Specialized Pro-Resolving Lipid Mediators in Cystic Fibrosis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Cystic Fibrosis
2. Persistent Inflammation in Cystic Fibrosis
3. Resolution of Inflammation
4. Abnormalities of Lipid Levels in Cystic Fibrosis
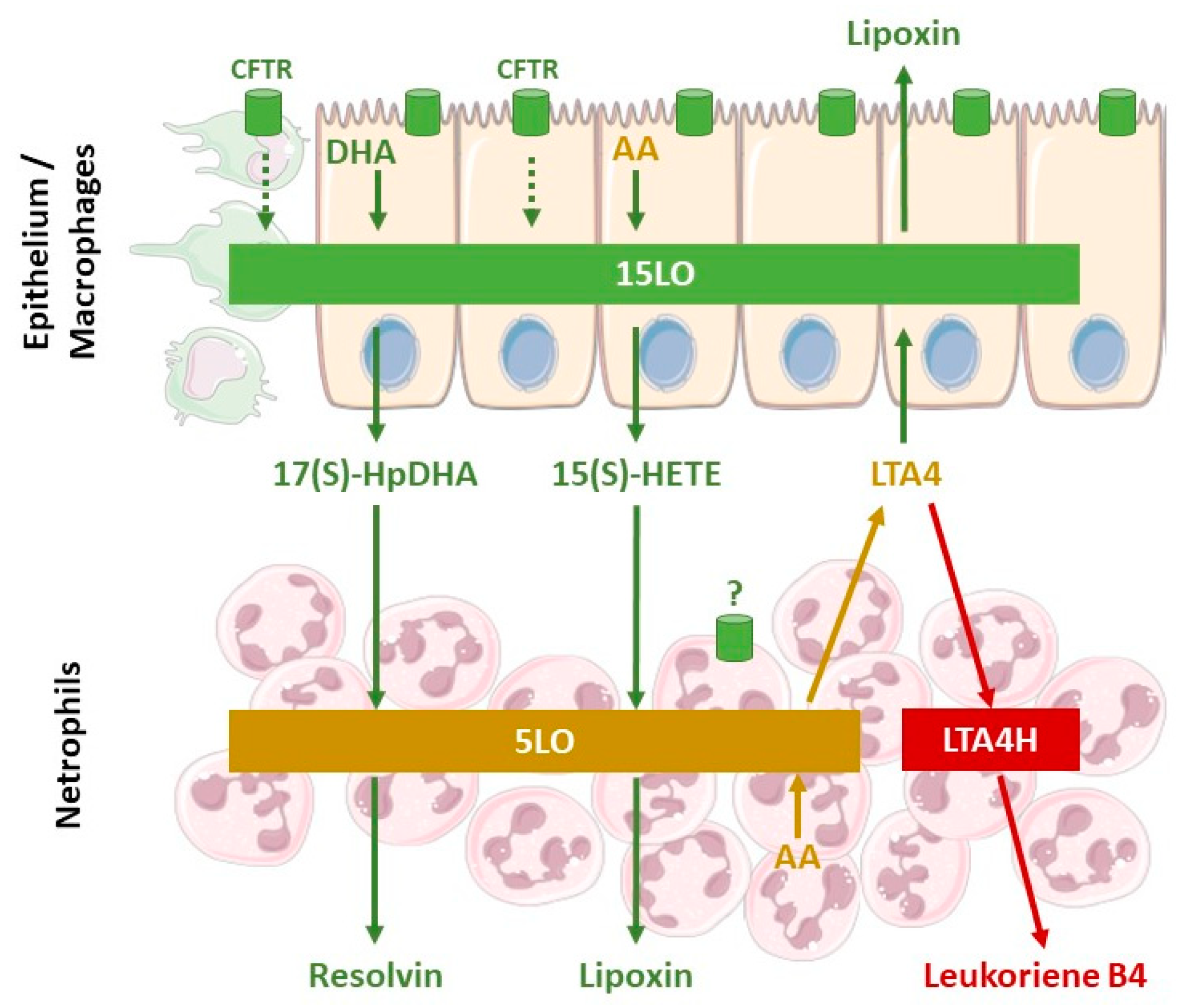
5. Cellular Mechanisms Involved in the Defective Production of SPM in Cystic Fibrosis
6. SPM Impact on CF Airway Epithelial Transport and ASL Height
7. SPMs’ Role in Bacterial Clearance and Limiting Tissue Damage
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pranke, I.M.; Sermet-Gaudelus, I. Biosynthesis of cystic fibrosis transmembrane conductance regulator. Int. J. Biochem. Cell Biol. 2014, 52, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Airway surface dehydration in cystic fibrosis: Pathogenesis and therapy. Annu. Rev. Med. 2007, 58, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Noah, T.L. Endotoxin activity and inflammatory markers in the airways of young patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2002, 165, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.P.; Chmiel, J.F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 2015, 50, S39–S56. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Berger, M.; Konstan, M.W. The role of inflammation in the pathophysiology of CF lung disease. Clin. Rev. Allergy Immunol. 2002, 23, 5–27. [Google Scholar] [CrossRef]
- Armstrong, D.S.; Grimwood, K.; Carlin, J.B.; Carzino, R.; Gutièrrez, J.P.; Hull, J.; Olinsky, A.; Phelan, E.M.; Robertson, C.F.; Phelan, P.D. Lower airway inflammation in infants and young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1997, 156, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.J.; Soong, G.; Bryan, R.; Saba, S.; Prince, A. Activation of NF-kappaB in airway epithelial cells is dependent on CFTR trafficking and Cl− channel function. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L71–L78. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Soltys, J.; Berger, M. Role of IL-10 deficiency in excessive nuclear factor-kappaB activation and lung inflammation in cystic fibrosis transmembrane conductance regulator knockout mice. J. Allergy Clin. Immunol. 2005, 115, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Cohen, T.S.; Alhede, M.; Harfenist, B.S.; Martin, F.J.; Prince, A. Induction of type I interferon signaling by Pseudomonas aeruginosa is diminished in cystic fibrosis epithelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Vandivier, R.W.; Richens, T.R.; Horstmann, S.A.; de Cathelineau, A.M.; Ghosh, M.; Reynolds, S.D.; Xiao, Y.Q.; Riches, D.W.; Plumb, J.; Vachon, E.; et al. Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L677–L686. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Jackson, P.L.; Liu, G.; Hardison, M.; Livraghi, A.; Solomon, G.M.; McQuaid, D.B.; Noerager, B.D.; Gaggar, A.; Clancy, J.P.; et al. Potential role of high-mobility group box 1 in cystic fibrosis airway disease. Am. J. Respir. Crit. Care Med. 2008, 178, 822–831. [Google Scholar] [CrossRef] [PubMed]
