Structure and Molecular Dynamics Simulations of Protein Tyrosine Phosphatase Non-Receptor 12 Provide Insights into the Catalytic Mechanism of the Enzyme

 , and
, and
Abstract

1. Introduction
2. Results
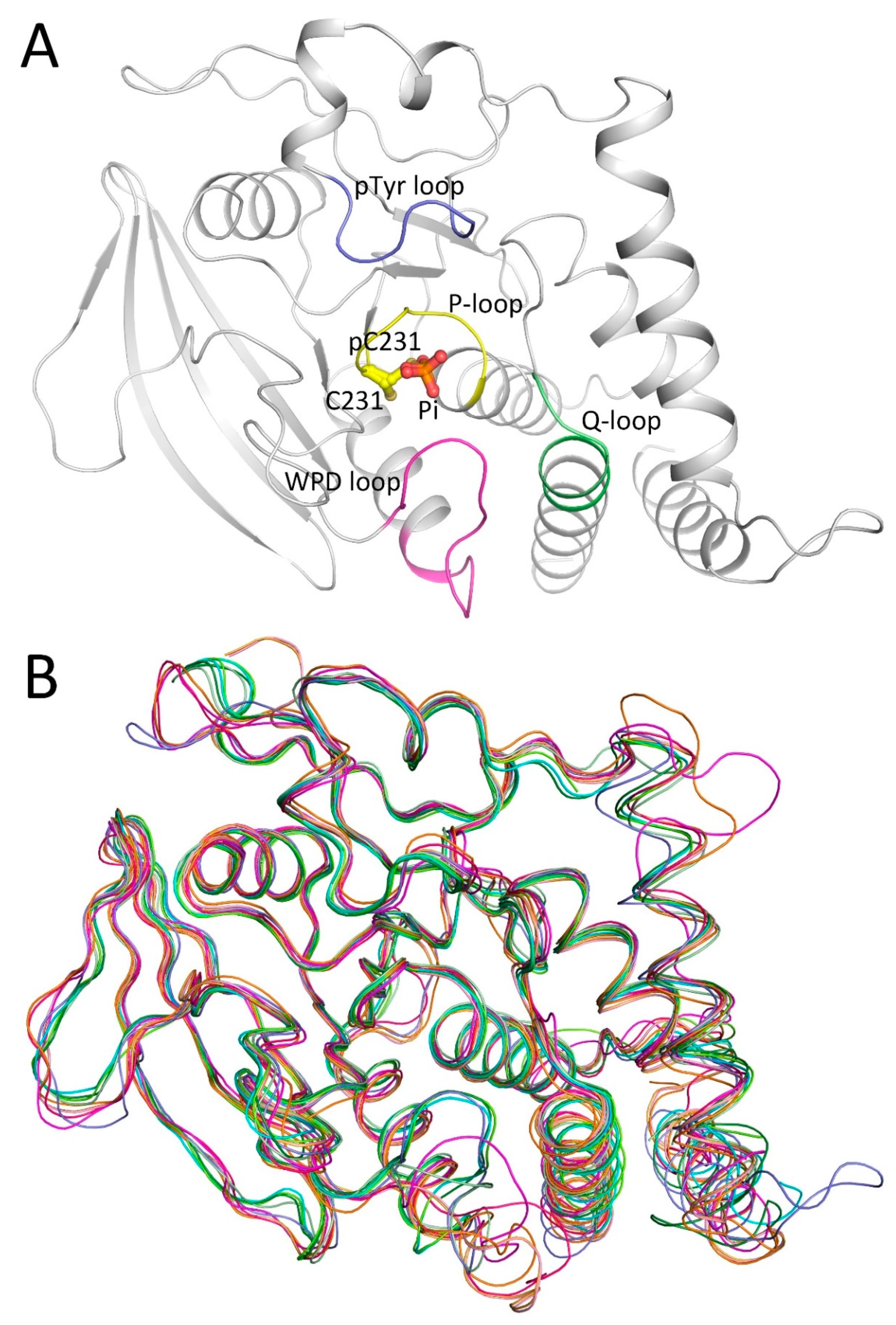
2.1. Overall Structure of PTPN12
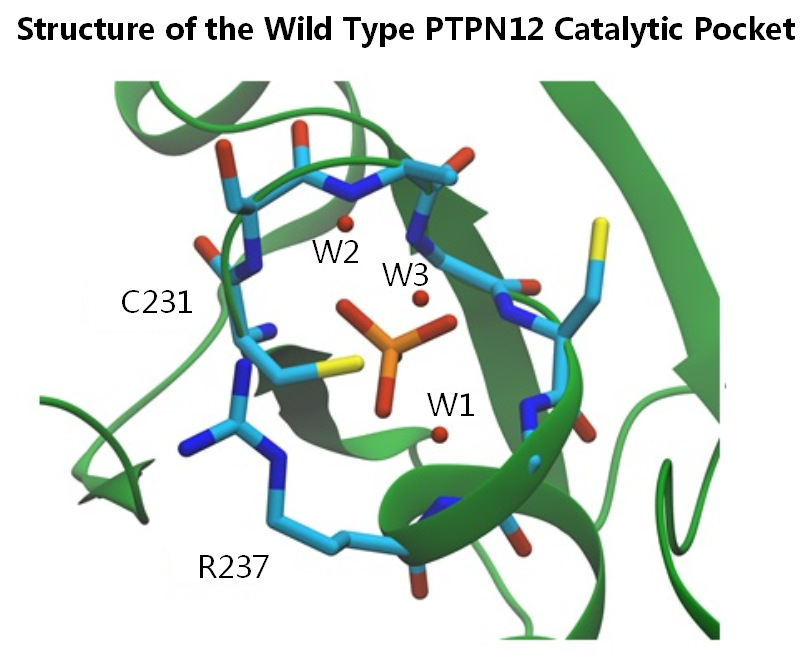
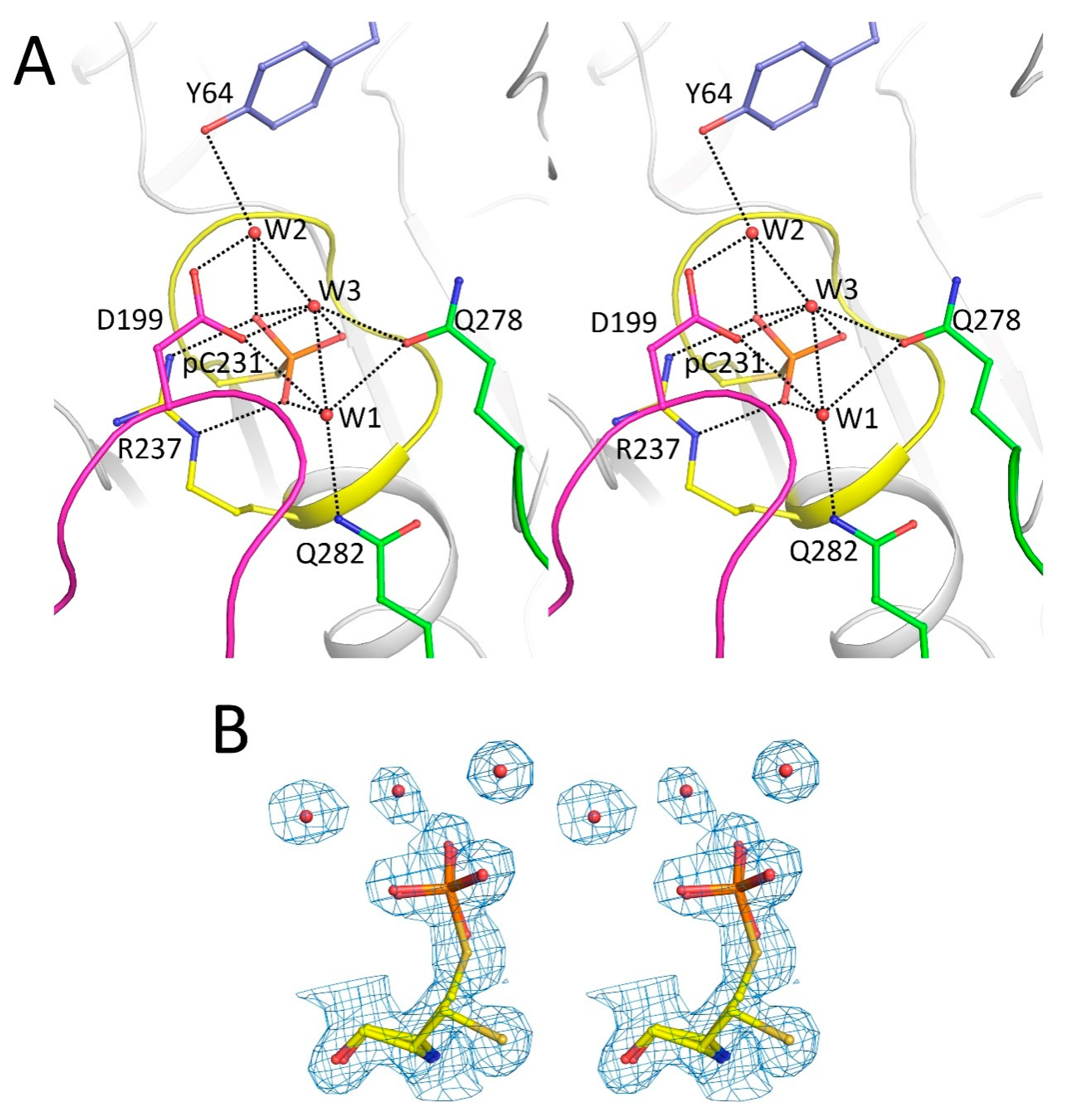
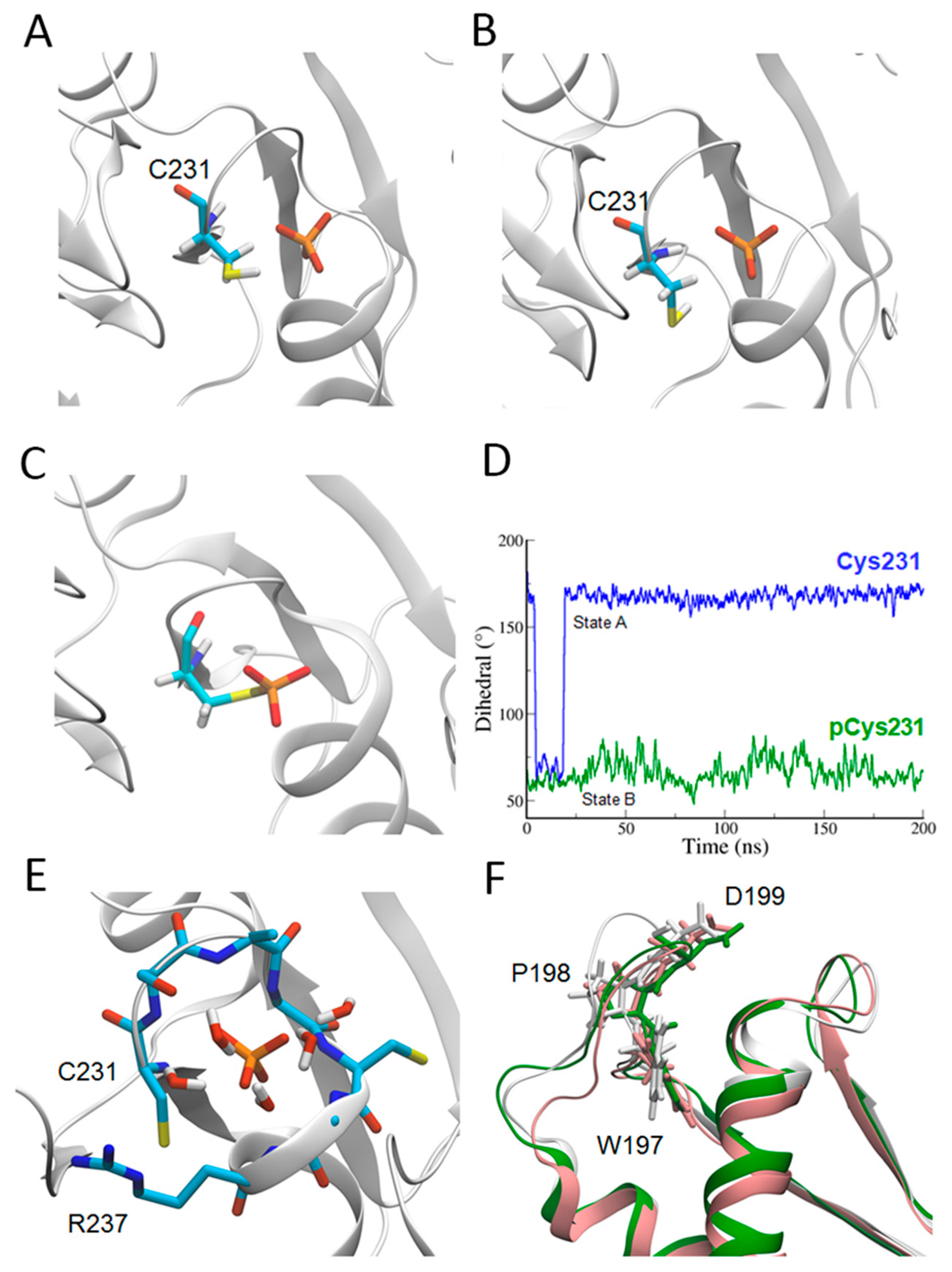
2.2. Catalytic Cys231 and Water Molecules
2.3. WPD Loop
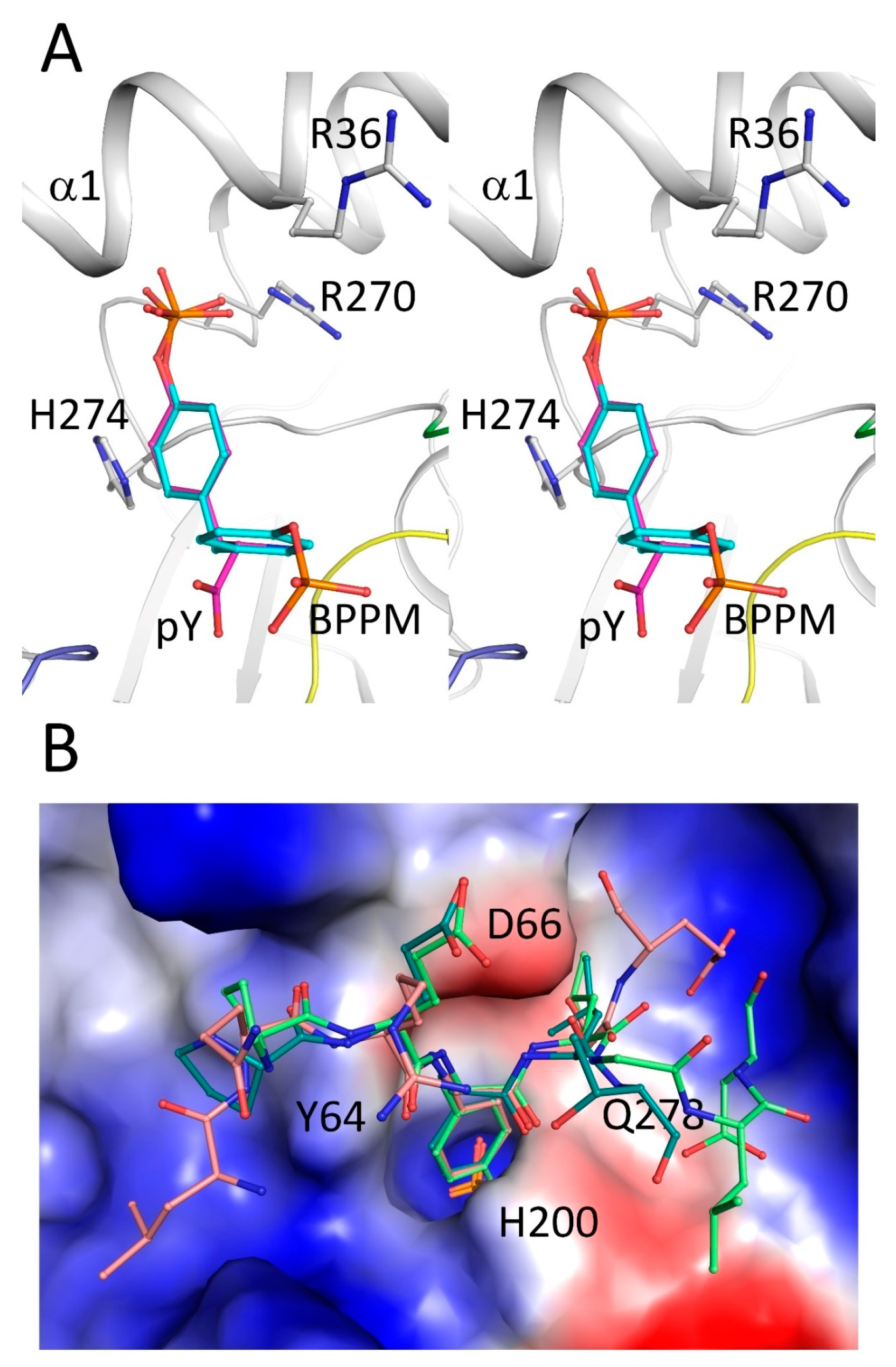
2.4. Secondary Binding Site
2.5. Substrate Recognition
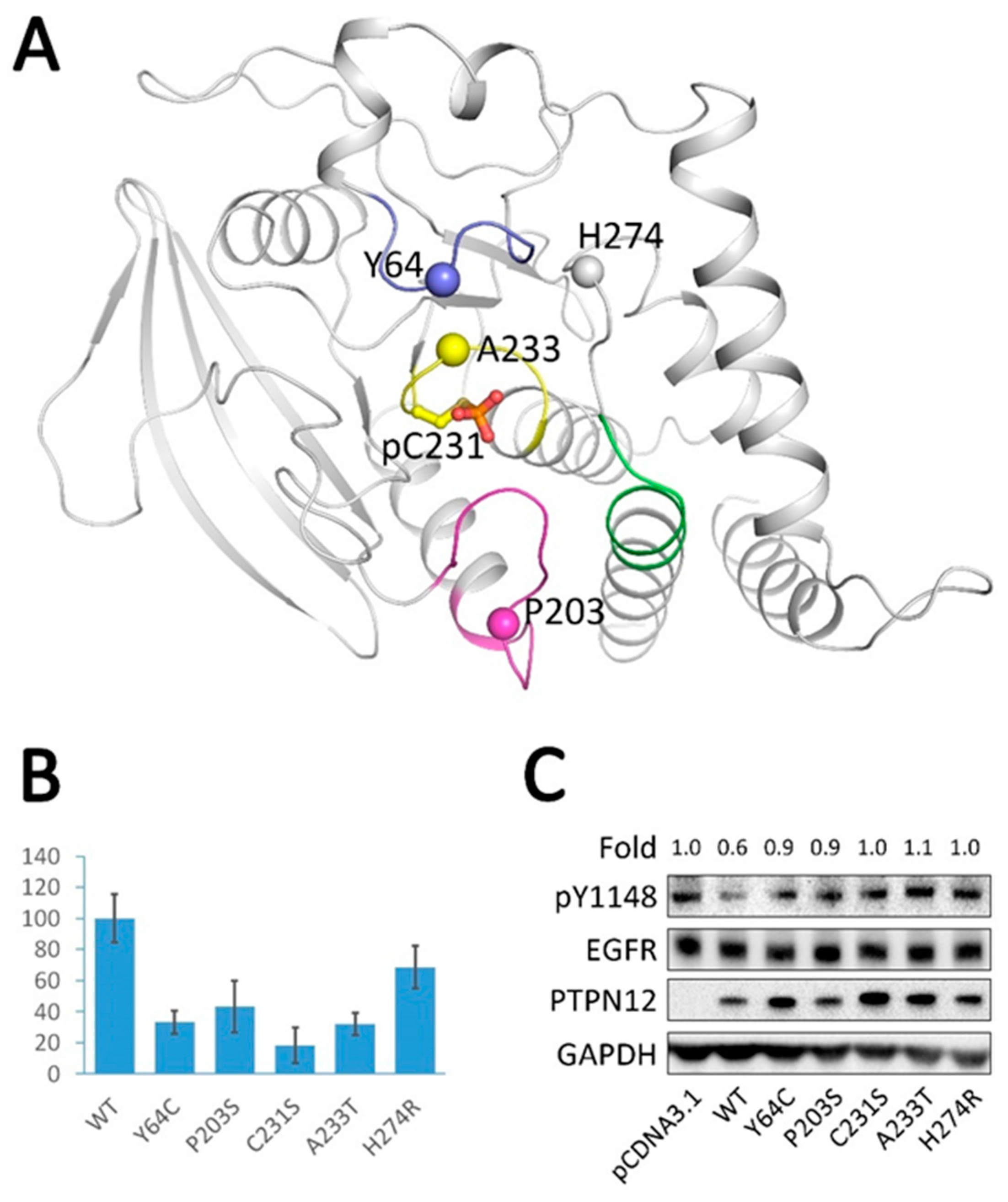
2.6. Analysis of Tumor-Derived Mutations
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of Recombinant PTPN12
4.2. Crystallization and Diffraction Data Collection
4.3. Structure Determinations and Refinement
4.4. Parametrization of Phosphoryl–Cysteine for Molecular Dynamics Simulations
4.5. Molecular Dynamics Simulations
4.6. Phosphatase Activity Assay
4.7. Cell Culture, Transfection, and Treatment
4.8. Accession Code
Acknowledgments
Author Contributions
Conflicts of Interest
References
- He, R.J.; Yu, Z.H.; Zhang, R.Y.; Zhang, Z.Y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin. 2014, 35, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yi, J.S.; Lawan, A.; Min, K.; Bennett, A.M. Mining the