Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members


Abstract
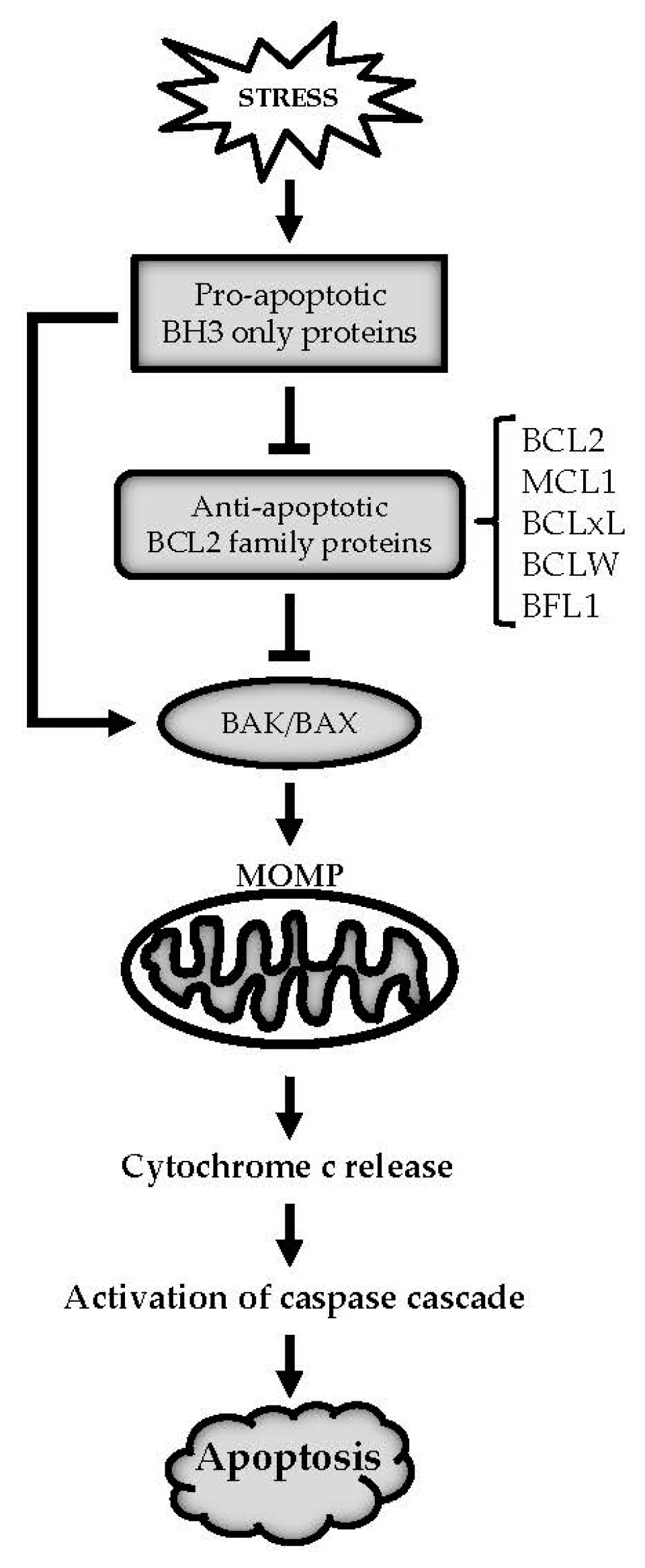
1. Introduction
2. RNA Binding Proteins (RBPs) and Anti-Apoptotic BCL2 Family Members
2.1. Regulation of Pre-mRNA Alternative Splicing
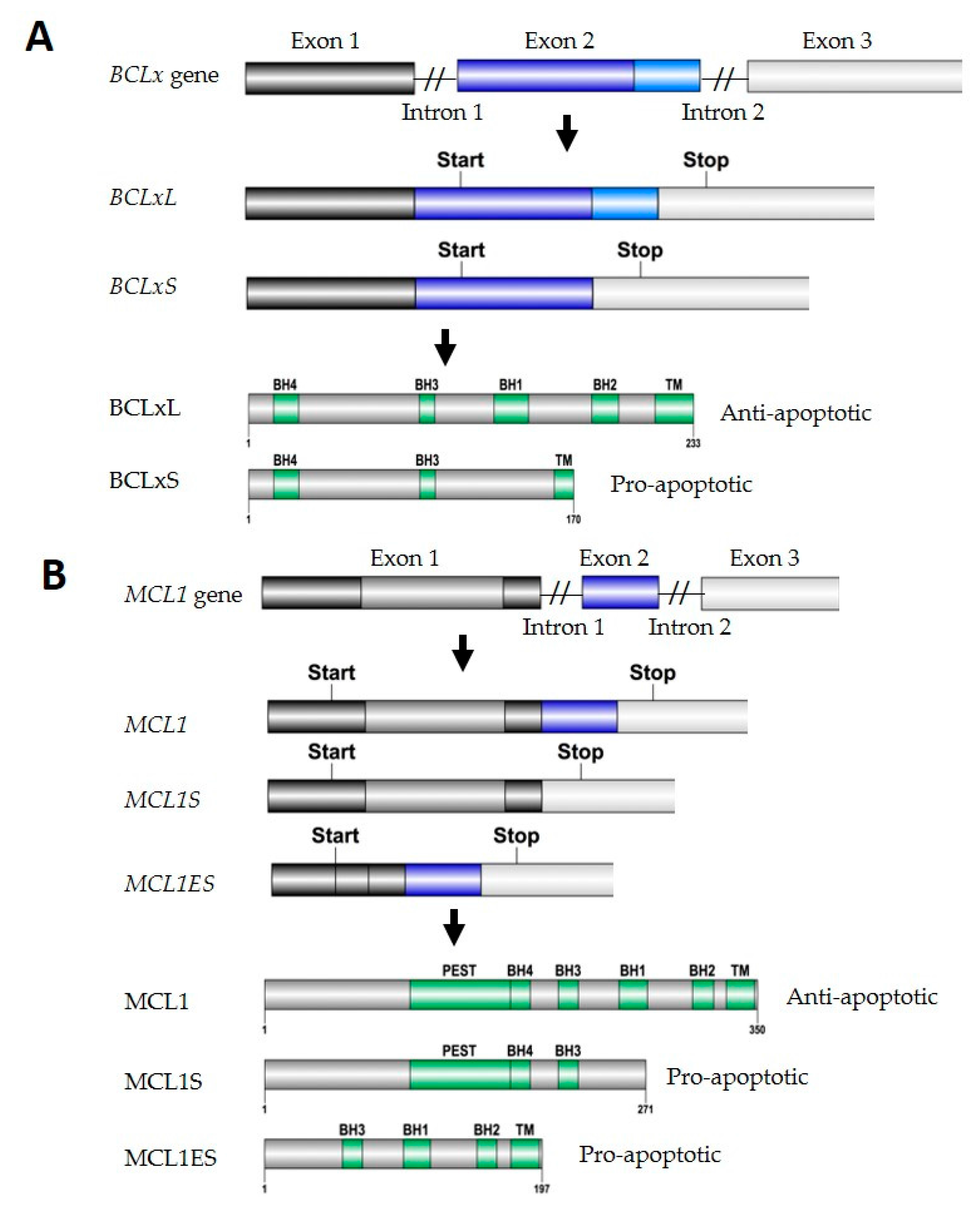
2.1.1. BCLx Splicing
hnRNP Proteins
SR Proteins
STAR Proteins
RBM Proteins
2.1.2. MCL1 Splicing
2.2. Regulation of mRNA Stability
2.2.1. AU-Rich-Element (ARE) Mediated RNA Decay

{kind=link}
{kind=link}
{kind=link}
| mRNA | Half-Life | Cell Type | mRNA Stabilizer | mRNA Destabilizer |
|---|---|---|---|---|
| BCL2 | >6 h [74] | U251 | HuR [74], nucleolin [76], ζ-crystallin [81], TRA2β [82], LARP1 [83] | AUF1 [85], ZFP36L1 [87] |
| 5 h [81] | Jurkat | |||
| 7.5 h [81] | HEK 293 | |||
| 8 h [82] | HCT116 | |||
| 2.5 h [87] | Murine leukemia BCL1 cells | |||
| MCL1 | 1.4 h [88] | PC3 | HuR [74], CUGBP2 [89] | PTBP1 [88], TTP [84] |
| 2.0 h [88] | H1299 | |||
| 4 h [74] | U251 | |||
| 2.3 h [84] | Mice peritoneal neutrophil stimulated with LPS | |||
| 0.5 h [89] | HCT116 | |||
| BCLxL | 3.4 h [74] | U251 | HuR [74], nucleolin [77] | n.a |
| 4 h [77] | HELA | |||
| >6 h [78] | PAEC |
2.2.2. CU-Rich-Element Mediated RNA Decay
2.2.3. GU-Rich-Element (GRE) Mediated RNA Decay
2.3. Regulation of mRNA Subcellular Localization
3. MicroRNAs (miRNAs) and Anti-Apoptotic BCL2 Family Members
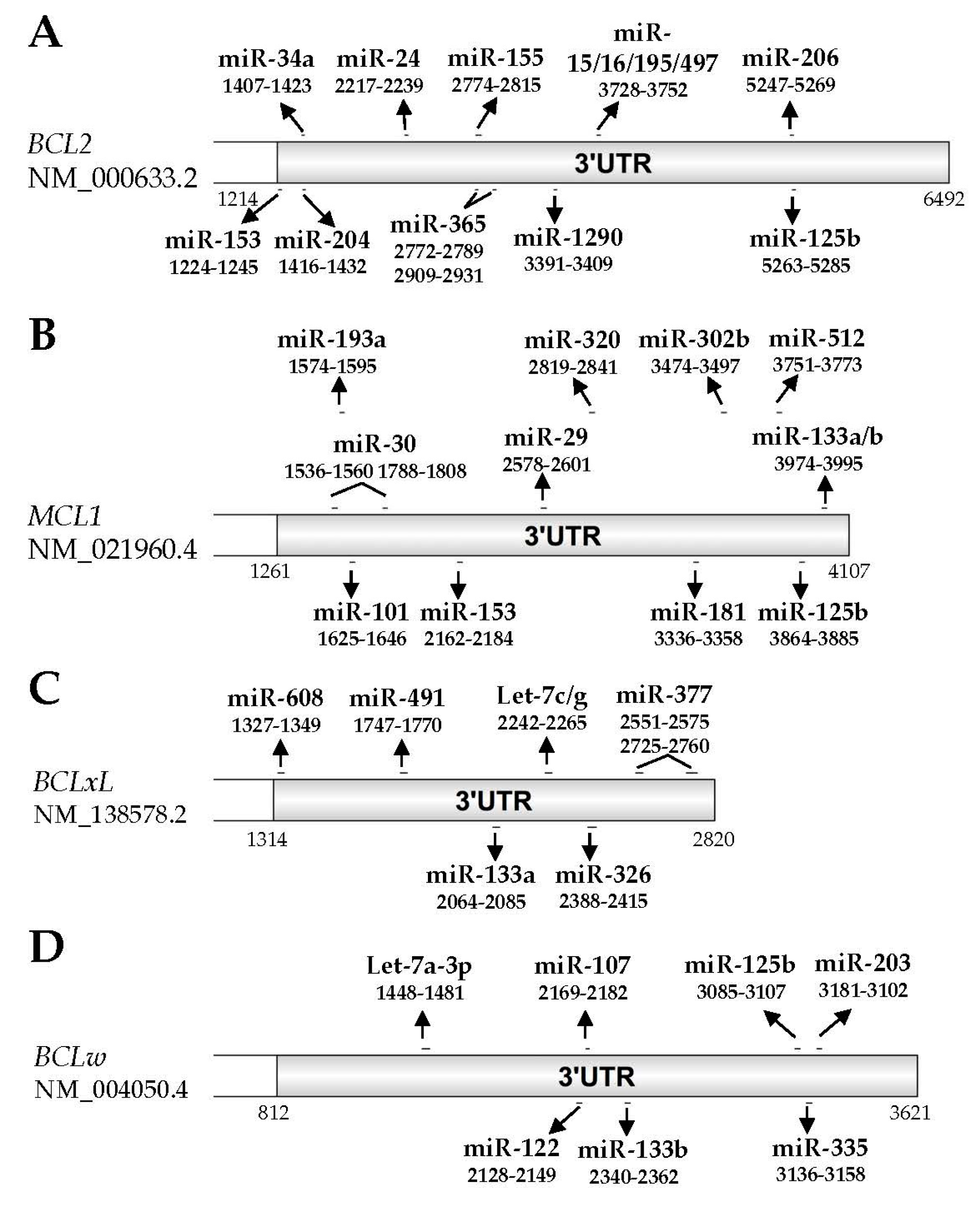
3.1. BCL2
3.2. MCL1
3.3. BCLxL
3.4. BCLW
3.5. BFL1
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGO | Argonaute |
| ALK | Anaplastic lymphoma kinase |
| ARE | AU rich element |
| ASD-2 | Alternative splicing defective protein 2 |
| AUF1 | AU-rich element RNA-binding protein 1 |
| BAD | BCL2-associated death promoter |
| BAK | BCL2 homologous antagonist/killer |
| BAX | BCL2-associated X |
| BCLW | BCL2-like protein 2 |
| BCLxL | B-cell lymphoma extra large |
| BCL2 | B cell lymphoma 2 |
| BFL1 | BCL2-related protein A1 |
| BH | BCL2 homology |
| BID | BH3 interacting-domain death agonist |
| BIM | BCL2-like protein 11 |
| BNIP3L | BCL2/adenovirus E1B 19kDa protein-interacting protein 3-like |
| BOK | BCL2-related ovarian killer |
| CLL | Chronic lymphoid leukemia |
| CPT | Camptothecin |
| CUGBP2 | CUG triplet repeat RNA-binding protein 2 |
| EGFR | Epidermal growth factor receptor |
| FBI-1 | Factor binding IST protein 1 |
| GEMIN4 | Gem-associated protein 4 |
| GRE | GU rich element |
| H3 | Histone 3 |
| H3K4 | Histone 3 lysine 4 |
| H4 | Histone 4 |
| HCC | Hepatocellular carcinoma |
| HDAC | Histone deacetylase |
| HIF | Hypoxia inducible factor |
| hnRNP | Heterogeneous nuclear ribonucleoprotein |
| HuR | Human antigen R |
| KAT | Lysine acetyltransferase |
| LARP1 | La-related protein 1 |
| lncRNA | Long non-coding RNA |
| LPS | Lipopolysaccharide |
| LSM | Like-spliceosomal |
| MCL1 | Myeloid cell leukemia 1 |
| MET | Hepatocyte growth factor receptor |
| miRISC | microRNA induced silencing complex |
| miRNA | microRNA |
| MOMP | Mitochondrial outer membrane permeabilization |
| NOXA | Phorbol-12-myristate-13-acetate-induced protein |
| NSCLC | Non-small cell lung cancer |
| PEST domain | Proline, glutamic acid, serine, threonine rich domain |
| PNN | Pinin |
| Pre-mRNA | Precursor messenger RNA |
| PRMT2 | Protein-arginine methyltransferase 2 |
| PRPF8 | Pre-mRNA-processing-splicing factor 8 |
| PTBP1 | Polypyrimidine tract binding protein 1 |
| PUMA | p53 upregulated modulator of apoptosis |
| qRRM | Quasi-RNA recognition motif |
| RBM | RNA binding motif |
| RBP | RNA binding protein |
| RNP | Ribonucleoprotein |
| ROS | Reactive oxygen species |
| RRM | RNA recognition motif |
| SAM68 | SRC associated in mitosis of 68 kDa |
| SART | Squamous cell carcinoma antigen recognized by T-cells |
| SF | Splicing factor |
| SFPQ | Splicing factor, proline and glutamine rich |
| siRNA | Small interfering RNA |
| Sm | Spliceosomal |
| SMN | Survival motor neuron |
| snRNP | Spliceosomal small nuclear ribonuclear protein |
| SR protein | Serine/arginine-rich protein |
| STAR | Signal transduction and activation of RNA |
| STAT | Signal transducer and activator of transcription |
| TNBC | Triple-negative breast cancer |