- McKeon, D.J.; Condliffe, A.M.; Cowburn, A.S.; Cadwallader, K.C.; Farahi, N.; Bilton, D.; Chilvers, E.R. Prolonged survival of neutrophils from patients with Delta F508 CFTR mutations. Thorax 2008, 63, 660–661. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Bonvillain, R.W.; Valentine, V.G.; Lombard, G.A.; LaPlace, S.G.; Nauseef, W.M.; Wang, G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J. Leukoc. Biol. 2008, 83, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm.-Lond. 2017, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Gotlinger, K.; Hong, S.; Arita, M. Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: An overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat. 2004, 73, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Kohli, P.; Gotlinger, K.; Haworth, O.; Hong, S.; Kazani, S.; Israel, E.; Haley, K.J.; Serhan, C.N. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J. Immunol. 2007, 178, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Okamoto, S.; Ishikawa, Y.; Matsushima, K. Molecular mechanism of interleukin-8 gene expression. J. Leukoc. Biol. 1994, 56, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yacoubian, S.; Yang, R. Anti-inflammatory and proresolving lipid mediators. Annu. Rev. Pathol. 2008, 3, 279–312. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Fukunaga, K.; Levy, M.A.; Levy, B.D. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am. J. Pathol. 2006, 168, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Aspects Med. 2017, 58, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.T.; Huang, N.N.; Bassett, D.R. The fatty acid composition of the serum chylomicrons and adipose tissue of children with cystic fibrosis of the pancreas. J. Pediatr. 1962, 60, 394–403. [Google Scholar] [CrossRef]
- Underwood, B.A.; Denning, C.R.; Navab, M. Polyunsaturated fatty acids and tocopherol levels in patients with cystic fibrosis. Ann. N. Y. Acad. Sci. 1972, 203, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.D.; Blanco, P.G.; Zaman, M.M.; Shea, J.C.; Ollero, M.; Hopper, I.K.; Weed, D.A.; Gelrud, A.; Regan, M.M.; Laposata, M.; et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N. Engl. J. Med. 2004, 350, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Gilljam, H.; Strandvik, B.; Ellin, A.; Wiman, L.G. Increased mole fraction of arachidonic acid in bronchial phospholipids in patients with cystic fibrosis. Scand. J. Clin. Lab. Investig. 1986, 46, 511–518. [Google Scholar] [CrossRef]
- Elliott, R.B.; Robinson, P.G. Unusual clinical course in a child with cystic fibrosis treated with fat emulsion. Arch. Dis. Child. 1975, 50, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.B. A therapeutic trial of fatty acid supplementation in cystic fibrosis. Pediatrics 1976, 57, 474–479. [Google Scholar] [PubMed]
- Medjane, S.; Raymond, B.; Wu, Y.; Touqui, L. Impact of CFTR DeltaF508 mutation on prostaglandin E2 production and type IIA phospholipase A2 expression by pulmonary epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L816–L824. [Google Scholar] [CrossRef] [PubMed]
- László, A.; Németh, M.; Gyukovits, K. Activity of phospholipase A in serum from patients with cystic fibrosis. Clin. Chim. Acta 1993, 214, 105–107. [Google Scholar] [CrossRef]
