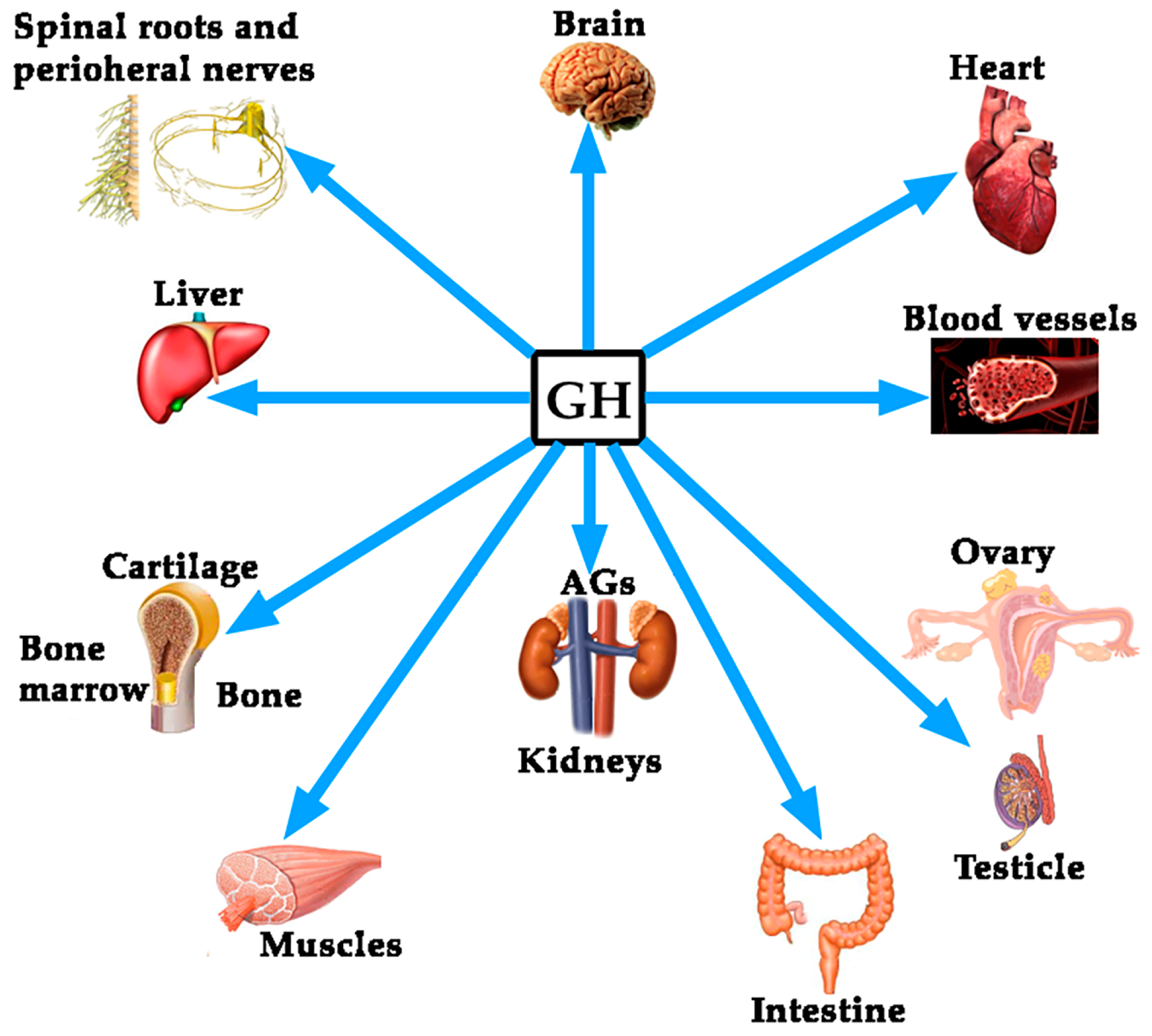
Growth Hormone (GH) and Cardiovascular System




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Role of the Vascular Endothelium as an Internal Secretion Gland and the Effects of GH on It
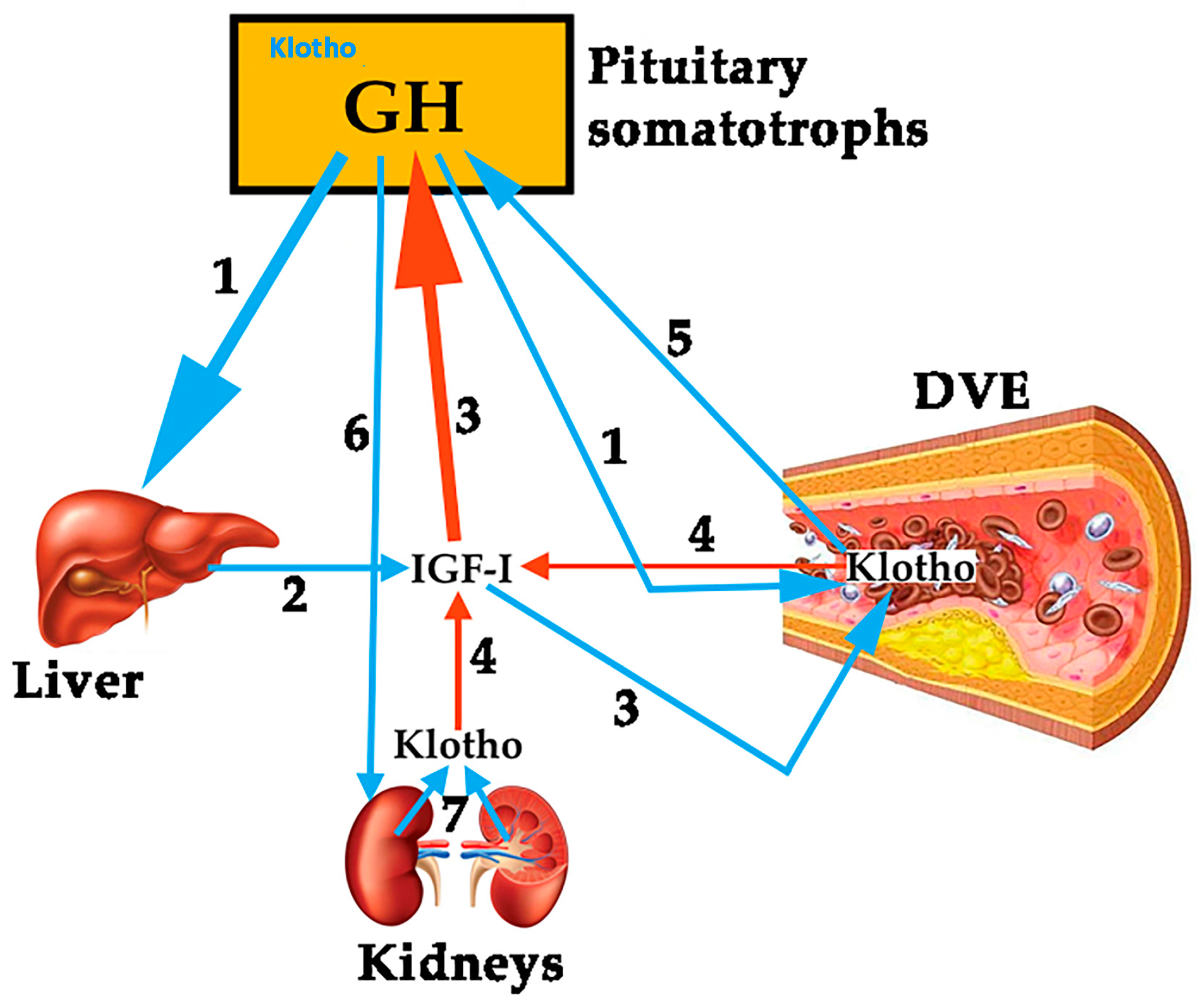
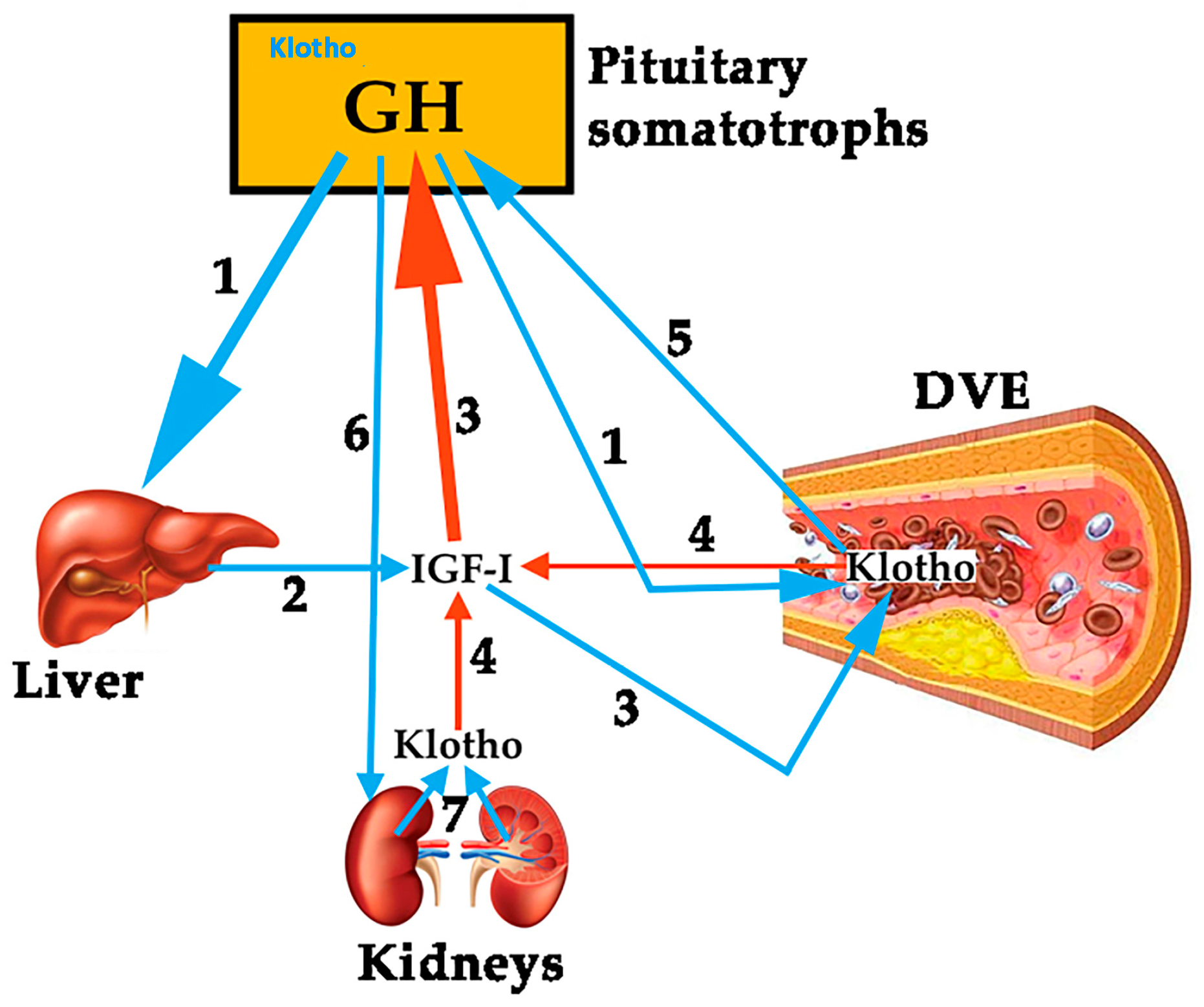
1.1.1. GH, IGF-I, Klotho and the Vascular Endothelium
1.1.2. GH, IGF-I and Ghrelin
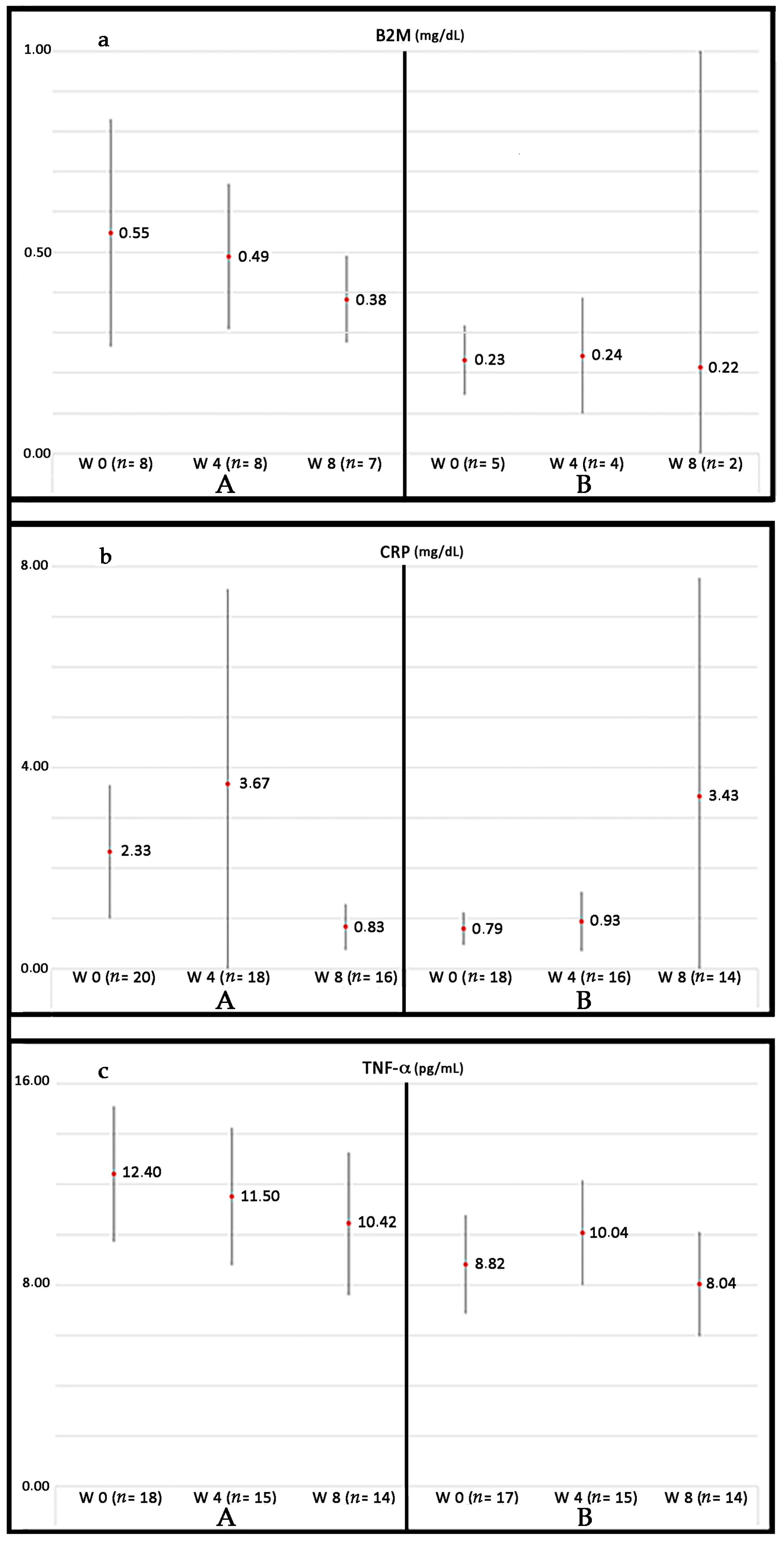
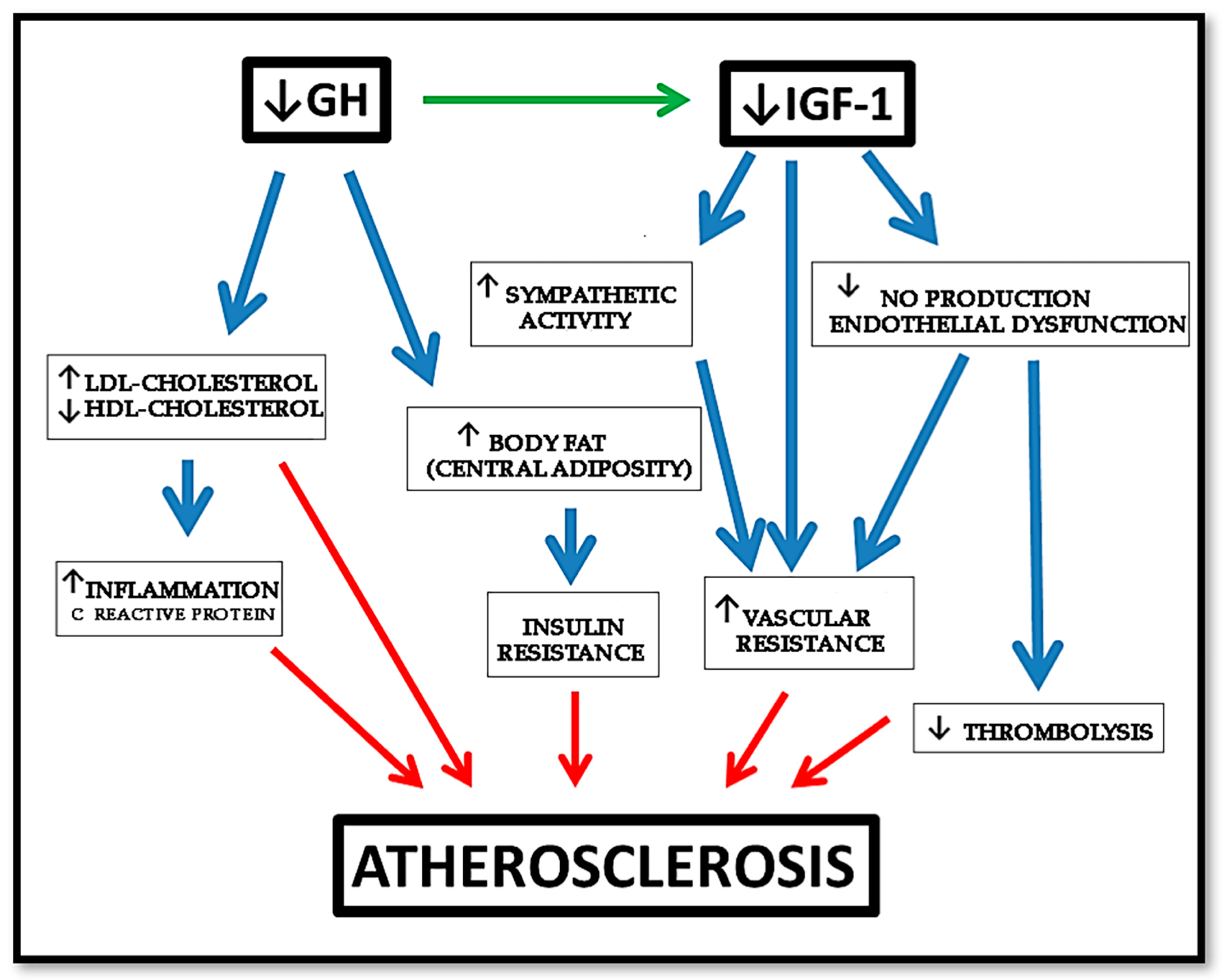
1.2. Cardiovascular Disease as an Inflammatory Condition
1.3. Coronary Arterial Disease (CAD) and Heart Failure
1.4. Peripheral Arterial Disease
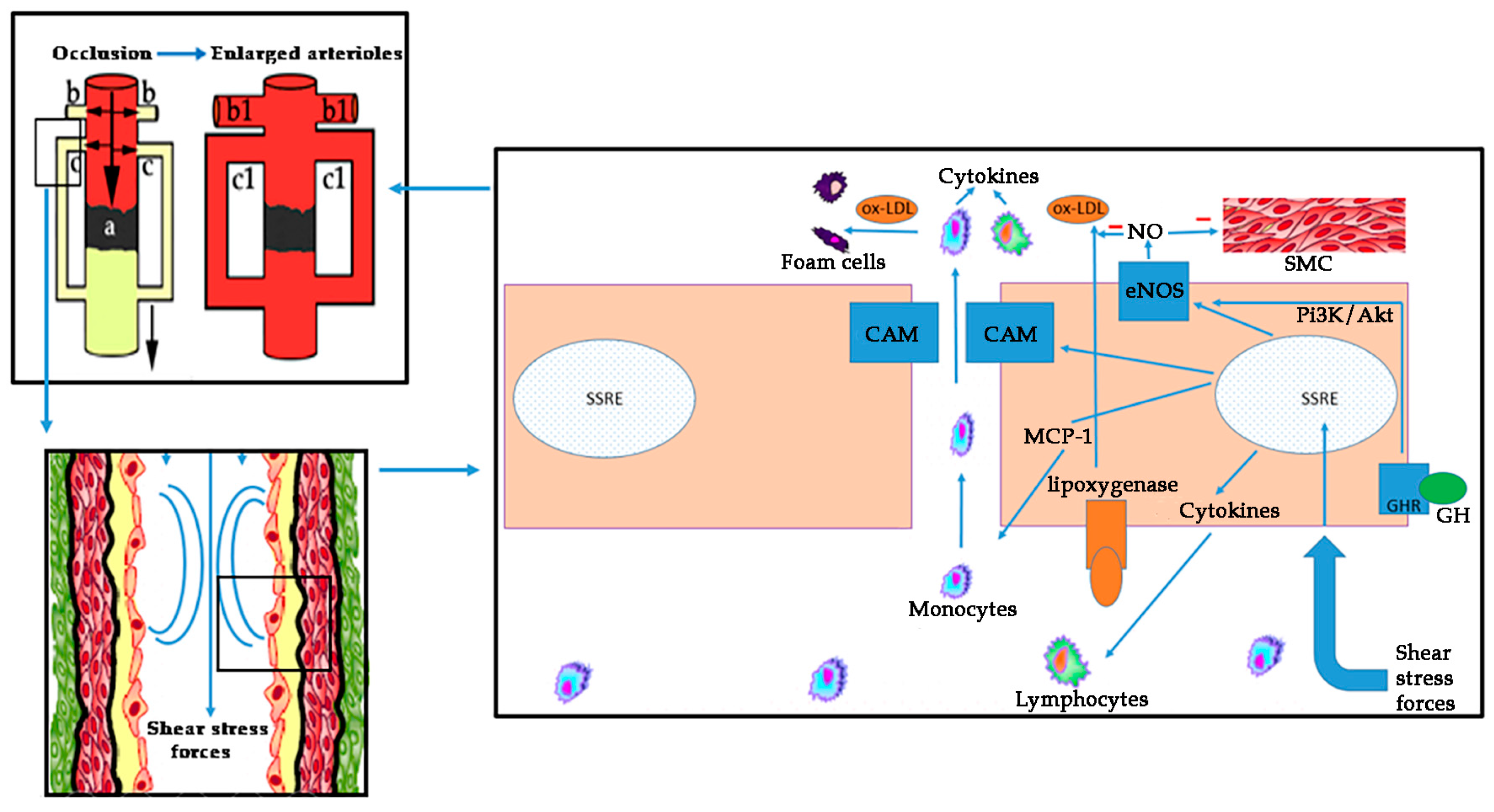
1.4.1. Endothelial and Mitochondrial Dysfunction in PAD: The Role of Oxidative Stress
1.4.2. Endothelin (ET) and PAD
2. Discussion
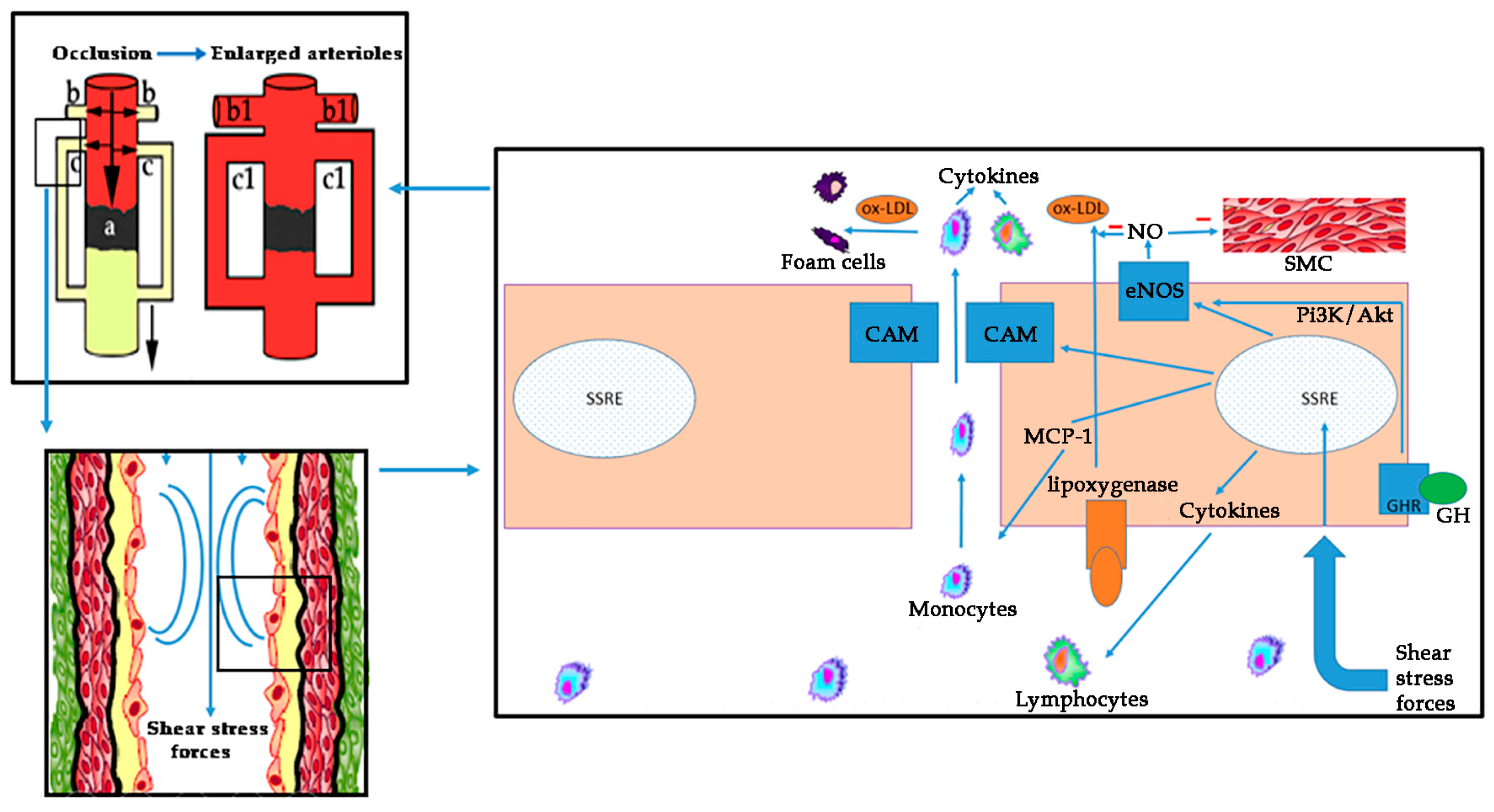
2.1. The Role of GH in the Vascular Endothelium
2.2. GH and Coronary Arterial Disease
2.3. GH and Heart Failure
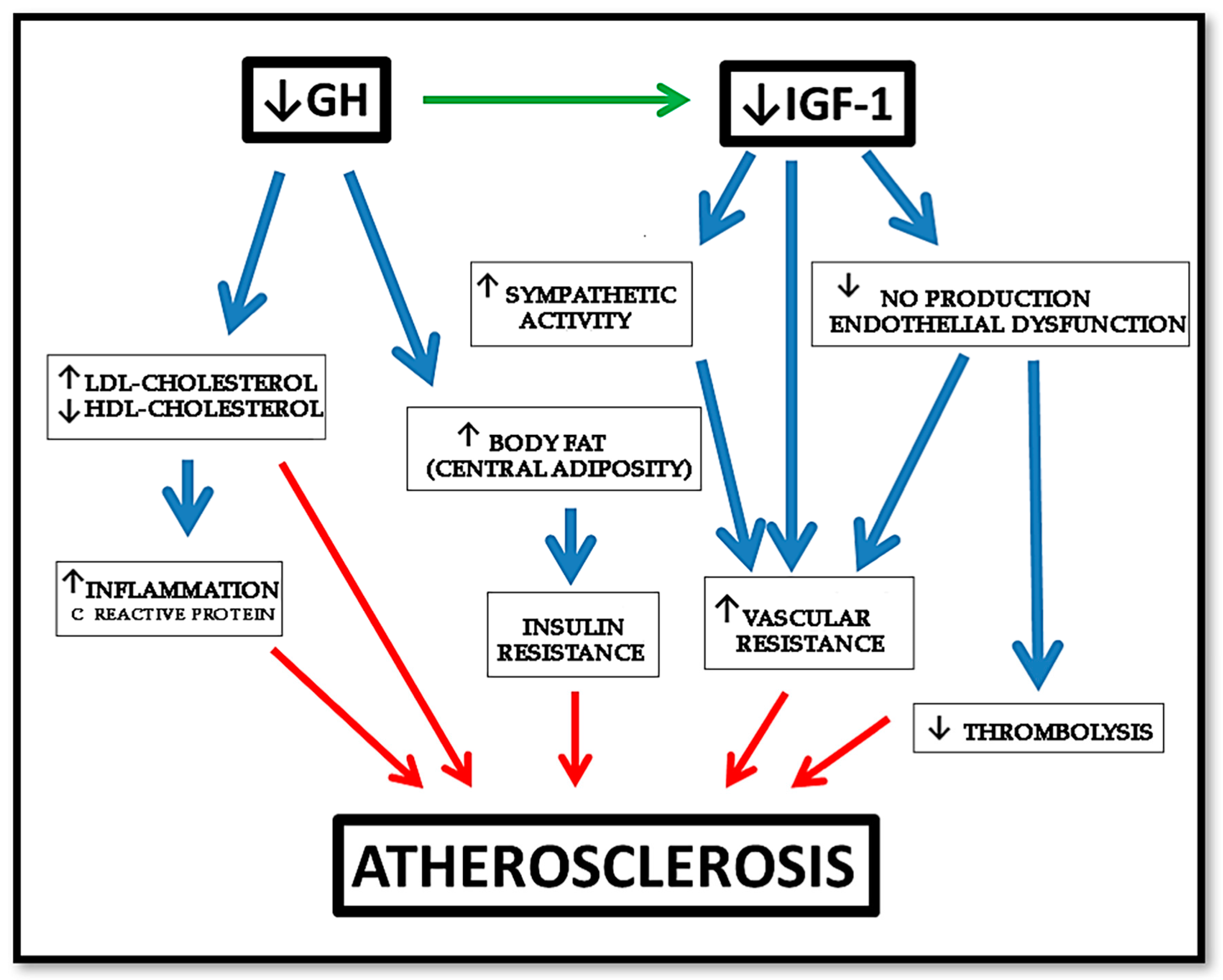
2.4. GH and Molecular Aspects of Cardiovascular Risk Factors in PAD
2.5. GH, Age, and Cardiovascular Disease
2.6. GH and Neovascularization: Experimental and Clinical Evidences
2.7. Effects of GH on Nerve Dysfunction and Other Abnormalities in the Ischemic Muscle
2.8. Why GH Treatment Could not Work Properly in Cardiovascular Disease?
2.9. Adverse Effects of GH Treatment
2.10. Controversies Regarding GH Treatments
2.11. Future Perspectives
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hirt, H.; Kimelman, J.; Birnbaum, M.J.; Chen, E.Y.; Seeburg, P.H.; Eberhardt, N.L.; Barta, A. The human growth hormone gene locus: Structure, evolution, and allelic variations. DNA 1987, 6, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.J.; Dominguez, F.; Haro, L.S.; Ling, N.; Devesa, J. A 12-kilodalton N-glycosylated growth hormone-related peptide is present in human pituitary extracts. J. Clin. Endocrinol. Metab. 1993, 77, 134–138. [Google Scholar] [CrossRef] [PubMed]
- García-Barros, M.; Costoya, J.; Ríos, R.; Arce, V.; Devesa, J. N-Glycosylated Variants of Growth Hormone in Human Pituitary Extracts. Horm. Res. Paediatr. 2000, 53, 40–45. [Google Scholar] [CrossRef] [PubMed]
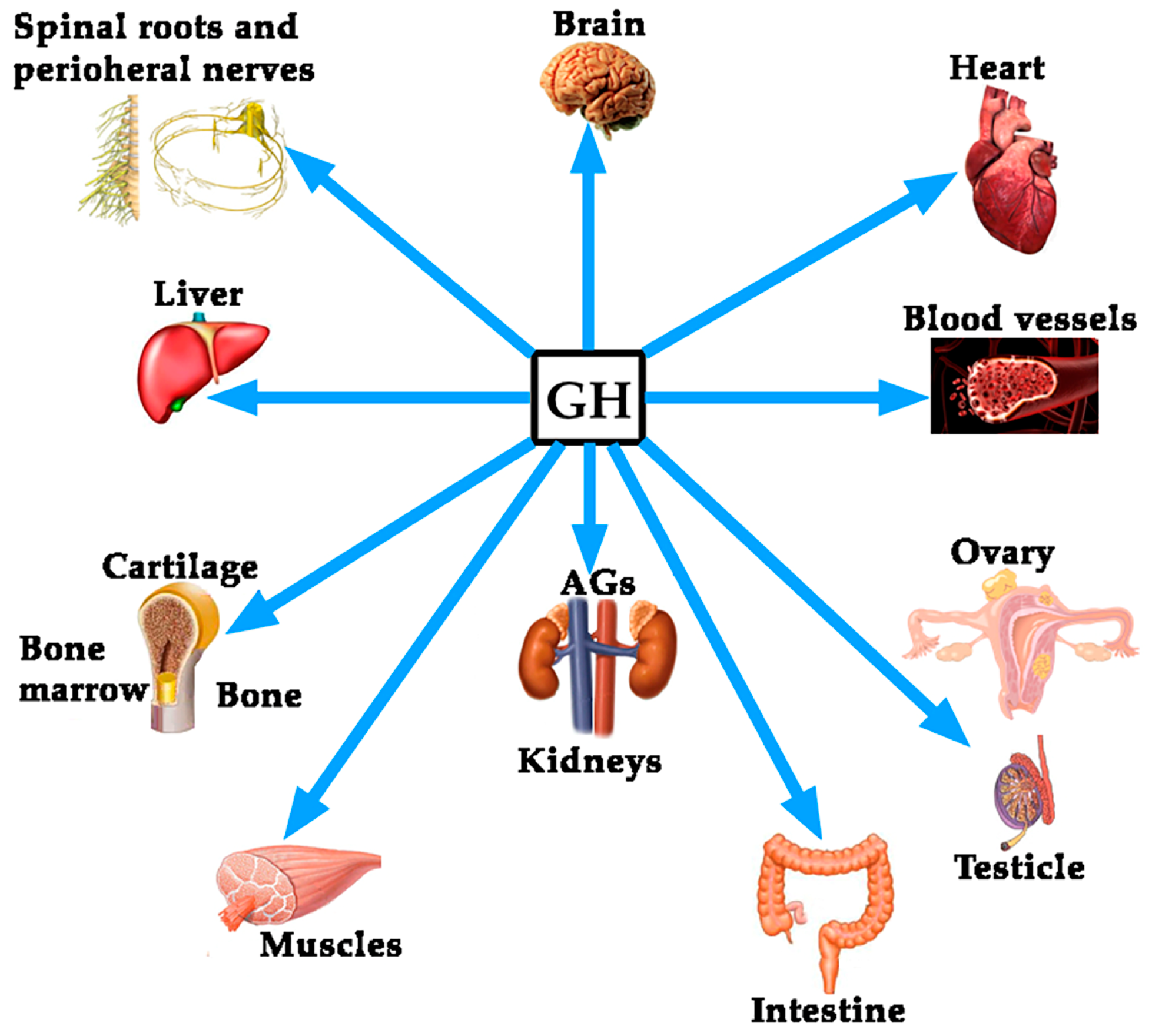
- Devesa, J.; Almengló, C.; Devesa, P. Multiple Effects of Growth Hormone in the Body: Is it Really the Hormone for Growth? Clin. Med. Insights. Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Lima, L.; Tresguerres, J.A. Neuroendocrine control of growth hormone secretion in humans. Trends Endocrinol. Metab. 1992, 3, 175–183. [Google Scholar] [CrossRef]
- Steyn, F.J.; Tolle, V.; Chen, C.; Epelbaum, J. Neuroendocrine regulation of growth hormone secretion. Compr. Physiol. 2016, 6, 687–735. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Modan-Moses, D. Klotho and the Growth Hormone/Insulin-Like Growth Factor 1 Axis: Novel Insights into Complex Interactions. In Vitamins and Hormones; Elsevier Academic Press: Amsterdam, The Netherlands, 2016; Volume 101, pp. 85–118. ISBN 9780128048191. [Google Scholar]
- Wang, T.; Liu, J.; McDonald, C.; Lupino, K.; Zhai, X.; Wilkins, B.J.; Hakonarson, H.; Pei, L. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol. Med. 2017, 9, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Del Rincon, J.P.; Jahn, L.A.; Wu, Y.; Gaylinn, B.; Thorner, M.O.; Liu, Z. Growth hormone exerts acute vascular effects independent of systemic or muscle insulin-like growth factor I. J. Clin. Endocrinol. Metab. 2008, 93, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Tsikas, D.; Frölich, J.C.; Borlak, J. Growth hormone induces eNOS expression and nitric oxide release in a cultured human endothelial cell line. FEBS Lett. 2003, 555, 567–571. [Google Scholar] [CrossRef]
- Colao, A. The GH-IGF-I axis and the cardiovascular system: Clinical implications. Clin. Endocrinol. (Oxf). 2008, 69, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Wickman, A.; Isgaard, J.; Adams, M.A.; Friberg, P. Inhibition of nitric oxide in rats. Regulation of cardiovascular structure and expression of insulin-like growth factor I and its receptor messenger RNA. J. Hypertens. 1997, 15, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Brunet-Dunand, S.E.; Vouyovitch, C.; Araneda, S.; Pandey, V.; Vidal, L.J.; Print, C.; Mertani, H.C.; Lobie, P.E.; Perry, J.K. Autocrine human growth hormone promotes tumor angiogenesis in mammary carcinoma. Endocrinology 2009, 150, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Lilien, M.R.; Schroder, C.H.; Levtchenko, E.N.; Koomans, H.A. Growth hormone therapy influences endothelial function in children with renal failure. Pediatr. Nephrol. 2004, 19, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Lanes, R.; Soros, A.; Flores, K.; Gunczler, P.; Carrillo, E.; Bandel, J. Endothelial function, carotid artery intima-media thickness, epicardial adipose tissue, and left ventricular mass and function in growth hormone-deficient adolescents: Apparent effects of growth hormone treatment on these parameters. J. Clin. Endocrinol. Metab. 2005, 90, 3978–3982. [Google Scholar] [CrossRef] [PubMed]
- Smotkin-Tangorra, M.; Anhalt, H.; Ten, S. Growth hormone and premature atherosclerosis in childhood obesity. J. Pediatr. Endocrinol. Metab. 2006, 19, 455–465. [Google Scholar] [PubMed]
- McCallum, R.W.; Sainsbury, C.A.R.; Spiers, A.; Dominiczak, A.F.; Petrie, J.R.; Sattar, N.; Connell, J.M.C. Growth hormone replacement reduces C-reactive protein and large-artery stiffness but does not alter endothelial function in patients with adult growth hormone deficiency. Clin. Endocrinol. (Oxf). 2005, 62, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Bollerslev, J.; Ueland, T.; Jørgensen, A.P.; Fougner, K.J.; Wergeland, R.; Schreiner, T.; Burman, P. Positive effects of a physiological dose of GH on markers of atherogenesis: A placebo-controlled study in patients with adult-onset GH defiency. Eur. J. Endocrinol. 2006, 154, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Oflaz, H.; Sen, F.; Elitok, A.; Cimen, A.O.; Onur, I.; Kasikcioglu, E.; Korkmaz, S.; Demirturk, M.; Kutluturk, F.; Pamukcu, B.; et al. Coronary flow reserve is impaired in patients with adult growth hormone (GH) deficiency. Clin. Endocrinol. (Oxf). 2007, 66, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Saccà, L. Growth hormone: A newcomer in cardiovascular medicine. Cardiovasc. Res. 1997, 36, 3–9. [Google Scholar] [CrossRef]
- Setola, E.; Monti, L.D.; Lanzi, R.; Lucotti, P.; Losa, M.; Gatti, E.; Galluccio, E.; Oldani, M.; Fermo, I.; Giovannelli, M.; et al. Effects of growth hormone treatment on arginine to asymmetric dimethylarginine ratio and endothelial function in patients with growth hormone deficiency. Metabolism 2008, 57, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.J.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Civin, C.I.; Strauss, L.C.; Brovall, C.; Fackler, M.J.; Schwartz, J.F.; Shaper, J.H. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-1a cells. J. Immunol. 1984, 133, 157–165. [Google Scholar] [PubMed]