| TNF | Tumor necrosis factor |
| TRA2β | Transformer 2β |
| TTP | Tristetraprolin |
| TM domain | Transmembrane domain |
| UBL5 | Ubiquitin-like protein 5 |
| UTR | Untranslated region |
| ZFP36L1 | Zinc finger protein 36, C3H1 type-like 1 |
References
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2016, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Kothari, A. Anti-apoptotic BCL2 family members in development. Cell Death Differ. 2017, 25, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T. Apoptosis in the development of the immune system. Cell Death Differ. 2008, 15, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Creagh, E.M. Caspase crosstalk: Integration of apoptotic and innate immune signalling pathways. Trends Immunol. 2014, 35, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.M.; Izumo, S. Apoptosis in heart: Basic mechanisms and implications in cardiovascular diseases. Trends Mol. Med. 2003, 9, 177–182. [Google Scholar] [CrossRef]
- Teringova, E.; Tousek, P. Apoptosis in ischemic heart disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, I.; Johnstone, R.W.; Smyth, M.J. SnapShot: Extrinsic apoptosis pathways. Cell 2010, 143, 1192–1192.e2. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Finger, L.; Yunis, J.; Nowell, P.; Croce, C. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Vervliet, T.; Parys, J.B.; Bultynck, G. BCL2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079–5092. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of BCL2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Juin, P.; Geneste, O.; Gautier, F.; Depil, S.; Campone, M. Decoding and unlocking the BCL2 dependency of cancer cells. Nat. Rev. Cancer 2013, 13, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.S.; Toombs, J.; Kuo, Y.C.; Piazza, J.T.; Tuladhar, R.; Barrett, Q.; Fan, C.W.; Zhang, X.; Walensky, L.D.; et al. Extra-mitochondrial prosurvival BCL2 proteins regulate gene transcription by inhibiting the SUFU tumour suppressor. Nat. Cell Biol. 2017, 19, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Kutuk, O. Regulation of BCL2 Family Proteins by Posttranslational Modifications. Curr. Mol. Med. 2008, 8, 102–118. [Google Scholar] [CrossRef]
- Ascano, M.; Gerstberger, S.; Tuschl, T. Multi-disciplinary methods to define RNA-protein interactions and regulatory networks. Curr. Opin. Genet. Dev. 2013, 23, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. BCLx, a BCL2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Xiao, Q.; Ford, A.L.; Xu, J.; Yan, P.; Lee, K.Y.; Gonzales, E.; West, T.; Holtzman, D.M.; Lee, J.M. BCLx pre-mRNA splicing regulates brain injury after neonatal hypoxia-ischemia. J. Neurosci. 2012, 32, 13587–13596. [Google Scholar] [CrossRef] [PubMed]
- Scherr, A.L.; Gdynia, G.; Salou, M.; Radhakrishnan, P.; Duglova, K.; Heller, A.; Keim, S.; Kautz, N.; Jassowicz, A.; Elssner, C.; et al. BCLxL is an oncogenic driver in colorectal cancer. Cell Death Dis. 2016, 7, e2342. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Iervolino, A.; Santilli, G.; Trotta, R.; Guerzoni, C.; Cesi, V.; Bergamaschi, A.; Gambacorti-Passerini, C.; Calabretta, B.; Perrotti, D. hnRNP A1 Nucleocytoplasmic Shuttling Activity Is Required for Normal Myelopoiesis and BCR/ABL Leukemogenesis. Mol. Cell. Biol. 2002, 22, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gupta, S.C.; Peng, W.X.; Zhou, N.; Pochampally, R.; Atfi, A.; Watabe, K.; Lu, Z.; Mo, Y.Y. Regulation of alternative splicing of BCLx by BC200 contributes to breast cancer pathogenesis. Cell Death Dis. 2016, 7, e2262. [Google Scholar] [CrossRef] [PubMed]
- Leu, S.; Lin, Y.M.; Wu, C.H.; Ouyang, P. Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of BCLxS and ICAD. J. Cell Sci. 2012, 125, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, P.; Toutant, J.; Shkreta, L.; Goekjian, S.; Revil, T.; Chabot, B. Antagonistic effects of the SRp30c protein and cryptic 5′ splice sites on the alternative splicing of the apoptotic regulator BCLx. J. Biol. Chem. 2008, 283, 21315–21324. [Google Scholar] [CrossRef] [PubMed]
- Garneau, D.; Revil, T.; Fisette, J.F.; Chabot, B. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator BCLx. J. Biol. Chem. 2005, 280, 22641–22650. [Google Scholar] [CrossRef] [PubMed]