function of protein tyrosine phosphatases in health and disease. Semin. Cell Dev. Biol. 2015, 37, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, W.J.; Elson, A.; Harroch, S.; Pulido, R.; Stoker, A.; den Hertog, J. Protein tyrosine phosphatases in health and disease. FEBS J. 2013, 280, 708–730. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; van Huijsduijnen, R.H. Protein tyrosine phosphatases: Dual-specificity phosphatases in health and disease. FEBS J. 2008, 275, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dube, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Bialy, L.; Waldmann, H. Inhibitors of protein tyrosine phosphatases: Next-generation drugs? Angew. Chem. Int. Ed. 2005, 44, 3814–3839. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, M.; Wu, L.; Keng, Y.F.; Song, L.; Luo, Z.; Huang, Z.; Wu, G.Z.; Yuan, A.K.; Zhang, Z.Y. Structure-based discovery of small molecule inhibitors targeted to protein tyrosine phosphatase 1B. J. Med. Chem. 2000, 43, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Co, D.; Sommercorn, J.; Tonks, N.K. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J. Biol. Chem. 1993, 268, 6622–6628. [Google Scholar] [PubMed]
- Veillette, A.; Rhee, I.; Souza, C.M.; Davidson, D. PEST family phosphatases in immunity, autoimmunity, and autoinflammatory disorders. Immunol. Rev. 2009, 228, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Sirois, J.; Cote, J.F.; Charest, A.; Uetani, N.; Bourdeau, A.; Duncan, S.A.; Daniels, E.; Tremblay, M.L. Essential function of PTP-PEST during mouse embryonic vascularization, mesenchyme formation, neurogenesis and early liver development. Mech. Dev. 2006, 123, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Crowe, P.; Yang, J.L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 671–689. [Google Scholar] [CrossRef] [PubMed]
- Bermudez Brito, M.; Goulielmaki, E.; Papakonstanti, E.A. Focus on PTEN Regulation. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, L.; Noh, S.J.; Guan, K.L.; Zhang, Z.Y. Altering the nucleophile specificity of a protein-tyrosine phosphatase-catalyzed reaction. Probing the function of the invariant glutamine residues. J. Biol. Chem. 1998, 273, 5484–5492. [Google Scholar] [CrossRef] [PubMed]
- Scrima, M.; De Marco, C.; De Vita, F.; Fabiani, F.; Franco, R.; Pirozzi, G.; Rocco, G.; Malanga, D.; Viglietto, G. The nonreceptor-type tyrosine phosphatase PTPN13 is a tumor suppressor gene in non-small cell lung cancer. Am. J. Pathol. 