- Nielsen, J.S.; McNagny, K.M. Novel functions of the CD34 family. J. Cell Sci. 2008, 121, 3683–3692. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Miyamoto, Y.; Kikuchi-Taura, A.; Soma, T.; Taguchi, A.; Kishimoto, I. Decreased levels of circulating CD34 + cells are associated with coronary heart disease in Japanese patients with type 2 diabetes. J. Diabetes Investig. 2015, 6, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Sohmiya, M.; Kanazawa, I.; Kato, Y. Effect of recombinant human GH on circulating granulocyte colony-stimulating factor and neutrophils in patients with adult GH deficiency. Eur. J. Endocrinol. 2005, 152, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Heeschen, C.; Mildner-Rihm, C.; Urbich, C.; Ihling, C.; Technau-Ihling, K.; Zeiher, A.M.; Dimmeler, S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 2003, 9, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Van der Klaauw, A.A.; Pereira, A.M.; Rabelink, T.J.; Corssmit, E.P.M.; Zonneveld, A.-J.; Pijl, H.; de Boer, H.C.; Smit, J.W.A.; Romijn, J.A.; de Koning, E.J.P. Recombinant human GH replacement increases CD34+ cells and improves endothelial function in adults with GH deficiency. Eur. J. Endocrinol. 2008, 159, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Devin, J.K.; Vaughan, D.E.; Blevins, L.S., Jr.; Chen, Q.; Covington, J.; Verity, D.K.; Young, P.P. Low-dose growth hormone administration mobilizes endothelial progenitor cells in healthy adults. Growth Horm. IGF Res. 2008, 18, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Miljic, D.; Miljic, P.; Doknic, M.; Pekic, S.; Stojanovic, M.; Cvijovic, G.; Micic, D.; Popovic, V. Growth hormone replacement normalizes impaired fibrinolysis: New insights into endothelial dysfunction in patients with hypopituitarism and growth hormone deficiency. Growth Horm. IGF Res. 2013, 23, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.M.; Sahun, M.; Vila, R.; Domenech, P.; Catalina, P.; Soler, J.; Badimon, L. Elevation of E-selectin concentrations may correlate with potential endothelial dysfunction in individuals with hypopituitarism during therapy with growth hormone. Curr. Neurovasc. Res. 2007, 4, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yanagi, K.; Shimizu, M.; Wakamatsu, S.; Niitani, T.; Hosonuma, S.; Sagara, M.; Aso, Y. Effect of growth hormone replacement therapy on plasma diacron-reactive oxygen metabolites and endothelial function in Japanese patients: The GREAT clinical study. Endocr. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.K.; Fisker, S.; Dall, R.; Ledet, T.; Jørgensen, J.O.L.; Rasmussen, L.M. Growth hormone increases vascular cell adhesion molecule 1 expression: In vivo and in vitro evidence. J. Clin. Endocrinol. Metab. 2004, 89, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Takala, J.; Ruokonen, E.; Webster, N.R.; Nielsen, M.S.; Zandstra, D.F.; Vundelinckx, G.; Hinds, C.J. Increased Mortality Associated with Growth Hormone Treatment in Critically Ill Adults. N. Engl. J. Med. 1999, 341, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.P. Growth hormone (GH) treatment does not restore endothelial function in children with GH deficiency. J. Pediatr. Endocrinol. Metab. 2008, 21, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the Human Klotho Gene and Its Two Transcripts Encoding Membrane and SecretedKlothoProtein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Nagai, R.; Hoshino, Y. [Molecular genetics of cardiovascular diseases]. Rinsho Byori 1998, 46, 249–257. [Google Scholar] [PubMed]
- Saito, Y.; Yamagishi, T.; Nakamura, T.; Ohyama, Y.; Aizawa, H.; Suga, T.; Matsumura, Y.; Masuda, H.; Kurabayashi, M.; Kuro-o, M.; et al. Klotho protein protects against endothelial dysfunction. Biochem. Biophys. Res. Commun. 1998, 248, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Nagai, R.; Saito, Y.; Ohyama, Y.; Aizawa, H.; Suga, T.; Nakamura, T.; Kurabayashi, M.; Kuro-o, M. Endothelial dysfunction in the klotho mouse and downregulation of klotho gene expression in various animal models of vascular and metabolic diseases. Cell. Mol. Life Sci. 2000, 57, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-o, M.; Nabeshima, Y.; Kurabayashi, M.; Nagai, R. In Vivo klotho Gene Delivery Protects against Endothelial Dysfunction in Multiple Risk Factor Syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Takeshita, Y.; Murohara, T.; Sasaki, K.I.; Egami, K.; Shintani, S.; Katsuda, Y.; Ikeda, H.; Nabeshima, Y.I.; Imaizumi, T. Angiogenesis and vasculogenesis are impaired in the precocious-aging klotho mouse. Circulation 2004, 110, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, M.; Rakugi, H.; Ishikawa, K.; Maekawa, Y.; Yamamoto, K.; Ohta, J.; Chihara, Y.; Kida, I.; Ogihara, T. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem. Biophys. Res. Commun. 2006, 339, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.J.; Oh, K.W.; Lee, W.Y.; Kim, S.W.Y.; Jung, C.H.; Kim, B.J.S.; Sung, K.C.; Kim, B.J.S.; Kang, J.H.; Lee, M.H.; et al. The differential effects of age on the association of Klotho gene polymorphisms with coronary artery disease. Metabolism 2006, 55, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappa B activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Chang, Y.C.; Ding, Y.; Lim, K.; Liu, Q.; Zhu, L.; Zhang, W.; Lu, T.S.; Molostvov, G.; Zehnder, D.; et al. α-Klotho expression determines nitric oxide synthesis in response to FGF-23 in human aortic endothelial cells. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. α-Klotho and Vascular Calcification: An Evolving Paradigm. Curr. Opin. Nephrol. Hypertens. 2014, 23, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Shahmoon, S.; Rubinfeld, H.; Wolf, I.; Cohen, Z.R.; Hadani, M.; Shimon, I.; Rubinek, T. The aging suppressor klotho: A potential regulator of growth hormone secretion. AJP Endocrinol. Metab. 2014, 307, E326–E334. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.; Stein, D.; Shahmoon, S.; Ziv, S.I.; Hemi, R.; Kanety, H.; Rubinek, T.; Modan-Moses, D. Alteration in serum klotho levels in anorexia nervosa patients. Clin. Nutr. 2016, 35, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, A.E.; Pincelli, A.I.; Corrà, B.; Viarengo, R.; Bonomo, S.M.; Galimberti, D.; Scacchi, M.; Scarpini, E.; Cavagnini, F.; Müller, E.E. Plasma ghrelin concentrations in elderly subjects: Comparison with anorexic and obese patients. J. Endocrinol. 2002, 175, R1–R5. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, N.; Kojima, M.; Uematsu, M.; Yamagishi, M.; Hosoda, H.; Oya, H.; Hayashi, Y.; Kangawa, K. Hemodynamic and hormonal effects of human ghrelin in healthy volunteers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1483–R1487. [Google Scholar] [CrossRef] [PubMed]
- Dembiński, A.; Warzecha, Z.; Ceranowicz, P.; Cieszkowski, J.; Pawlik, W.W.; Tomaszewska, R.; Kuśnierz-Cabala, B.; Naskalski, J.W.; Kuwahara, A.; Kato, I. Role of growth hormone and insulin-like growth factor-1 in the protective effect of ghrelin in ischemia/reperfusion-induced acute pancreatitis. Growth Horm. IGF Res. 2006, 16, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, D.; Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Cieszkowski, J.; Kusnierz-Cabala, B.; Tomaszewska, R.; Kuwahara, A.; Kato, I. Role of hormonal axis, growth hormone—IGF-1, in the therapeutic effect of ghrelin in the course of cerulein-induced acute pancreatitis. J. Physiol. Pharmacol. 2010, 61, 599–606. [Google Scholar] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Kuśnierz-Cabala, B.; Bonior, J.; Jaworek, J.; Ambroży, T.; Gil, K.; Olszanecki, R.; et al. Essential Role of Growth Hormone and IGF-1 in Therapeutic Effect of Ghrelin in the Course of Acetic Acid-Induced Colitis. Int. J. Mol. Sci. 2017, 18, 1118. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, W.R.; Armstrong, E.J.; Larson, C.J.; Brass, E.P. Pathogenesis of the Limb Manifestations and Exercise Limitations in Peripheral Artery Disease. Circ. Res. 2015, 116, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Fragoso Lona, J.M.; Martínez, M.S.; Vargas Alarcón, G.; Barrios Rodas, A.; Ramírez Bello, J. El factor de necrosis tumoral α(TNF-α) en las enfermedades cardiovasculares: Biología molecular y genética. Gac. Med. Mex. 2013, 149, 521–530. [Google Scholar] [PubMed]