- Revil, T.; Pelletier, J.; Toutant, J.; Cloutier, A.; Chabot, B. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic BCLxS splice isoform. J. Biol. Chem. 2009, 284, 21458–21467. [Google Scholar] [CrossRef] [PubMed]
- Bielli, P.; Bordi, M.; Di Biasio, V.; Sette, C. Regulation of BCLX splicing reveals a role for the polypyrimidine tract binding protein (PTBP1/hnRNP I) in alternative 5′ splice site selection. Nucleic Acids Res. 2014, 42, 12070–12081. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Toutant, J.; Durand, M.; Manley, J.L.; Chabot, B. SRSF10 Connects DNA Damage to the Alternative Splicing of Transcripts Encoding Apoptosis, Cell-Cycle Control, and DNA Repair Factors. Cell Rep. 2016, 17, 1990–2003. [Google Scholar] [CrossRef] [PubMed]
- Merdzhanova, G.; Edmond, V.; De Seranno, S.; Van den Broeck, A.; Corcos, L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008, 15, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein Sam68 modulates the alternative splicing of BCLx. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, D.; Qian, H.; Tsai, Y.S.; Shao, S.; Liu, Q.; Dominguez, D.; Wang, Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 2014, 26, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Yamamoto, N.; Kimura, M.; Nishio, K.; Yamane, H.; Nakajima, K. RBM10 regulates alternative splicing. FEBS Lett. 2014, 588, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, S.; Busa, R.; Compagnucci, C.; Sette, C. The RNA recognition motif protein RBM11 is a novel tissue-specific splicing regulator. Nucleic Acids Res. 2012, 40, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Ou, A.C.; Cho, A.; Benz, E.J., Jr.; Huang, S.C. Novel splicing factor RBM25 modulates BCLx pre-mRNA 5′ splice site selection. Mol. Cell. Biol. 2008, 28, 5924–5936. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.L.; Tyson-Capper, A.J. Regulation of Mcl-1 by SRSF1 and SRSF5 in cancer cells. PLoS ONE 2012, 7, e51497. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, H.; Poplawski, P.; Hoser, G.; Rybicka, B.; Rodzik, K.; Sokol, E.; Boguslawska, J.; Tanski, Z.; Fogtman, A.; Koblowska, M.; et al. Decreased Expression of SRSF2 Splicing Factor Inhibits Apoptotic Pathways in Renal Cancer. Int. J. Mol. Sci. 2016, 17, 1598. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; Liu, X.; Vu, A.; Sliozberg, M.; Vido, M.; Elci, O.U.; Goldsmith, K.C.; Hogarty, M.D. Multiple components of the spliceosome regulate Mcl1 activity in neuroblastoma. Cell Death Dis. 2014, 5, e1072. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.; Cook, M.E.; Blanchette, M. SR proteins control a complex network of RNA-processing events. RNA 2015, 21, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Vernet, C.; Artzt, K. STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet. 1997, 13, 479–484. [Google Scholar] [CrossRef]
- Feracci, M.; Foot, J.N.; Grellscheid, S.N.; Danilenko, M.; Stehle, R.; Gonchar, O.; Kang, H.S.; Dalgliesh, C.; Meyer, N.H.; Liu, Y.; et al. Structural basis of RNA recognition and dimerization by the STAR proteins T-STAR and Sam68. Nat. Commun. 2016, 7, 10355. [Google Scholar] [CrossRef] [PubMed]
- Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.; Boisvert, F.M.; Boulanger, M.C.; Bedford, M.T.; Richard, S. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell 2003, 14, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Babic, I.; Jakymiw, A.; Fujita, D.J. The RNA binding protein Sam68 is acetylated in tumor cell lines, and its acetylation correlates with enhanced RNA binding activity. Oncogene 2004, 23, 3781–3789. [Google Scholar] [CrossRef] [PubMed]
- Babic, I.; Cherry, E.; Fujita, D.J. SUMO modification of Sam68 enhances its ability to repress cyclin D1 expression and inhibits its ability to induce apoptosis. Oncogene 2006, 25, 4955–4964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Cai, L.; Zhu, J.; Chen, M.; Chen, J.; Li, Z.H.; Liu, X.D.; Wang, S.G.; Bie, P.; Jiang, P.; et al. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis 2011, 32, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Vhuiyan, M.I.; Pak, M.L.; Park, M.A.; Thomas, D.; Lakowski, T.M.; Chalfant, C.E.; Frankel, A. PRMT2 interacts with splicing factors and regulates the alternative splicing of BCLX. J. Biochem. 2017, 162, 17–25. [Google Scholar] [PubMed]