2012, 180, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Muller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ryu, S.E. Structure and catalytic mechanism of human protein tyrosine phosphatome. BMB Rep. 2012, 45, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Moller, N.P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.L.; Dixon, J.E. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 1991, 266, 17026–17030. [Google Scholar] [PubMed]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 268, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, J.A.; Schubert, H.L.; Fauman, E.B.; Zhang, Z.Y.; Dixon, J.E.; Saper, M.A. Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature 1994, 370, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Pannifer, A.D.; Flint, A.J.; Tonks, N.K.; Barford, D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by X-ray crystallography. J. Biol. Chem. 1998, 273, 10454–10462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 209–234. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal structure of human protein tyrosine phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Sun, J.P.; He, Y.; Guo, X.; Liu, S.; Zhou, B.; Hudmon, A.; Zhang, Z.Y. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 19767–19772. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.J.; Sen, U.; Zhao, L.; Greenleaf, W.B.; Dasgupta, J.; Fiorillo, E.; Orru, V.; Bottini, N.; Chen, X.S. Crystal structure of the human lymphoid tyrosine phosphatase catalytic domain: Insights into redox regulation. Biochemistry 2009, 48, 4838–4845. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Xu, Y.F.; Ning, S.L.; Yang, D.X.; Li, Y.; Du, Y.J.; Yang, F.; Zhang, Y.; Liang, N.; Yao, W.; et al. The catalytic region and PEST domain of PTPN18 distinctly regulate the HER2 phosphorylation and ubiquitination barcodes. Cell Res. 2014, 24, 1067–1090. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, S.; Menon, A.; Stanford, S.; Oppong, E.; Gunawan, A.M.; Wu, L.; Wu, D.J.; Barrios, A.M.; Bottini, N.; et al. A potent and selective small-molecule inhibitor for the lymphoid-specific tyrosine phosphatase (LYP), a target associated with autoimmune diseases. J. Med. Chem. 2013, 56, 4990–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Chen, Y.Z.; Luo, R.Z.; Zhang, L.; Zhang, S.L.; Zeng, J.; Jiang, Y.C.; Han, Y.J.; Wen, Z.S. Tyrosine-protein phosphatase non-receptor type 12 expression is a good prognostic factor in resectable non-small cell lung cancer. Oncotarget 2015, 6, 11704–11713. [Google Scholar] [CrossRef] [PubMed]