- Mazidi, M.; Toth, P.P.; Banach, M. C-reactive Protein Is Associated With Prevalence of the Metabolic Syndrome, Hypertension, and Diabetes Mellitus in US Adults. Angiology 2017, 3319717729288. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.; Lloyd-Jones, D.M. The Role of Biomarkers and Genetics in Peripheral Arterial Disease. J. Am. Coll. Cardiol. 2009, 54, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of interleukin-1ß inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen a phase IIb randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.S.; Kumari, M.; Humphries, S.E.; Mohamed-Ali, V. Inflammation, obesity, stress and coronary heart disease: Is interleukin-6 the link? Atherosclerosis 2000, 148, 209–214. [Google Scholar] [CrossRef]
- Kerr, R.; Stirling, D.; Ludlam, C.A. Interleukin 6 and haemostasis. Br. J. Haematol. 2001, 115, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.; Briones, A. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef] [PubMed]
- Ooi, B.; Goh, B.; Yap, W. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Schiano, V.; Chiariello, M. Cellular adhesion molecules and peripheral arterial disease. Vasc. Med. 2006, 11, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Enlimomab Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434. [Google Scholar] [CrossRef]
- Comi, G. Treatment of multiple sclerosis: Role of natalizumab. Neurol. Sci. 2009, 30, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Tanguay, J.-F.; Wright, S.R.; Duchatelle, V.; Petroni, T.; Grégoire, J.C.; Ibrahim, R.; Heinonen, T.M.; Robb, S.; Bertrand, O.F.; et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage After Percutaneous Coronary Intervention for Non–ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2013, 61, 2048–2055. [Google Scholar] [CrossRef] [PubMed]
- Kling, D.; Stucki, C.; Kronenberg, S.; Tuerck, D.; Rhéaume, E.; Tardif, J.-C.; Gaudreault, J.; Schmitt, C. Pharmacological control of platelet-leukocyte interactions by the human anti-P-selectin antibody inclacumab—Preclinical and clinical studies. Thromb. Res. 2013, 131, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Nylaende, M.; Kroese, A.; Stranden, E.; Morken, B.; Sandbaek, G.; Lindahl, A.K.; Arnesen, H.; Seljeflot, I. Markers of vascular inflammation are associated with the extent of atherosclerosis assessed as angiographic score and treadmill walking distances in patients with peripheral arterial occlusive disease. Vasc. Med. 2006, 11, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, R.C.; Salomon, F.; Wiles, C.M.; Hesp, R.; Sonksen, P.H. Growth hormone treatment in growth hormone-deficient adults. II. Effects on exercise performance. J. Appl. Physiol. 1991, 70, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Arce, V.; Lois, N.; Tresguerres, J.A.F.; Lima, L. α2-adrenergic agonism enhances the growth hormone (GH) response to GH-releasing hormone through an inhibition of hypothalamic somatostatin release in normal men. J. Clin. Endocrinol. Metab. 1990, 71, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Grenon, S.M.; Chong, K.; Alley, H.; Nosova, E.; Gasper, W.; Hiramoto, J.; Boscardin, W.J.; Owens, C.D. Walking disability in patients with peripheral artery disease is associated with arterial endothelial function. J. Vasc. Surg. 2014, 59, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Naldini, A.; Carraro, F. Role of inflammatory mediators in angiogenesis. Curr. Drug Targets. Inflamm. Allergy 2005, 4, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.W.; Parker, D.E.; Montgomery, P.S.; Sosnowska, D.; Casanegra, A.I.; Esponda, O.L.; Ungvari, Z.; Csiszar, A.; Sonntag, W.E. Impaired Vascular Endothelial Growth Factor A and Inflammation in Patients With Peripheral Artery Disease. Angiology 2014, 65, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Wexler, T.L.; Nachtigall, L.; Tritos, N.; Swearingen, B.; Hemphill, L.; Loeffler, J.; Biller, B.M.K.; Klibanski, A.; Miller, K.K. Effects of growth hormone deficiency on body composition and biomarkers of cardiovascular risk after definitive therapy for acromegaly. Clin. Endocrinol. (Oxf). 2012, 77, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Devesa, P.; Reimunde, P. Growth hormone revisited. Med. Clin. 2010, 135, 665–670. [Google Scholar] [CrossRef] [PubMed]
- GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet (London, England) 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Bloom, D.E.; Cafiero, E.; Jané-Llopis, E.; Abrahams-Gessel, S.; Reddy Bloom, L.; Fathima, S.; Feigl, A.B.; Gaziano, T.; Hamandi, A.; Mowafi, M.; et al. The Global Economic Burden of Noncommunicable Diseases. World Econ. Forum 2011, 1–46. [Google Scholar] [CrossRef]
- Roth, G.A.; Forouzanfar, M.H.; Moran, A.E.; Barber, R.; Nguyen, G.; Feigin, V.L.; Naghavi, M.; Mensah, G.A.; Murray, C.J.L. Demographic and Epidemiologic Drivers of Global Cardiovascular Mortality. N. Engl. J. Med. 2015, 372, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Benziger, C.P.; Roth, G.A.; Moran, A.E. The Global Burden of Disease Study and the Preventable Burden of NCD. Glob. Heart 2016, 11, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Bleumink, G.S.; Knetsch, A.M.; Sturkenboom, M.C.J.M.; Straus, S.M.J.M.; Hofman, A.; Deckers, J.W.; Witteman, J.C.M.; Stricker, B.H.C. Quantifying the heart failure epidemic: Prevalence, incidence rate, lifetime risk and prognosis of heart failure. Eur. Heart J. 2004, 25, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; De Ferranti, S.; Després, J.P.; Fullerton, H.J.; et al. Executive summary: Heart disease and stroke statistics-2016 update: A Report from the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States: A Policy Statement From the American Heart Association. Circ. Hear. Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Marleau, S.; Mulumba, M.; Lamontagne, D.; Ong, H. Cardiac and peripheral actions of growth hormone and its releasing peptides: Relevance for the treatment of cardiomyopathies. Cardiovasc. Res. 2006, 69, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barros, M.; Devesa, J.; Arce, V.M. Proteolytic processing of human growth hormone (GH) by rat tissues in vitro: Influence of sex and age. J. Endocrinol. Investig. 2000, 23, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Saccà, L.; Cittadini, A.; Fazio, S. Growth hormone and the heart. Endocr. Rev. 1994, 15, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Samson, W.K.; Sowers, J.R. Insulin-like growth factor I as a cardiac hormone: Physiological and pathophysiological implications in heart disease. J. Mol. Cell. Cardiol. 1999, 31, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Rosén, T.; Bengtsson, B.A. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet (Lond., Engl.) 1990, 336, 285–288. [Google Scholar] [CrossRef]
- Tomlinson, J.W.; Holden, N.; Hills, R.K.; Wheatley, K.; Clayton, R.N.; Bates, A.S.; Sheppard, M.C.; Stewart, P.M. Association between premature mortality and hypopituitarism. Lancet 2001, 357, 425–431. [Google Scholar] [CrossRef]
- Fowkes, F.G.R.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Blanes, J.I.; Cairols, M.A.; Marrugat, J. Prevalence of peripheral artery disease and its associated risk factors in Spain: The ESTIME Study. Int. Angiol. 2009, 28, 20–25. [Google Scholar] [PubMed]