- Bielli, P.; Busa, R.; Di Stasi, S.M.; Munoz, M.J.; Botti, F.; Kornblihtt, A.R.; Sette, C. The transcription factor FBI-1 inhibits SAM68-mediated BCLX alternative splicing and apoptosis. EMBO Rep. 2014, 15, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Ehrmann, I.; Dalgliesh, C.; Liu, Y.; Danilenko, M.; Crosier, M.; Overman, L.; Arthur, H.M.; Lindsay, S.; Clowry, G.J.; Venables, J.P.; et al. The tissue-specific RNA binding protein T-STAR controls regional splicing patterns of neurexin pre-mRNAs in the brain. PLoS Genet. 2013, 9, e1003474. [Google Scholar] [CrossRef] [PubMed]
- Ohno, G.; Hagiwara, M.; Kuroyanagi, H. STAR family RNA-binding protein ASD-2 regulates developmental switching of mutually exclusive alternative splicing in vivo. Genes Dev. 2008, 22, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, L.C.; Rintala-Maki, N.D.; White, R.D.; Morin, C.D. RNA binding motif (RBM) proteins: A novel family of apoptosis modulators? J. Cell. Biochem. 2005, 94, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Leo, C.P.; Hsu, S.Y.; Hsueh, A.J. MCL-1S, a splicing variant of the antiapoptotic BCL2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J. Biol. Chem. 2000, 275, 25255–25261. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Sim, S.H.; Ha, H.J.; Ko, J.J.; Lee, K.; Bae, J. MCL-1ES, a novel variant of MCL-1, associates with MCL-1L and induces mitochondrial cell death. FEBS Lett. 2009, 583, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.W. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia 2002, 16, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Yecies, D.; Carlson, N.E.; Deng, J.; Letai, A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010, 115, 3304–3313. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.R.; Wei, J.; Konopleva, M.; Reed, J.C.; Pellecchia, M.; Andreeff, M.; Ruvolo, P.P. Inhibition of Mcl-1 with the pan-BCL2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood 2015, 126, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCLXL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Khan, D.H.; Gonzalez, C.; Cooper, C.; Sun, J.M.; Chen, H.Y.; Healy, S.; Xu, W.; Smith, K.T.; Workman, J.L.; Leygue, E.; et al. RNA-dependent dynamic histone acetylation regulates MCL1 alternative splicing. Nucleic Acids Res. 2014, 42, 1656–1670. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Filippova, N.; Yang, X.; Wang, Y.; Gillespie, G.Y.; Langford, C.; King, P.H.; Wheeler, C.; Nabors, L.B. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol. Cancer Res. 2011, 9, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Lal, A.; Kim, H.H.; Gorospe, M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007, 6, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, T.K.; Bandyopadhyay, S.; Fernandes, D.J.; Spicer, E.K. Identification of nucleolin as an AU-rich element binding protein involved in BCL2 mRNA stabilization. J. Biol. Chem. 2004, 279, 10855–10863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tsaprailis, G.; Bowden, G.T. Nucleolin stabilizes BCLx L messenger RNA in response to UVA irradiation. Cancer Res. 2008, 68, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Mungunsukh, O.; Tutino, R.L.; Marquez, A.P.; Day, R.M. Angiotensin-II-induced apoptosis requires regulation of nucleolin and BCLxL by SHP-2 in primary lung endothelial cells. J. Cell Sci. 2010, 123, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Li, H.Y.; Hsu, T.I.; Chen, S.H.; Wu, C.J.; Chang, W.C.; Hung, J.J. Heat shock protein 90 stabilizes nucleolin to increase mRNA stability in mitosis. J. Biol. Chem. 2011, 286, 43816–43829. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Childress, M.O.; Geahlen, R.L. Syk interacts with and phosphorylates nucleolin to stabilize BCLx(L) mRNA and promote cell survival. Mol. Cell. Biol. 2014, 34, 3788–3799. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, A.; Lulli, M.; Amedei, A.; Papucci, L.; Witort, E.; Di Gesualdo, F.; Bertolini, F.; Brewer, G.; Nicolin, A.; Bevilacqua, A.; et al. zeta-Crystallin is a BCL2 mRNA binding protein involved in BCL2 overexpression in T-cell acute lymphocytic leukemia. FASEB J. 2010, 24, 1852–1865. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, Y.; Nishida, K.; Kajita, K.; Satake, Y.; Akaike, Y.; Fujita, K.; Kano, S.; Masuda, K.; Rokutan, K. Transformer 2beta and miR-204 regulate apoptosis through competitive binding to 3′ UTR of BCL2 mRNA. Cell Death Differ. 2015, 22, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.G.; Mura, M.; Al-Ashtal, H.A.; Lahr, R.M.; Abd-Latip, N.; Sweeney, K.; Lu, H.; Weir, J.; El-Bahrawy, M.; Steel, J.H.; et al. The RNA-binding protein LARP1 is a post-transcriptional regulator of survival and tumorigenesis in ovarian cancer. Nucleic Acids Res. 2016, 44, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Ebner, F.; Sedlyarov, V.; Tasciyan, S.; Ivin, M.; Kratochvill, F.; Gratz, N.; Kenner, L.; Villunger, A.; Sixt, M.; Kovarik, P. The RNA-binding protein tristetraprolin schedules apoptosis of pathogen-engaged neutrophils during bacterial infection. J. Clin. Investig. 2017, 127, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, A.; Donnini, M.; Papucci, L.; Witort, E.; Tempestini, A.; Bevilacqua, A.; Nicolin, A.; Brewer, G.; Schiavone, N.; Capaccioli, S. AUF1 Is a BCL2 A + U-rich element-binding protein involved in BCL2 mRNA destabilization during apoptosis. J. Biol. Chem. 2002, 277, 16139–16146. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, D.; Zuraw, L.; Ramalingam, S.; Sengupta, T.K.; Bandyopadhyay, S.; Reuben, A.; Fernandes, D.J.; Spicer, E.K. Mechanism of regulation of BCL2 mRNA by nucleolin and A+U-rich element-binding factor 1 (AUF1). J. Biol. Chem. 2010, 285, 27182–27191. [Google Scholar] [CrossRef] [PubMed]
- Zekavati, A.; Nasir, A.; Alcaraz, A.; Aldrovandi, M.; Marsh, P.; Norton, J.D.; Murphy, J.J. Post-transcriptional regulation of BCL2 mRNA by the RNA-binding protein ZFP36L1 in malignant B cells. PLoS ONE 2014, 9, e102625. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Placzek, W.J. PTBP1 modulation of MCL1 expression regulates cellular apoptosis induced by antitubulin chemotherapeutics. Cell Death Differ. 2016, 23, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; Natarajan, G.; Ramalingam, S.; Ramachandran, I.; May, R.; Queimado, L.; Houchen, C.W.; Anant, S. Translation inhibition during cell cycle arrest and apoptosis: Mcl-1 is a novel target for RNA binding protein CUGBP2. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1025–G1032. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.M.; Davidson, A.; Davis, I.; Weil, T.T. Subcellular mRNA localisation at a glance. J. Cell Sci. 2014, 127, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Mofatteh, M.; Bullock, S.L. SnapShot: Subcellular mRNA Localization. Cell 2017, 169, 178–178.e1. [Google Scholar] [CrossRef] [PubMed]
- Cosker, K.E.; Fenstermacher, S.J.; Pazyra-Murphy, M.F.; Elliott, H.L.; Segal, R.A. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 2016, 19, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Ciafre, S.A.; Galardi, S. microRNAs and RNA-binding proteins: A complex network of interactions and reciprocal regulations in cancer. RNA Biol. 2013, 10, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Saini, N. Downregulation of BCL2 by miRNAs augments drug-induced apoptosis—A combined computational and experimental approach. J. Cell Sci. 2012, 125 Pt 6, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, Q.J.; Gong, Z.B.; Zhou, L.; You, N.; Wang, S.; Li, X.L.; Li, J.J.; An, J.Z.; Wang, D.S.; et al. MicroRNA-34a targets BCL2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol. Cancer Res. Treat. 2014, 13, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Zeng, Q.; Xie, X.; Zhou, J.; Yue, W.; Li, Y.; Pei, X. MicroRNA-125b induces cancer cell apoptosis through suppression of BCL2 expression. J. Genet. Genom. 