- Berguerand, M.; Klapisz, E.; Thomas, G.; Humbert, L.; Jouniaux, A.M.; Olivier, J.L.; Béréziat, G.; Masliah, J. Differential stimulation of cytosolic phospholipase A2 by bradykinin in human cystic fibrosis cell lines. Am. J. Respir. Cell Mol. Biol. 1997, 17, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Borot, F.; Vieu, D.L.; Faure, G.; Fritsch, J.; Colas, J.; Moriceau, S.; Baudouin-Legros, M.; Brouillard, F.; Ayala-Sanmartin, J.; Touqui, L.; et al. Eicosanoid release is increased by membrane destabilization and CFTR inhibition in Calu-3 cells. PLoS ONE 2009, 4, e7116. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. Prostaglandins Leukot. Essent. Fatty Acids 2010, 83, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B.; O’Neal, W.K.; Ali, M.A.; Hammar, U. Low linoleic and high docosahexaenoic acids in a severe phenotype of transgenic cystic fibrosis mice. Exp. Biol. Med. 2018, 243, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B.; Brönnegård, M.; Gilljam, H.; Carlstedt-Duke, J. Relation between defective regulation of arachidonic acid release and symptoms in cystic fibrosis. Scand. J. Gastroenterol. Suppl. 1988, 143, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.K.; Kelly, F.J. Role of free radicals in the pathogenesis of cystic fibrosis. Thorax 1994, 49, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinter, M.; Shank, S.; Cotton, C.; Kelley, T.J.; Ziady, A.G. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS ONE 2008, 3, e3367. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Still, J.D.; Powers, C.A.; Hoffman, D.R.; Boyd-Trull, K.; Lester, L.A.; Benisek, D.C.; Arterburn, L.M. Bioavailability and safety of a high dose of docosahexaenoic acid triacylglycerol of algal origin in cystic fibrosis patients: A randomized, controlled study. Nutrition 2006, 22, 36–46. [Google Scholar] [CrossRef] [PubMed]
- McKarney, C.; Everard, M.; N’Diaye, T. Omega-3 fatty acids (from fish oils) for cystic fibrosis. Cochrane Database Syst. Rev. 2007, CD002201. [Google Scholar] [CrossRef] [PubMed]
- Coste, T.C.; Armand, M.; Lebacq, J.; Lebecque, P.; Wallemacq, P.; Leal, T. An overview of monitoring and supplementation of omega 3 fatty acids in cystic fibrosis. Clin. Biochem. 2007, 40, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Ringholz, F.C.; Buchanan, P.J.; Clarke, D.T.; Millar, R.G.; McDermott, M.; Linnane, B.; Harvey, B.J.; McNally, P.; Urbach, V. Reduced 15-lipoxygenase 2 and lipoxin A4/leukotriene B4 ratio in children with cystic fibrosis. Eur. Respir. J. 2014, 44, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Karp, C.L.; Flick, L.M.; Park, K.W.; Softic, S.; Greer, T.M.; Keledjian, R.; Yang, R.; Uddin, J.; Guggino, W.B.; Atabani, S.F.; et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat. Immunol. 2004, 5, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Starosta, V.; Ratjen, F.; Rietschel, E.; Paul, K.; Griese, M. Anti-inflammatory cytokines in cystic fibrosis lung disease. Eur. Respir. J. 2006, 28, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Eickmeier, O.; Fussbroich, D.; Mueller, K.; Serve, F.; Smaczny, C.; Zielen, S.; Schubert, R. Pro-resolving lipid mediator Resolvin D1 serves as a marker of lung disease in cystic fibrosis. PLoS ONE 2017, 12, e0171249. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Ozcan, L.; Spolitu, S.; Hellmann, J.; Spite, M.; Backs, J.; Tabas, I. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl. Acad Sci. USA 2014, 111, 14530–14535. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Evangelista, V.; De Cristofaro, R.; Recchiuti, A.; Pandolfi, A.; Di Silvestre, S.; Manarini, S.; Martelli, N.; Rocca, B.; Petrucci, G.; et al. Cystic fibrosis transmembrane conductance regulator (CFTR) expression in human platelets: Impact on mediators and mechanisms of the inflammatory response. FASEB J. 2010, 24, 3970–3980. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, L.; Guerrera, I.C.; Papon, J.F.; Chhuon, C.; Zadigue, P.; Prulière-Escabasse, V.; Amselem, S.; Escudier, E.; Coste, A.; Edelman, A. Proteomic analysis of nasal epithelial cells from cystic fibrosis patients. PLoS ONE 2014, 9, e108671. [Google Scholar] [CrossRef] [PubMed]
- Jame, A.J.; Lackie, P.M.; Cazaly, A.M.; Sayers, I.; Penrose, J.F.; Holgate, S.T.; Sampson, A.P. Human bronchial epithelial cells express an active and inducible biosynthetic pathway for leukotrienes B4 and C4. Clin. Exp. Allergy 2007, 37, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Salomon, J.J.; Leitz, D.; Feigenbutz, D.; Korsch, L.; Lisewski, I.; Schrimpf, K.; Millar-Büchner, P.; Mall, M.A.; Frings, S.; et al. Expression and function of Anoctamin 1/TMEM16A calcium-activated chloride channels in airways of in vivo mouse models for cystic fibrosis research. Pflugers Arch. 2018, 470, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Mall, M. Pharmacotherapy of the ion transport defect in cystic fibrosis: Role of purinergic receptor agonists and other potential therapeutics. Am. J. Respir. Med. 2003, 2, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M. The role of intracellular calcium signals in inflammatory responses of polarised cystic fibrosis human airway epithelia. Drugs R. D. 2006, 7, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Salomon, J.J.; Sheppard, D.N.; Mall, M.A.; Galietta, L.J. Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr. Opin. Pharmacol. 2017, 34, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Strug, L.J.; Gonska, T.; He, G.; Keenan, K.; Ip, W.; Boëlle, P.Y.; Lin, F.; Panjwani, N.; Gong, J.; Li, W.; et al. Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet. 2016, 25, 4590–4600. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, C.A.; Mitra, S.; Mishra, S.K.; Wang, X.; Zhao, Y.; Pilewski, J.M.; Madden, D.R.; Frizzell, R.A. The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L912–L925. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Mainprice, B.; Chanez, P.; Bousquet, J.; Urbach, V. Lipoxin A(4) stimulates a cytosolic Ca2+ increase in human bronchial epithelium. J. Biol. Chem. 2003, 278, 10879–10884. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.; Fustero Torre, C.; Tyrrell, J.; McNally, P.; Harvey, B.J.; Urbach, V. Lipoxin A4 prevents tight junction disruption and delays the colonization of cystic fibrosis bronchial epithelial cells by Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L1053–L1061. [Google Scholar] [CrossRef] [PubMed]