- Criqui, M.H.; Aboyans, V. Epidemiology of Peripheral Artery Disease. Circ. Res. 2015, 116, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The Role of Oxidative Stress and Inflammation in Cardiovascular Aging. Biomed. Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.G. Lower Extremity Manifestations of Peripheral Artery Disease: The Pathophysiologic and Functional Implications of Leg Ischemia. Circ. Res. 2015, 116, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Judge, A.R.; Zhu, Z.; Selsby, J.T.; Swanson, S.A.; Johanning, J.M.; Baxter, B.T.; Lynch, T.G.; Dodd, S.L. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med. 2006, 41, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Sharov, V.G.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A.; Todor, A.; Filis, K.A.; Sabbah, H.N. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J. Vasc. Surg. 2003, 38, 827–832. [Google Scholar] [CrossRef]
- Weiss, D.J.; Casale, G.P.; Koutakis, P.; Nella, A.A.; Swanson, S.A.; Zhu, Z.; Miserlis, D.; Johanning, J.M.; Pipinos, I.I. Oxidative damage and myofiber degeneration in the gastrocnemius of patients with peripheral arterial disease. J. Transl. Med. 2013, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. Molecular Mechanisms of Superoxide Production by the Mitochondrial Respiratory Chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [PubMed]
- Hickman, P.; Harrison, D.K.; Hill, A.; McLaren, M.; Tamei, H.; McCollum, P.T.; Belch, J.J. Exercise in patients with intermittent claudication results in the generation of oxygen derived free radicals and endothelial damage. Adv. Exp. Med. Biol. 1994, 361, 565–570. [Google Scholar] [PubMed]
- Melov, S.; Shoffner, J.M.; Kaufman, A.; Wallace, D.C. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995, 23, 4122–4126. [Google Scholar] [CrossRef] [PubMed]
- Bhat, H.K.; Hiatt, W.R.; Hoppel, C.L.; Brass, E.P. Skeletal muscle mitochondrial DNA injury in patients with unilateral peripheral arterial disease. Circulation 1999, 99, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Rouslin, W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am. J. Physiol. 1983, 244, H743–H748. [Google Scholar] [CrossRef] [PubMed]
- Brass, E.P.; Hiatt, W.R.; Gardner, A.W.; Hoppel, C.L. Decreased NADH dehydrogenase and ubiquinol-cytochrome c oxidoreductase in peripheral arterial disease. Am. J. Physiol. Hear. Circ. Physiol. 2001, 280, H603–H609. [Google Scholar] [CrossRef] [PubMed]
- Barker, G.A.; Green, S.; Green, A.A.; Walker, P.J. Walking performance, oxygen uptake kinetics and resting muscle pyruvate dehydrogenase complex activity in peripheral arterial disease. Clin. Sci. 2004, 106, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Ardail, D.; Debon, A.; Perret-Vivancos, C.; Biol-N’Garagba, M.-C.; Krantic, S.; Lobie, P.E.; Morel, G. Growth hormone internalization in mitochondria decreases respiratory chain activity. Neuroendocrinology 2010, 91, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Nylander, E.; Grönbladh, A.; Zelleroth, S.; Diwakarla, S.; Nyberg, F.; Hallberg, M. Growth hormone is protective against acute methadone-induced toxicity by modulating the NMDA receptor complex. Neuroscience 2016, 339, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Keane, J.; Tajouri, L.; Gray, B. The effect of growth hormone administration on the regulation of mitochondrial apoptosis in-vivo. Int. J. Mol. Sci. 2015, 16, 12753–12772. [Google Scholar] [CrossRef] [PubMed]
- Scacchi, M.; Valassi, E.; Pincelli, A.I.; Fatti, L.M.; Pecori Giraldi, F.; Ascoli, P.; Viarengo, R.; Cestaro, B.; Cavagnini, F.; Cazzola, R. Increased lipid peroxidation in adult GH-deficient patients: Effects of short-term GH administration. J. Endocrinol. Investig. 2006, 29, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Rojanathammanee, L.; Rakoczy, S.; Brown-Borg, H.M. Growth hormone alters the glutathione S-transferase and mitochondrial thioredoxin systems in long-living Ames dwarf mice. J. Gerontol. A. Biol. Sci. Med. Sci. 2014, 69, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Kokoszko, A.; Lewiński, A.; Karbownik-Lewińska, M. The role of growth hormone and insulin-like growth factor I in oxidative processes. Endokrynol. Pol. 2008, 59, 496–501. [Google Scholar] [PubMed]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.E.; Huribal, M.; Bala, R.J.; McMillen, M.A. Endothelin-1 and endothelin-4 stimulate monocyte production of cytokines. Crit. Care Med. 1997, 25, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Babaei, S.; Picard, P.; Ravandi, A.; Monge, J.C.; Lee, T.C.; Cernacek, P.; Stewart, D.J. Blockade of endothelin receptors markedly reduces atherosclerosis in LDL receptor deficient mice: Role of endothelin in macrophage foam cell formation. Cardiovasc. Res. 2000, 48, 158–167. [Google Scholar] [CrossRef]
- Zhao, H.; Thanthan, S.; Yannaing, S.; Kuwayama, H. Involvement of endothelin B receptors in the endothelin-3-induced increase of ghrelin and growth hormone in Holstein steers. Peptides 2010, 31, 938–943. [Google Scholar] [CrossRef] [PubMed]
- ThanThan, S.; Mekaru, C.; Seki, N.; Hidaka, K.; A Ueno; ThidarMyint, H.; Kuwayama, H. Endogenous ghrelin released in response to endothelin stimulates growth hormone secretion in cattle. Domest. Anim. Endocrinol. 2010, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pathipati, P.; Surus, A.; Williams, C.E.; Scheepens, A. Delayed and chronic treatment with growth hormone after endothelin-induced stroke in the adult rat. Behav. Brain Res. 2009, 204, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kirilov, G.; Zacharieva, S.; Alexandrov, A.S.; Lozanov, V.; Mitev, V. Increased plasma endothelin level as an endothelial marker of cardiovascular risk in patients with active acromegaly: A comparison with plasma homocysteine. Methods Find Exp. Clin. Pharmacol. 2009, 31, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Isgaard, J.; Arcopinto, M.; Karason, K.; Cittadini, A. GH and the cardiovascular system: An update on a topic at heart. Endocrine 2014, 48, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Hoeber, S.; Froese, S.; Klink, I.; Stichtenoth, D.O.; Galuppo, P.; Jakob, M.; Tsikas, D.; Anker, S.D.; Poole-Wilson, P.A.; et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ. Res. 2007, 100, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.R.; Evans, P.; Thomas, N.E.; Davies, B.; Baker, J.S. Changes in endothelial dysfunction and associated cardiovascular disease morbidity markers in GH-IGF axis pathology. Am. J. Cardiovasc. Drugs 2009, 9, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Tivesten, Å.; Barlind, A.; Caidahl, K.; Klintland, N.; Cittadini, A.; Ohlsson, C.; Isgaard, J. Growth hormone-induced blood pressure decrease is associated with increased mRNA levels of the vascular smooth muscle KATP channel. J. Endocrinol. 2004, 183, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Andersson, I.J.; Johansson, M.E.; Wickman, A.; Bohlooly-Y, M.; Klintland, N.; Caidahl, K.; Gustafsson, M.; Borén, J.; Gan, L.; Bergström, G. Endothelial dysfunction in growth hormone transgenic mice. Clin. Sci. (Lond). 2006, 110, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Perez, V.; Recchia, F.A.; Podlutsky, A.; Mukhopadhyay, P.; Losonczy, G.; Pacher, P.; Austad, S.N.; Bartke, A.; et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. AJP Hear. Circ. Physiol. 2008, 295, H1882–H1894. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Gautam, T.; Koncz, P.; Henthorn, J.C.; Pinto, J.T.; Ballabh, P.; Yan, H.; Mitschelen, M.; Farley, J.; Sonntag, W.E.; et al. Vasoprotective effects of life span-extending peripubertal GH replacement in lewis dwarf rats. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Li, M.; Reynolds, C.M.; Vickers, M.H. Pre-weaning growth hormone treatment reverses hypertension, endothelial dysfunction and alters heart development in adult male offspring induced as a consequence of maternal undernutrition. Endocr. Rev. 2013, 34, 34. [Google Scholar]