2012, 39, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Willimott, S.; Wagner, S.D. miR-125b and miR-155 contribute to BCL2 repression and proliferation in response to CD40 ligand (CD154) in human leukemic B-cells. J. Biol. Chem. 2012, 287, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liao, X.; Wong, C. Downregulations of B-cell lymphoma 2 and myeloid cell leukemia sequence 1 by microRNA 153 induce apoptosis in a glioblastoma cell line DBTRG-05MG. Int. J. Cancer 2010, 126, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Tivnan, A.; Fay, J.; Bryan, K.; Meehan, M.; Creevey, L.; Lynch, J.; Bray, I.M.; O’Meara, A.; Tracey, L.; et al. MicroRNA-204 increases sensitivity of neuroblastoma cells to cisplatin and is associated with a favourable clinical outcome. Br. J. Cancer 2012, 107, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, Z.; Li, S.; Yang, C.; Xue, R.; Xi, Y.; Wang, L.; Wang, S.; He, Q.; Huang, J.; et al. Down-regulation of c-Met and BCL2 by microRNA-206, activates apoptosis, and inhibits tumor cell proliferation, migration and colony formation. Oncotarget 2015, 6, 25533–25574. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Luo, W.; Bai, M.; Li, J.; Bai, X.; Guo, J.; Wu, J.; Wang, M. MicroRNA-206 Inhibited the Progression of Glioblastoma Through BCL2. J. Mol. Neurosci. 2016, 60, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Wei C, L.Q.; Sun, X.; Li, D.; Song, H.; Li, X.; Song, J.; Hua, K.; Fang, L. microRNA-497 induces cell apoptosis by negatively regulating BCL2 protein expression at the posttranscriptional level in human breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 7729–7739. [Google Scholar] [PubMed]
- Kim, K.B.; Kim, K.; Bae, S.; Choi, Y.; Cha, H.J.; Kim, S.Y.; Lee, J.H.; Jeon, S.H.; Jung, H.J.; Ahn, K.J.; et al. MicroRNA-1290 promotes asiatic acidinduced apoptosis by decreasing BCL2 protein level in A549 nonsmall cell lung carcinoma cells. Oncol. Rep. 2014, 32, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Fang, J.H.; Yun, J.P.; Yang, J.; Zhang, Y.; Jia, W.H.; Zhuang, S.M. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 2010, 51, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef] [PubMed]
- Desjobert, C.; Renalier, M.H.; Bergalet, J.; Dejean, E.; Joseph, N.; Kruczynski, A.; Soulier, J.; Espinos, E.; Meggetto, F.; Cavaille, J.; et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 2011, 117, 6627–6637. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Ha, C.T.; Xiao, M. MicroRNA-30 inhibits antiapoptotic factor Mcl-1 in mouse and human hematopoietic cells after radiation exposure. Apoptosis 2016, 21, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tang, H.; Chen, J.; Song, C.; Yang, L.; Liu, P.; Wang, N.; Xie, X.; Lin, X.; Xie, X. MicroRNA-101 inhibits cell progression and increases paclitaxel sensitivity by suppressing MCL-1 expression in human triple-negative breast cancer. Oncotarget 2015, 6, 20070–20083. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Ruan, H.J.; He, X.J.; Ma, Y.Y.; Jiang, X.T.; Xia, Y.J.; Ye, Z.Y.; Tao, H.Q. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer 2010, 46, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Dong, P.; Xiong, Y.; Suzuki, F.; Lu, J.; Cai, M.; Watari, H.; Mitamura, T.; Hosaka, M.; Hanley, S.J.; et al. MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation, invasion and stem cell-like phenotype of aggressive endometrial cancer cells. Oncotarget 2014, 5, 6049–6062. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, J.R.; Xu, T.; Huang, J.; Xu, L.; Yuan, Y.; Zhuang, S.M. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009, 69, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhang, J.P.; Li, B.; Zeng, C.; You, K.; Chen, M.X.; Yuan, Y.; Zhuang, S.M. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, BCLW and IL-6R. Oncogene 2013, 32, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Zhang, H.; Wang, Y.; Li, M.; Xu, W.; Kang, Y.; Wang, Z.; Wang, Z.; Cheng, P.; Tong, D.; et al. MicroRNA-133a, downregulated in osteosarcoma, suppresses proliferation and promotes apoptosis by targeting BCLxL and Mcl-1. Bone 2013, 56, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.; Batte, K.; Yu, L.; Wu, X.; Nuovo, G.J.; Marsh, C.B.; Otterson, G.A.; Nana-Sinkam, S.P. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem. Biophys. Res. Commun. 