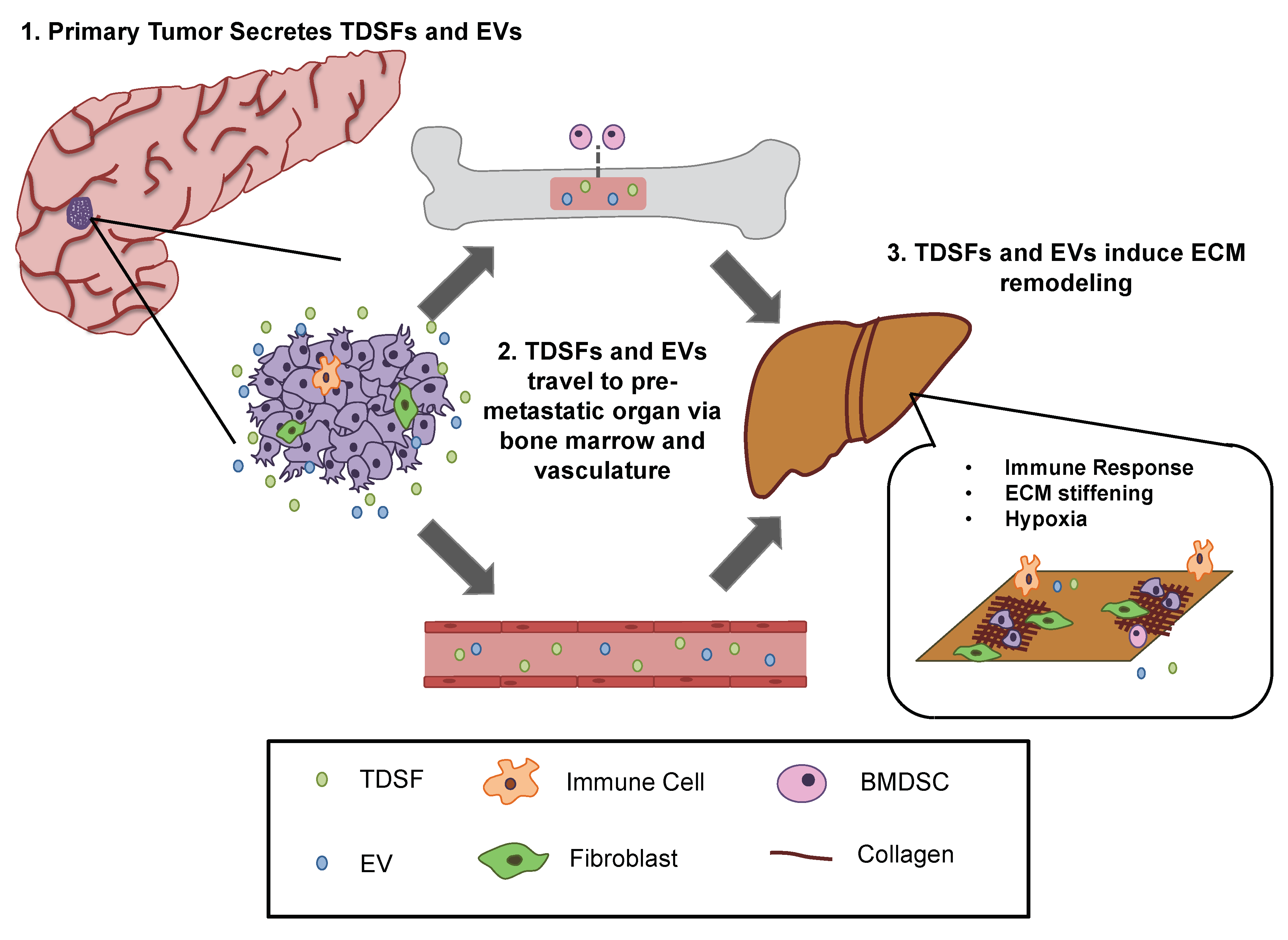
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. Vegfr1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- Lachowski, D.; Cortes, E.; Pink, D.; Chronopoulos, A.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A.E. Substrate rigidity controls activation and durotaxis in pancreatic stellate cells. Sci. Rep. 2017, 7, 2506. [Google Scholar] [CrossRef] [PubMed]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Rio Hernandez, A.E. Fak controls the mechanical activation of yap, a transcriptional regulator required for durotaxis. FASEB J. 2017, 32, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. Atra mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sarper, M.; Cortes, E.; Lieberthal, T.J.; Del Rio Hernandez, A. Atra modulates mechanical activation of tgf-beta by pancreatic stellate cells. Sci.. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.P. The lymph node pre-metastatic niche. J. Mol. Med. (Berl) 2015, 93, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Moran, P.; Huelsken, J. Complex metastatic niches: Already a target for therapy? Curr. Opin. Cell Biol. 2014, 31, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. Lox-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Hoye, A.M.; Agrawal, A.; Bird, D.; Latif, N.A.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.; Pickup, M.; Pang, Y.; Gorska, A.E.; Li, Z.; Chytil, A.; Geng, Y.; Gray, J.W.; Moses, H.L.; Yang, L. Gr-1+cd11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010, 70, 6139–6149. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, C.; Mojares, E.; Del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. https://doi.org/10.3390/ijms19103028
Walker C, Mojares E, Del Río Hernández A. Role of Extracellular Matrix in Development and Cancer Progression. International Journal of Molecular Sciences. 2018; 19(10):3028. https://doi.org/10.3390/ijms19103028
Chicago/Turabian StyleWalker, Cameron, Elijah Mojares, and Armando Del Río Hernández. 2018. "Role of Extracellular Matrix in Development and Cancer Progression" International Journal of Molecular Sciences 19, no. 10: 3028. https://doi.org/10.3390/ijms19103028
APA StyleWalker, C., Mojares, E., & Del Río Hernández, A. (2018). Role of Extracellular Matrix in Development and Cancer Progression. International Journal of Molecular Sciences, 19(10), 3028. https://doi.org/10.3390/ijms19103028