- Delafontaine, P.; Song, Y.H.; Li, Y. Expression, Regulation, and Function of IGF-1, IGF-1R, and IGF-1 Binding Proteins in Blood Vessels. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Pathipati, P.; Gorba, T.; Scheepens, A.; Goffin, V.; Sun, Y.; Fraser, M. Growth hormone and prolactin regulate human neural stem cell regenerative activity. Neuroscience 2011, 190, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Napoli, R.; Guardasole, V.; Angelini, V.; D’Amico, F.; Zarra, E.; Matarazzo, M.; Saccà, L. Acute effects of growth hormone on vascular function in human subjects. J. Clin. Endocrinol. Metab. 2003, 88, 2817–2820. [Google Scholar] [CrossRef] [PubMed]
- Michell, B.J.; Griffiths, J.E.; Mitchelhill, K.I.; Rodriguez-Crespo, I.; Tiganis, T.; Bozinovski, S.; Ortiz de Montellano, P.R.; Kemp, B.E.; Pearson, R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 1999, 9, 845–848. [Google Scholar] [CrossRef]
- Costoya, J.A.; Finidori, J.; Moutoussamy, S.; Señaris, R.; Devesa, J.; Arce, V.M. Activation of growth hormone receptor delivers an antiapoptotic signal: Evidence for a role of Akt in this pathway. Endocrinology 1999, 140, 5937–5943. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Agasse, F.; Xapelli, S.; Almengló, C.; Devesa, J.; Malva, J.O.; Arce, V.M. Growth hormone pathways signaling for cell proliferation and survival in hippocampal neural precursors from postnatal mice. BMC Neurosci. 2014, 15, 100. [Google Scholar] [CrossRef] [PubMed]
- Carter-Su, C.; Schwartz, J.; Argetsinger, L.S. Growth hormone signaling pathways. Growth Horm. IGF Res. 2016, 28, 11–15. [Google Scholar] [CrossRef] [PubMed]
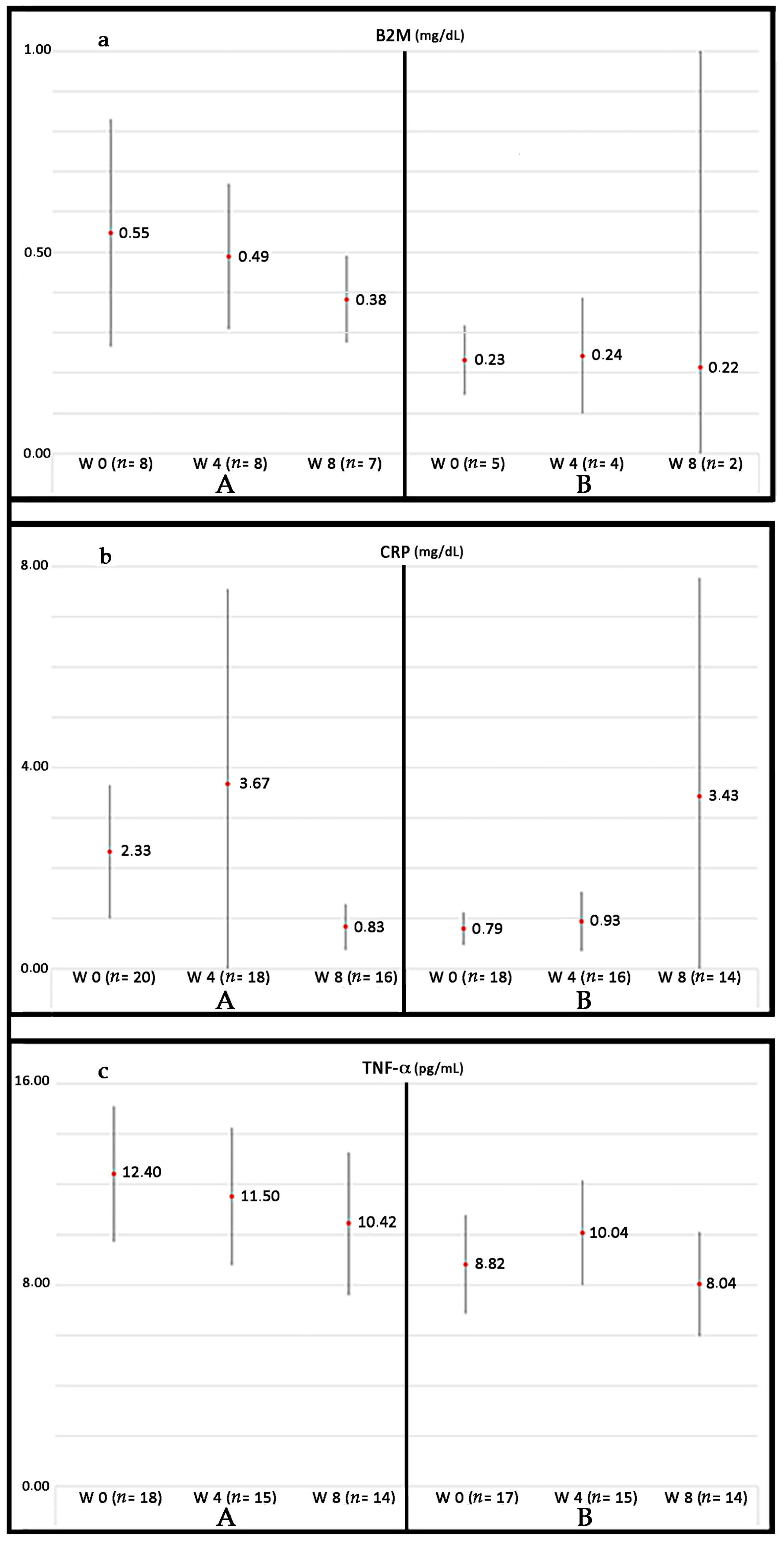
- Deepak, D.; Daousi, C.; Javadpour, M.; Clark, D.; Perry, Y.; Pinkney, J.; Macfarlane, I.A. The influence of growth hormone replacement on peripheral inflammatory and cardiovascular risk markers in adults with severe growth hormone deficiency. Growth Horm. IGF Res. 2010, 20, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Cao, Y.; Mao, S.H.; Liu, H.; Ji, L.L.; Xu, S.Y.; Zhang, M.; Huang, Y. Recombinant human growth hormone improves survival and protects against acute lung injury in murine Staphylococcus aureus sepsis. Inflamm. Res. 2009, 58, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Masternak, M.M.; Bartke, A. Growth hormone, inflammation and aging. Pathobiol. Aging Age-Related Dis. 2012, 2, 17293. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Moon, S.O.; Kim, S.H.; Kim, H.J.; Koh, Y.S.; Koh, G.Y. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 2001, 276, 7614–7620. [Google Scholar] [CrossRef] [PubMed]
- Torzewski, M.; Rist, C.; Mortensen, R.F.; Zwaka, T.P.; Bienek, M.; Waltenberger, J.; Koenig, W.; Schmitz, G.; Hombach, V.; Torzewski, J. C-Reactive Protein in the Arterial Intima: Role of C-Reactive Protein Receptor-Dependent Monocyte Recruitment in Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Sesmilo, G.; Biller, B.M.; Llevadot, J.; Hayden, D.; Hanson, G.; Rifai, N.; Klibanski, A. Effects of growth hormone administration on inflammatory and other cardiovascular risk markers in men with growth hormone deficiency. A randomized, controlled clinical trial. Ann. Intern. Med. 2000, 133, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Beaudeux, J.-L. Serum Plasma Pregnancy-Associated Protein A: A Potential Marker of Echogenic Carotid Atherosclerotic Plaques in Asymptomatic Hyperlipidemic Subjects at High Cardiovascular Risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 7e–e10. [Google Scholar] [CrossRef]
- Heeschen, C.; Dimmeler, S.; Hamm, C.W.; Fichtlscherer, S.; Simoons, M.L.; Zeiher, A.M. Pregnancy-associated plasma protein-A levels in patients with acute coronary syndromes: Comparison with markers of systemic inflammation, platelet activation, and myocardial necrosis. J. Am. Coll. Cardiol. 2005, 45, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ren, W.; Li, J.; Liu, J.; Wang, L.; Zheng, X.; Liu, D.; Li, S.; Souvenir, R.; Tang, J. Increase in serum pregnancy-associated plasma protein-A is correlated with increase in cardiovascular risk factors in adult patients with growth hormone deficiency. Endocrine 2012, 42, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Joaquin, C.; Aguilera, E.; Granada, M.L.; Pastor, M.C.; Salinas, I.; Alonso, N.; Sanmartí, A. Effects of GH treatment in GH-deficient adults on adiponectin, leptin and pregnancy-associated plasma protein-A. Eur. J. Endocrinol. 2008, 158, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.M.; Bobbio, E.; D’Assante, R.; Salzano, A.; Arcopinto, M.; Bossone, E.; Cittadini, A. Growth Hormone as Biomarker in Heart Failure. Heart Fail. Clin. 2018, 14, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Gazzaruso, C.; Gola, M.; Karamouzis, I.; Giubbini, R.; Giustina, A. Cardiovascular risk in adult patients with growth hormone (GH) deficiency and following substitution with GH-An update. J. Clin. Endocrinol. Metab. 2014, 99, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Maison, P.; Griffin, S.; Nicoue-Beglah, M.; Haddad, N.; Balkau, B.; Chanson, P. Impact of growth hormone (GH) treatment on cardiovascular risk factors in GH-deficient adults: A Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. J. Clin. Endocrinol. Metab. 2004, 89, 2192–2199. [Google Scholar] [CrossRef] [PubMed]
- Elbornsson, M.; Gotherstrom, G.; Bosaeus, I.; Bengtsson, B.A.; Johannsson, G.; Svensson, J. Fifteen years of Growth Hormone (GH) Replacement Improves Body Composition and Cardiovascular Risk Factors. Eur. J. Endocrinol. 2013, 168, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, R.; Walsh, M.F.; Rangi, J.S.; Zayas, R.M.; Standley, P.R.; Ram, J.L.; Sowers, J.R. Insulin like growth factor 1 increases vascular smooth muscle nitric oxide production. Life Sci. 1997, 61, 925–931. [Google Scholar] [CrossRef]
- Sverrisdóttir, Y.B.; Elam, M.; Caidahl, K.; Söderling, A.-S.; Herlitz, H.; Johannsson, G. The effect of growth hormone (GH) replacement therapy on sympathetic nerve hyperactivity in hypopituitary adults: A double-blind, placebo-controlled, crossover, short-term trial followed by long-term open GH replacement in hypopituitary adults. J. Hypertens. 2003, 21, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E.; Clemmons, D.R.; Malozowski, S.; Merriam, G.R.; Vance, M.L. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1587–1609. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Di Somma, C.; Filippella, M.; Rota, F.; Pivonello, R.; Orio, F.; Vitale, G.; Lombardi, G. Insulin-like growth factor-1 deficiency determines increased intima-media thickness at common carotid arteries in adult patients with growth hormone deficiency. Clin. Endocrinol. (Oxf). 2004, 61, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Lekakis, J.P.; Papamichael, C.M.; Cimponeriu, A.T.; Stamatelopoulos, K.S.; Papaioannou, T.G.; Kanakakis, J.; Alevizaki, M.K.; Papapanagiotou, A.; Kalofoutis, A.T.; Stamatelopoulos, S.F. Atherosclerotic changes of extracoronary arteries are associated with the extent of coronary atherosclerosis. Am. J. Cardiol. 2000, 85, 949–952. [Google Scholar] [CrossRef]