- Verrière, V.; Higgins, G.; Al-Alawi, M.; Costello, R.W.; McNally, P.; Chiron, R.; Harvey, B.J.; Urbach, V. Lipoxin A4 stimulates calcium-activated chloride currents and increases airway surface liquid height in normal and cystic fibrosis airway epithelia. PLoS ONE 2012, 7, e37746. [Google Scholar] [CrossRef] [PubMed]
- Al-Alawi, M.; Buchanan, P.; Verriere, V.; Higgins, G.; McCabe, O.; Costello, R.W.; McNally, P.; Urbach, V.; Harvey, B.J. Physiological levels of lipoxin A4 inhibit ENaC and restore airway surface liquid height in cystic fibrosis bronchial epithelium. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Ringholz, F.C.; Higgins, G.; Hatton, A.; Sassi, A.; Moukachar, A.; Fustero-Torre, C.; Hollenhorst, M.; Sermet-Gaudelus, I.; Harvey, B.J.; McNally, P.; et al. Resolvin D1 regulates epithelial ion transport and inflammation in cystic fibrosis airways. J. Cyst. Fibros. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B. The challenges and promises of new therapies for cystic fibrosis. J. Exp. Med. 2012, 209, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Bragonzi, A.; Worlitzsch, D.; Pier, G.B.; Timpert, P.; Ulrich, M.; Hentzer, M.; Andersen, J.B.; Givskov, M.; Conese, M.; Doring, G. Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. J. Infect. Dis. 2005, 192, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, C.; O′Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Grumbach, Y.; Quynh, N.V.; Chiron, R.; Urbach, V. LXA4 stimulates ZO-1 expression and transepithelial electrical resistance in human airway epithelial (16HBE14o-) cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L101–L108. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, P.J.; McNally, P.; Harvey, B.J.; Urbach, V. Lipoxin A₄-mediated KATP potassium channel activation results in cystic fibrosis airway epithelial repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L193–L201. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.; Buchanan, P.; Perriere, M.; Al-Alawi, M.; Costello, R.W.; Verriere, V.; McNally, P.; Harvey, B.J.; Urbach, V. Activation of P2RY11 and ATP release by lipoxin A4 restores the airway surface liquid layer and epithelial repair in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Codagnone, M.; Cianci, E.; Lamolinara, A.; Mari, V.C.; Nespoli, A.; Isopi, E.; Mattoscio, D.; Arita, M.; Bragonzi, A.; Iezzi, M.; et al. Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection. Mucosal Immunol. 2018, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philippe, R.; Urbach, V. Specialized Pro-Resolving Lipid Mediators in Cystic Fibrosis. Int. J. Mol. Sci. 2018, 19, 2865. https://doi.org/10.3390/ijms19102865
Philippe R, Urbach V. Specialized Pro-Resolving Lipid Mediators in Cystic Fibrosis. International Journal of Molecular Sciences. 2018; 19(10):2865. https://doi.org/10.3390/ijms19102865
Chicago/Turabian StylePhilippe, Réginald, and Valerie Urbach. 2018. "Specialized Pro-Resolving Lipid Mediators in Cystic Fibrosis" International Journal of Molecular Sciences 19, no. 10: 2865. https://doi.org/10.3390/ijms19102865
APA StylePhilippe, R., & Urbach, V. (2018). Specialized Pro-Resolving Lipid Mediators in Cystic Fibrosis. International Journal of Molecular Sciences, 19(10), 2865. https://doi.org/10.3390/ijms19102865