- Villa-Moruzzi, E. PTPN12 controls PTEN and the AKT signalling to FAK and HER2 in migrating ovarian cancer cells. Mol. Cell. Biochem. 2013, 375, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. Part A 1997, 276, 307–326. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Cianci, M.; Folli, C.; Zonta, F.; Florio, P.; Berni, R.; Zanotti, G. Structural evidence for asymmetric ligand binding to transthyretin. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Zonta, F.; Polles, G.; Sanasi, M.F.; Bortolozzi, M.; Mammano, F. The 3.5 angstrom X-ray structure of the human connexin26 gap junction channel is unlikely that of a fully open channel. Cell Commun. Signal. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10093. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | Data Statistics |
|---|---|
| space group | C2 |
| Cell parameters | |
| a, b, c (Å); β° | 136.01, 40.97, 75.69; 116.63 |
| Resolution (Å) | 50.00–1.62 (1.65–1.62) a |
| Redundancy | 6.1 (5.3) a |
| Completeness (%) | 99.9 (99.2) a |
| Rmerge b | 9.6 (66.2) a |
| Average I/σ (I) | 37.7 (3.0) a |
| Refinement | |
| Rwork(%)/Rfree(%) c | 16.3/21.2 |
| Protein atoms (phosphate/water) | 2517 (5/354) |
| RMSD bond length (Å) | 0.012 |
| RMSD angle (°) | 1.146 |
| Average B-factor | 22.70 |
| Protein | 21.00 |
| PO4 | 24.42 |
| pCys | 16.88 |
| water | 34.75 |
| Ramachandran plot | |
| Allowed (%) | 99.6 |
| Generously allowed (%) | 0.4 |