- Colao, A.; Di Somma, C.; Rota, F.; Di Maio, S.; Salerno, M.; Klain, A.; Spiezia, S.; Lombardi, G. Common carotid intima-media thickness in growth hormone (GH)-deficient adolescents: A prospective study after GH withdrawal and restarting GH replacement. J. Clin. Endocrinol. Metab. 2005, 90, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Svensson, J.; Bengtsson, B.; Rosén, T.; Odén, A.; Johannsson, G. Malignant disease and cardiovascular morbidity in hypopituitary adults with or without growth hormone replacement therapy. J. Clin. Endocrinol. Metab. 2004, 89, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Dobrucki, L.W.; Tsutsumi, Y.; Kalinowski, L.; Dean, J.; Gavin, M.; Sen, S.; Mendizabal, M.; Sinusas, A.J.; Aikawa, R. Analysis of angiogenesis induced by local IGF-1 expression after myocardial infarction using microSPECT-CT imaging. J. Mol. Cell. Cardiol. 2010, 48, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Omerovic, E.; Bollano, E.; Mobini, R.; Kujacic, V.; Madhu, B.; Soussi, B.; Fu, M.; Hjalmarson, A.; Waagstein, F.; Isgaard, J. Growth hormone improves bioenergetics and decreases catecholamines in postinfarct rat hearts. Endocrinology 2000, 141, 4592–4599. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yang, R.; Lu, H.; Ogasawara, A.K.; Li, W.; Ryan, A.; Peale, F.; Paoni, N.F. Effects of early treatment with growth hormone on infarct size, survival, and cardiac gene expression after acute myocardial infarction. Growth Horm. IGF Res. 2002, 12, 208–215. [Google Scholar] [CrossRef]
- Shen, Y.T.; Wiedmann, R.T.; Lynch, J.J.; Grossman, W.; Johnson, R.G. GH replacement fails to improve ventricular function in hypophysectomized rats with myocardial infarction. Am. J. Physiol. 1996, 271, H1721–H1727. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, S.; Cuocolo, A.; Merola, B.; Di Rella, F.; Colao, A.; Nicolai, E.; Cardei, S.; Salvatore, M.; Lombardi, G. Left ventricular function in young adults with childhood and adulthood onset growth hormone deficiency. Clin. Endocrinol. (Oxf). 1998, 48, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Colao, A.; Cuocolo, A.; Di Somma, C.; Cerbone, G.; Della Morte, A.M.; Nicolai, E.; Lucci, R.; Salvatore, M.; Lombardi, G. Impaired cardiac performance in elderly patients with growth hormone deficiency. J. Clin. Endocrinol. Metab. 1999, 84, 3950–3955. [Google Scholar] [CrossRef] [PubMed]
- Losa, M.; von Werder, K. Growth Hormone and the Heart. The Heart in Acromegaly; Manelli, F., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 2001. [Google Scholar]
- Castellano, G.; Affuso, F.; Di Conza, P.; Fazio, S. The GH/IGF-1 Axis and Heart Failure. Curr. Cardiol. Rev. 2009, 5, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.; Oxlund, H. Biosynthetic growth hormone increases the collagen deposition rate in rat aorta and heart. Eur. J. Endocrinol. 1995, 132, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, B.; Wang, X.; Leri, A.; Jana, K.P.; Liu, Y.; Kajstura, J.; Baserga, R.; Anversa, P. Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J. Clin. Investig. 1997, 100, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Napoli, R.; Guardasole, V.; Matarazzo, M.; Palmieri, E.A.; Oliviero, U.; Fazio, S.; Saccà, L. Growth hormone corrects vascular dysfunction in patients with chronic heart failure. J. Am. Coll. Cardiol. 2002, 39, 90–95. [Google Scholar] [CrossRef]
- Le Corvoisier, P.; Hittinger, L.; Chanson, P.; Montagne, O.; Macquin-Mavier, I.; Maison, P. Cardiac effects of growth hormone treatment in chronic heart failure: A meta-analysis. J. Clin. Endocrinol. Metab. 2007, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, M.S.; van Golde, J.M.; Schaper, N.C.; Stehouwer, C.D.; Huijberts, M.S. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin. Sci. (Lond). 2010, 119, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Hiatt, W.R. Peripheral Arterial Disease in Patients with Diabetes. J. Am. Coll. Cardiol. 2006, 47, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Eton, D.; Zhou, G.; He, T.C.; Elsorady, M.; Syed, Z.A. SS18. Enhancing Neovascularization in Chronic Limb-Threatening Ischemia. J. Vasc. Surg. 2015, 61, 106S. [Google Scholar] [CrossRef]
- Leinninger, G.M.; Vincent, A.M.; Feldman, E.L. The role of growth factors in diabetic peripheral neuropathy. J. Peripher. Nerv. Syst. 2004, 9, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Arita, J.; Kojima, Y.; Yamamoto, I.; Mazawa, S.; Kimura, F. Somatotropes and thyrotropes in the rat anterior pituitary gland cosecrete substance P: Analysis by the sandwich cell immunoblot assay. Neuroendocrinology 1994, 60, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Coiro, V.; Volpi, R.; Capretti, L.; Speroni, G.; Bocchi, R.; Caffarri, G.; Colla, R.; Rossi, G.; Chiodera, P. Intravenously infused substance P enhances basal and growth hormone (GH) releasing hormone-stimulated GH secretion in normal men. Peptides 1992, 13, 843–846. [Google Scholar] [CrossRef]
- Thorey, I.S.; Hinz, B.; Hoeflich, A.; Kaesler, S.; Bugnon, P.; Elmlinger, M.; Wanke, R.; Wolf, E.; Werner, S. Transgenic mice reveal novel activities of growth hormone in wound repair, angiogenesis, and myofibroblast differentiation. J. Biol. Chem. 2004, 279, 26674–26684. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Rabuel, C.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Protection in the aged heart: Preventing the heart-break of old age? Cardiovasc. Res. 2005, 66, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Weinsaft, J.W.; Edelberg, J.M. Aging-associated changes in vascular activity: A potential link to geriatric cardiovascular disease. Am. J. Geriatr. Cardiol. 2001, 10, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Yang, H. Effect of Aging on Angiogenesis and Arteriogenesis. Curr. Cardiol. Rev. 2007, 3, 65–74. [Google Scholar] [CrossRef]
- Hoffmann, J.; Haendeler, J.; Aicher, A.; Rössig, L.; Vasa, M.; Zeiher, A.M.; Dimmeler, S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 2001, 89, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Agah, A.; Kyriakides, T.R.; Letrondo, N.; Björkblom, B.; Bornstein, P. Thrombospondin 2 levels are increased in aged mice: Consequences for cutaneous wound healing and angiogenesis. Matrix Biol. 2004, 22, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.T.; Feng, Y. bFGF increases collateral blood flow in aged rats with femoral artery ligation. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H85–H93. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.H.M.; Sadoun, E.; Reed, M.J. Defects in activation of nitric oxide synthases occur during delayed angiogenesis in aging. Mech. Ageing Dev. 2005, 126, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.; DiPietro, L.A. Aging and Wound Healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Pislaru, S.V.; Simari, R.D. Gene transfer for ischemic cardiovascular disease: Is this the end of the beginning or the beginning of the end? Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Sane, D.C.; Wannenburg, T.; Sonntag, W.E. Growth hormone, insulin-like growth factor-1 and the aging cardiovascular system. Cardiovasc. Res. 2002, 54, 25–35. [Google Scholar] [CrossRef]
- Rudman, D.; Feller, A.G.; Nagraj, H.S.; Gergans, G.A.; Lalitha, P.Y.; Goldberg, A.F.; Schlenker, R.A.; Cohn, L.; Rudman, I.W.; Mattson, D.E. Effects of human growth hormone in men over 60 years old. N. Engl. J. Med. 1990, 323, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Paredes, S.D.; Forman, K.A.; García, C.; Vara, E.; Escames, G.; Tresguerres, J.A.F. Protective actions of melatonin and growth hormone on the aged cardiovascular system. Horm. Mol. Biol. Clin. Investig. 2014, 18, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, W.E.; Lynch, C.D.; Cooney, P.T.; Hutchins, P.M. Decreases in cerebral microvasculature with age are associated with the decline in growth hormone and insulin-like growth factor 1. Endocrinology 1997, 138, 3515–3520. [Google Scholar] [CrossRef] [PubMed]