2009, 388, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.I.; Dollins, C.M.; Crawford, M.; Grant, S.; Nana-Sinkam, S.P.; Richards, K.L.; Hammond, S.M.; Graves, L.M. Lyn kinase-dependent regulation of miR181 and myeloid cell leukemia-1 expression: Implications for drug resistance in myelogenous leukemia. Mol. Pharmacol. 2010, 78, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; Kim, B.Y.; Kwak, S.Y.; Bae, I.H.; Han, Y.H. Ionizing radiation-inducible microRNA miR-193a-3p induces apoptosis by directly targeting Mcl-1. Apoptosis 2013, 18, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Khodayari, N.; Mohammed, K.A.; Lee, H.; Kaye, F.; Nasreen, N. MicroRNA-302b targets Mcl-1 and inhibits cell proliferation and induces apoptosis in malignant pleural mesothelioma cells. Am. J. Cancer Res. 2016, 6, 1996–2009. [Google Scholar] [PubMed]
- Zhang, T.; Zou, P.; Wang, T.; Xiang, J.; Cheng, J.; Chen, D.; Zhou, J. Down-regulation of miR-320 associated with cancer progression and cell apoptosis via targeting Mcl-1 in cervical cancer. Tumour Biol. 2016, 37, 8931–8940. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Suzuki, H.; Tsugawa, H.; Nakagawa, I.; Matsuzaki, J.; Kanai, Y.; Hibi, T. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 2009, 28, 2738–2744. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; Noda, T.; Nagano, H.; et al. The let-7 family of microRNAs inhibits BCLxL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J. Hepatol. 2010, 52, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Huang, H.; Deng, G.; Xie, Z.; Ye, Y.; Guo, R.; Cai, X.; Hong, J.; Qian, D.; Zhou, X.; et al. miR-326 targets antiapoptotic BCLxL and mediates apoptosis in human platelets. PLoS ONE 2015, 10, e0122784. [Google Scholar]
- Al-Harbi, S.; Choudhary, G.S.; Ebron, J.S.; Hill, B.T.; Vivekanathan, N.; Ting, A.H.; Radivoyevitch, T.; Smith, M.R.; Shukla, G.C.; Almasan, A. miR-377-dependent BCLxL regulation drives chemotherapeutic resistance in B-cell lymphoid malignancies. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Lambert, B.; Meryet-Figuiere, M.; Vigneron, N.; Brotin, E.; Lecerf, C.; Abeilard, E.; Giffard, F.; Louis, M.H.; Gauduchon, P.; et al. miR-491-5p-induced apoptosis in ovarian carcinoma depends on the direct inhibition of both BCLXL and EGFR leading to BIM activation. Cell Death Dis. 2014, 5, e1445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schiff, D.; Park, D.; Abounader, R. MicroRNA-608 and microRNA-34a regulate chordoma malignancy by targeting EGFR, BCLxL and MET. PLoS ONE 2014, 9, e91546. [Google Scholar]
- Dong, Z.; Lei, Q.; Yang, R.; Zhu, S.; Ke, X.X.; Yang, L.; Cui, H.; Yi, L. Inhibition of neurotensin receptor 1 induces intrinsic apoptosis via let-7a-3p/BCLW axis in glioblastoma. Br. J. Cancer 2017, 116, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xie, Z.; Peng, Q. MiRNA-107 enhances chemosensitivity to paclitaxel by targeting antiapoptotic factor BCLW in non small cell lung cancer. Am. J. Cancer Res. 2017, 7, 1863–1873. [Google Scholar] [PubMed]
- Cui, Y.H.; Li, H.Y.; Gao, Z.X.; Liang, N.; Ma, S.S.; Meng, F.J.; Li, Z.J.; Pan, H.W. Regulation of Apoptosis by miR-122 in Pterygium via Targeting BCLW. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Liu, X.; Fang, A.; Li, P.; Li, Z.; Liu, T.; Yang, Y.; Du, L.; Wang, C. MicroRNA-203 Is a Prognostic Indicator in Bladder Cancer and Enhances Chemosensitivity to Cisplatin via Apoptosis by Targeting BCLW and Survivin. PLoS ONE 2015, 10, e0143441. [Google Scholar]
- Bo, J.; Yang, G.; Huo, K.; Jiang, H.; Zhang, L.; Liu, D.; Huang, Y. MicroRNA-203 suppresses bladder cancer development by repressing BCLw expression. FEBS J. 2011, 278, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, X.; Zhan, Y.; Jiang, W.; Liu, X.; Wang, X.; Wu, B. miR-335 inhibits the proliferation and invasion of clear cell renal cell carcinoma cells through direct suppression of BCLW. Tumour Biol. 2015, 36, 6875–6882. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, T.; Liu, H.; Lv, T.; Yuan, D.; Yao, Y.; Lv, Y.; Song, Y. miR-101 and Mcl-1 in non-small-cell lung cancer: Expression profile and clinical significance. Med. Oncol. 2012, 29, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Lu, X.; Zhang, H.; Lesniewski, R.; Rosenberg, S.; Semizarov, D. A microRNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (navitoclax). Mol. Cancer Ther. 2010, 9, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Muller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [PubMed]