| Disallowed (%) | 0.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Zonta, F.; Wang, S.; Song, K.; He, X.; He, M.; Nie, Y.; Li, S. Structure and Molecular Dynamics Simulations of Protein Tyrosine Phosphatase Non-Receptor 12 Provide Insights into the Catalytic Mechanism of the Enzyme. Int. J. Mol. Sci. 2018, 19, 60. https://doi.org/10.3390/ijms19010060
Dong H, Zonta F, Wang S, Song K, He X, He M, Nie Y, Li S. Structure and Molecular Dynamics Simulations of Protein Tyrosine Phosphatase Non-Receptor 12 Provide Insights into the Catalytic Mechanism of the Enzyme. International Journal of Molecular Sciences. 2018; 19(1):60. https://doi.org/10.3390/ijms19010060
Chicago/Turabian StyleDong, Hui, Francesco Zonta, Shanshan Wang, Ke Song, Xin He, Miaomiao He, Yan Nie, and Sheng Li. 2018. "Structure and Molecular Dynamics Simulations of Protein Tyrosine Phosphatase Non-Receptor 12 Provide Insights into the Catalytic Mechanism of the Enzyme" International Journal of Molecular Sciences 19, no. 1: 60. https://doi.org/10.3390/ijms19010060
APA StyleDong, H., Zonta, F., Wang, S., Song, K., He, X., He, M., Nie, Y., & Li, S. (2018). Structure and Molecular Dynamics Simulations of Protein Tyrosine Phosphatase Non-Receptor 12 Provide Insights into the Catalytic Mechanism of the Enzyme. International Journal of Molecular Sciences, 19(1), 60. https://doi.org/10.3390/ijms19010060