- García-Esteo, F.; Pascual, G.; Gallardo, A.; San-Román, J.; Buján, J.; Bellón, J.M. A biodegradable copolymer for the slow release of growth hormone expedites scarring in diabetic rats. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 81, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Oomen, P.H.N.; Beentjes, J.A.M.; Bosma, E.; Smit, A.J.; Reitsma, W.D.; Dullaart, R.P.F. Reduced capillary permeability and capillary density in the skin of GH-deficient adults: Improvement after 12 months GH replacement. Clin. Endocrinol. (Oxf). 2002, 56, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, D.; Devesa, P.; Arce, V.M.; Requena, J.; Devesa, J. Chronic limb-threatening ischemia could benefit from growth hormone therapy for wound healing and limb salvage. Ther. Adv. Cardiovasc. Dis. 2017, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rabinovsky, E.D.; Draghia-Akli, R. Insulin-like growth factor I plasmid therapy promotes in vivo angiogenesis. Mol. Ther. 2004, 9, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Fang, J.; Kassik, K.A.; Gannon, J.; Cupesi, M.; MacGillivray, C.; Lee, R.T.; Rosenthal, N. Transgenic overexpression of locally acting insulin-like growth factor-1 inhibits ubiquitin-mediated muscle atrophy in chronic left-ventricular dysfunction. Circ. Res. 2005, 97, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, C.; LeRoith, D.; Torres-Aleman, I. Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc. Natl. Acad. Sci. USA 2004, 101, 9833–9838. [Google Scholar] [CrossRef] [PubMed]
- Su, E.J.; Cioffi, C.L.; Stefansson, S.; Mittereder, N.; Garay, M.; Hreniuk, D.; Liau, G. Gene therapy vector-mediated expression of insulin-like growth factors protects cardiomyocytes from apoptosis and enhances neovascularization. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1429–H1440. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Chequer, J.C.; Stouffer, R.L.; Hazzard, T.M.; Patton, P.E.; Molskness, T.A. Insulin-like growth factors-1 and -2, but not hypoxia, synergize with gonadotropin hormone to promote vascular endothelial growth factor-A secretion by monkey granulosa cells from preovulatory follicles. Biol. Reprod. 2003, 68, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Jabri, N.; Schalch, D.S.; Schwartz, S.L.; Fischer, J.S.; Kipnes, M.S.; Radnik, B.J.; Turman, N.J.; Marcsisin, V.S.; Guler, H.P. Adverse effects of recombinant human insulin-like growth factor I in obese insulin-resistant type II diabetic patients. Diabetes 1994, 43, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Clapp, C.; Thebault, S.; Jeziorski, M.C.; Martínez De La Escalera, G. Peptide hormone regulation of angiogenesis. Physiol. Rev. 2009, 89, 1177–1215. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, J.M.; Lee, E.J. Functional expression of CXCR4 in somatotrophs: CXCL12 activates GH gene, GH production and secretion, and cellular proliferation. J. Endocrinol. 2008, 199, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, F.; Bajetto, A.; Porcile, C.; Pattarozzi, A.; Schettini, G.; Florio, T. Role of stromal cell-derived factor 1 (SDF1/CXCL12) in regulating anterior pituitary function. J. Mol. Endocrinol. 2007, 38, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Smaniotto, S.; Martins-Neto, A.A.; Dardenne, M.; Savino, W. Growth hormone is a modulator of lymphocyte migration. Neuroimmunomodulation 2011, 18, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Eitenmüller, I.; Volger, O.; Kluge, A.; Troidl, K.; Barancik, M.; Cai, W.J.; Heil, M.; Pipp, F.; Fischer, S.; Horrevoets, A.J.G.; et al. The range of adaptation by collateral vessels after femoral artery occlusion. Circ. Res. 2006, 99, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Heil, M.; Schaper, W. Insights into Pathways of Arteriogenesis. Curr. Pharm. Biotechnol. 2007, 8, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.F.; Barazi, M.; Pete, G.; Muniyappa, R.; Dunbar, J.C.; Sowers, J.R. Insulin-like growth factor I diminishes in vivo and in vitro vascular contractility: Role of vascular nitric oxide. Endocrinology 1996, 137, 1798–1803. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, H.; Gordienko, D.V.; Tonshoff, B.; Gelato, M.C.; Goligorsky, M.S. Direct demonstration of insulin-like growth factor-I-induced nitric oxide production by endothelial cells. Kidney Int. 1994, 45, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, B.; Guardasole, V.; Pardo, F.; Matarazzo, M.; Di Rella, F.; Numis, F.; Merola, B.; Longobardi, S.; Sacca, L. Abnormal Vascular Reactivity in Growth Hormone Deficiency. Circulation 2001, 103, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Unthank, J.; Haas, T.L.; Miller, S. Impact of shear level and cardiovascular risk factors on bioavailable nitric oxide and outward vascular remodeling in mesenteric arteries. In Arteriogenesis-Molecular Regulation, Patophysiology and Therapeutics I; Schaper, W., Deindl, E., Eds.; Shaker Verlag: Herzogenrath, Germany, 2011; pp. 89–119. ISBN 978-3-8322-9797-8. [Google Scholar]
- Sverrisdottir, Y.B.; Elam, M.; Herlitz, H.; Bengtsson, B.A.; Johannsson, G. Intense sympathetic nerve activity in adults with hypopituitarism and untreated growth hormone deficiency. J. Clin. Endocrinol. Metab. 1998, 83, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Fasshauer, M.; Klein, J.; Kralisch, S.; Klier, M.; Lossner, U.; Bluher, M.; Paschke, R. Monocyte chemoattractant protein 1 expression is stimulated by growth hormone and interleukin-6 in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 2004, 317, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Meazza, C.; Pagani, S.; Travaglino, P.; Bozzola, M. Effect of growth hormone (GH) on the immune system. Pediatr. Endocrinol. Rev. 2004, 1 (Suppl. 3), 490–495. [Google Scholar] [PubMed]
- England, J.D.; Ferguson, M.A.; Hiatt, W.R.; Regensteiner, J.G. Progression of neuropathy in peripheral arterial disease. Muscle Nerve 1995, 18, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Gelabert, M.; González-Mosquera, T.; Gallego, R.; Relova, J.L.; Devesa, J.; Arce, V.M. Growth hormone treatment enhances the functional recovery of sciatic nerves after transection and repair. Muscle Nerve 2012, 45, 385–392. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, M.R.; Bronks, R.; Newton, R.U.; Sharman, M.J.; Graham, J.C.; Cody, D.V.; Kraemer, W.J. Muscle fiber characteristics in patients with peripheral arterial disease. Med. Sci. Sports Exerc. 2001, 33, 2016–2021. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.G.; Duscha, B.D.; Robbins, J.L.; Redfern, S.I.; Chung, J.; Bensimhon, D.R.; Kraus, W.E.; Hiatt, W.R.; Regensteiner, J.G.; Annex, B.H. Increased levels of apoptosis in gastrocnemius skeletal muscle in patients with peripheral arterial disease. Vasc. Med. 2007, 12, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.M. Effects of growth hormone on skeletal muscle. Horm. Res. 2002, 58 (Suppl. 3), 43–48. [Google Scholar] [CrossRef] [PubMed]
- Baum, H.B.; Biller, B.M.; Finkelstein, J.S.; Cannistraro, K.B.; Oppenhein, D.S.; Schoenfeld, D.A.; Michel, T.H.; Wittink, H.; Klibanski, A. Effects of physiologic growth hormone therapy on bone density and body composition in patients with adult-onset growth hormone deficiency. A randomized, placebo-controlled trial. Ann. Intern. Med. 1996, 125, 883–890. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J.; Esser, K.A. Anabolic and catabolic pathways regulating skeletal muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. mTOR as a key regulator in maintaining skeletal muscle mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed]