- Nakano, H.; Miyazawa, T.; Kinoshita, K.; Yamada, Y.; Yoshida, T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting BCLx(L) in colorectal cancer cells. Int. J. Cancer 2010, 127, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Granberg, K.J.; Wang, Q.; Moore, L.M.; Ji, P.; Gumin, J.; Sulman, E.P.; Calin, G.A.; Haapasalo, H.; et al. Two mature products of MIR-491 coordinate to suppress key cancer hallmarks in glioblastoma. Oncogene 2015, 34, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hong, Y.; Xiang, D.; Zhu, P.; Wu, E.; Li, W.; Mosenson, J.; Wu, W.S. MicroRNA-302/367 cluster governs hESC self-renewal by dually regulating cell cycle and apoptosis pathways. Stem Cell Rep. 2015, 4, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Davis-Dusenbery, B.N.; Kashima, R.; Jiang, X.; Marathe, N.; Sessa, R.; Louie, J.; Gu, W.; Lagna, G.; Hata, A. Acetylation of p53 stimulates miRNA processing and determines cell survival following genotoxic stress. EMBO J. 2013, 32, 3192–3205. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Gao, J.; Ding, S.L.; Wang, K.; Jiao, J.Q.; Wang, Y.; Sun, T.; Zhou, L.Y.; Long, B.; Zhang, X.J.; et al. Oxidative Modification of miR-184 Enables It to Target BCLxL and BCLW. Mol. Cell 2015, 59, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M. BCL2A1: The underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Greene, S. microRNA-10b Is Overexpressed and Critical for Cell Survival and Proliferation in Medulloblastoma. PLoS ONE 2015, 10, e0137845. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zeng, S.; Zhou, Z.W.; He, Z.X.; Zhou, S.F. Hsa-microRNA-181a is a regulator of a number of cancer genes and a biomarker for endometrial carcinoma in patients: A bioinformatic and clinical study and the therapeutic implication. Drug Des. Dev. Ther. 2015, 9, 1103–1175. [Google Scholar]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]



| Pre-mRNA | Splicing Variants | Spliceosome Components | hnRNP Proteins | SR Proteins | STAR Proteins | RBM Proteins |
|---|---|---|---|---|---|---|
| BCLx | Pro-BCLxL Anti-apoptotic | n.a. | hnRNP A1 [35], hnRNP A2/B1 [36] | SRSF1 [37], SRp30c [38] | n.a. | n.a. |
| Pro-BCLxS Pro-apoptotic | SF3B1 [34] | hnRNP F [39], hnRNP H [39], hnRNP K [40], hnRNP I [41] | SRSF10 [42], SC35 [43] | SAM68 [44] | RBM4 [45], RBM10 [46], RBM11 [47], RBM25 [48] | |
| MCL1 | Pro-MCL1 Anti-apoptotic | n.a. | n.a. | SRSF1 [49], SRSF2 [50] | n.a. | n.a. |
| Pro-MCL1S Pro-apoptotic | SF3B1 [34,51], UBL5 [51], PRPF8 [51], SART [51] | n.a. | n.a. | n.a. | n.a. |
| BCL2 | MCL1 | BCLxL | BCLW | |
|---|---|---|---|---|
| Validated miRNAs | miR-15/16 [96] miR-24 [97] miR-34a [98] miR-125b* [99,100] miR-153* [101] miR-155 [100] miR-195 [97] miR-204 [82,102] miR-206 [103,104] miR-365 [97] miR-497 [105] miR-1290 [106] | miR-29 [107,108,109] miR-30 [110] miR-101 [111,112,113,114] miR-125b* [115] miR-133a* [116] miR-133b* [117] miR-153* [101] miR-181 [118] miR-193a [119] miR-302b [120] miR-320 [121] miR-512 [122] | Let-7c/g [123] miR-133a* [116] miR-326 [124] miR-377 [125] miR-491 [126] miR-608 [127] | Let-7a-3p [128] miR-107 [129] miR-122 [130] miR-125b* [115] miR-133b* [117] miR-203 [131,132] miR-335 [133] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Placzek, W.J. Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. Int. J. Mol. Sci. 2018, 19, 308. https://doi.org/10.3390/ijms19010308
Cui J, Placzek WJ. Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. International Journal of Molecular Sciences. 2018; 19(1):308. https://doi.org/10.3390/ijms19010308
Chicago/Turabian StyleCui, Jia, and William J. Placzek. 2018. "Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members" International Journal of Molecular Sciences 19, no. 1: 308. https://doi.org/10.3390/ijms19010308
APA StyleCui, J., & Placzek, W. J. (2018). Post-Transcriptional Regulation of Anti-Apoptotic BCL2 Family Members. International Journal of Molecular Sciences, 19(1), 308. https://doi.org/10.3390/ijms19010308