- Clapp, C.; Aranda, J.; González, C.; Jeziorski, M.C.; Martínez de la Escalera, G. Vasoinhibins: Endogenous regulators of angiogenesis and vascular function. Trends Endocrinol. Metab. 2006, 17, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Jeffcoatte, W. Can growth hormone therapy cause diabetes? Lancet 2000, 355, 589–590. [Google Scholar] [CrossRef]
- Huang, Y.; Chang, Y. Regulation of pancreatic islet beta-cell mass by growth factor and hormone signaling. Prog. Mol. Biol. Transl. Sci. 2014, 121, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.U.; Monson, J.P.; Noonan, K.; John, W.G.; Edwards, A.; Evans, K.A.; Cunningham, J. The effect of low dose recombinant human growth hormone replacement on regional fat distribution, insulin sensitivity, and cardiovascular risk factors in hypopituitary adults. J. Clin. Endocrinol. Metab. 1995, 80, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Dagdelen, S.; Cinar, N.; Erbas, T. Increased thyroid cancer risk in acromegaly. Pituitary 2014, 17, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Montalto, G.; Rizvi, A.A.; Christ, E.R. The role of elevated growth hormone on the increased atherosclerosis in patients with acromegaly. Angiology 2012, 63, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Vilar, L.; Naves, L.A.; Costa, S.S.; Abdalla, L.F.; Coelho, C.E.; Casulari, L.A. Increase of classic and nonclassic cardiovascular risk factors in patients with acromegaly. Endocr. Pract. 2007, 13, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Boero, L.; Cuniberti, L.; Magnani, N.; Manavela, M.; Yapur, V.; Bustos, M.; Gómez Rosso, L.; Meroño, T.; Marziali, L.; Viale, L.; et al. Increased oxidized low density lipoprotein associated with high ceruloplasmin activity in patients with active acromegaly. Clin. Endocrinol. (Oxf). 2010, 72, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Palmeiro, C.R.; Anand, R.; Dardi, I.K.; Balasubramaniyam, N.; Schwarcz, M.D.; Weiss, I.A. Growth hormone and the cardiovascular system. Cardiol. Rev. 2012, 20, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Cittadini, A.; Strömer, H.; Katz, S.E.; Clark, R.; Moses, A.C.; Morgan, J.P.; Douglas, P.S. Differential cardiac effects of growth hormone and insulin-like growth factor-1 in the rat. A combined in vivo and in vitro evaluation. Circulation 1996, 93, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Hallengren, E.; Almgren, P.; Engström, G.; Hedblad, B.; Persson, M.; Suhr, J.; Bergmann, A.; Melander, O. Fasting levels of high-sensitivity growth hormone predict cardiovascular morbidity and mortality: The Malmö Diet and Cancer study. J. Am. Coll. Cardiol. 2014, 64, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- De Lemos, J.A.; Vigen, R. Keeping the genie in the bottle: Growth hormone and cardiovascular disease. J. Am. Coll. Cardiol. 2014, 64, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Lima, L.; Lois, N.; Fraga, C.; Lechuga, M.J.; Arce, V.; Tresguerres, J.A. Reasons for the variability in growth hormone (GH) responses to GHRH challenge: The endogenous hypothalamic-somatotroph rhythm (HSR). Clin. Endocrinol. (Oxf). 1989, 30, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Noiseux, N.; Gnecchi, M.; Lopez-Ilasaca, M.; Zhang, L.; Solomon, S.D.; Deb, A.; Dzau, V.J.; Pratt, R.E. Mesenchymal stem cells overexpressing Akt dramatically repair infarcted myocardium and improve cardiac function despite infrequent cellular fusion or differentiation. Mol. Ther. 2006, 14, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-Y.; Liu, D.; Zhong, Y.; Huang, R.-C. Effects of timing on intracoronary autologous bone marrow-derived cell transplantation in acute myocardial infarction: A meta-analysis of randomized controlled trials. Stem Cell Res. Ther. 2017, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-Y.; Cai, W.-Y.; Tian, M.; Liu, D.; Huang, R.-C. Stem cell transplantation dose in patients with acute myocardial infarction: A meta-analysis. Chronic Dis. Transl. Med. 2016, 2, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, V.; Sahebkar, A.; Kovanen, P.; Bagaglia, F.; Ricciuti, B.; Calabrò, P.; Patti, G.; Pirro, M. Endothelial and cardiac progenitor cells for cardiovascular repair: A controversial paradigm in cell therapy. Pharmacol. Ther. 2018, 181, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Soler-Botija, C.; Farré, J.; Sepúlveda, P.; Raya, A.; Roura, S.; Prat-Vidal, C.; Gálvez-Montón, C.; Montero, J.A.; Büscher, D.; et al. Human progenitor cells derived from cardiac adipose tissue ameliorate myocardial infarction in rodents. J. Mol. Cell. Cardiol. 2010, 49, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Gálvez-Montón, C.; Mirabel, C.; Vives, J.; Bayes-Genis, A. Mesenchymal stem cells for cardiac repair: Are the actors ready for the clinical scenario? Stem Cell Res. Ther. 2017, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Reimunde, P.; Gallego, R.; Devesa, J.; Arce, V.M. Growth hormone (GH) treatment may cooperate with locally-produced GH in increasing the proliferative response of hippocampal progenitors to kainate-induced injury. Brain Inj. 2011, 25, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Kullo, I.J.; Leeper, N.J. The Genetic Basis of Peripheral Arterial Disease: Current Knowledge, Challenges, and Future Directions. Circ. Res. 2015, 116, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Isner, J.M.; Baumgartner, I.; Rauh, G.; Schainfeld, R.; Blair, R.; Manor, O.; Razvi, S.; Symes, J.F. Treatment of thromboangiitis obliterans (Buerger’s disease) by intramuscular gene transfer of vascular endothelial growth factor: Preliminary clinical results. J. Vasc. Surg. 1998, 28, 964–965. [Google Scholar] [CrossRef]
- Hashimoto, T.; Koyama, H.; Miyata, T.; Hosaka, A.; Tabata, Y.; Takato, T.; Nagawa, H. Selective and Sustained Delivery of Basic Fibroblast Growth Factor (bFGF) for Treatment of Peripheral Arterial Disease: Results of a Phase I Trial. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lederman, R.J.; Mendelsohn, F.O.; Anderson, R.D.; Saucedo, J.F.; Tenaglia, A.N.; Hermiller, J.B.; Hillegass, W.B.; Rocha-Singh, K.; Moon, T.E.; Whitehouse, M.J.; et al. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): A randomised trial. Lancet 2002, 359, 2053–2058. [Google Scholar] [CrossRef]
- Li, F.; Sawada, J.; Komatsu, M. R-Ras-Akt axis induces endothelial lumenogenesis and regulates the patency of regenerating vasculature. Nat. Commun. 2017, 8, 1720. [Google Scholar] [CrossRef] [PubMed]
- Ackah, E.; Yu, J.; Zoellner, S.; Iwakiri, Y.; Skurk, C.; Shibata, R.; Ouchi, N.; Easton, R.M.; Galasso, G.; Birnbaum, M.J.; et al. Akt1/protein kinase Bα is critical for ischemic and VEGF-mediated angiogenesis. J. Clin. Investig. 2005, 115, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Martinez-Lemus, L.A.; Davis, G.E. EB1, p150Glued, and Clasp1 control endothelial tubulogenesis through microtubule assembly, acetylation, and apical polarization. Blood 2013, 121, 3521–3530. [Google Scholar] [CrossRef] [PubMed]
- Marte, B.M.; Rodriguez-Viciana, P.; Wennström, S.; Warne, P.H.; Downward, J. R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr. Biol. 1997, 7, 63–70. [Google Scholar] [CrossRef]
- Sawada, J.; Urakami, T.; Li, F.; Urakami, A.; Zhu, W.; Fukuda, M.; Li, D.Y.; Ruoslahti, E.; Komatsu, M. Small GTPase R-Ras Regulates Integrity and Functionality of Tumor Blood Vessels. Cancer Cell 2012, 22, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Ruoslahti, E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 2005, 11, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Layman, H.; Rahnemai-Azar, A.; Pham, S.; Tsechpenakis, G.; Andreopoulos, F.M. Synergistic angiogenic effect of co-delivering fibroblast growth factor 2 and granulocyte-colonystimulating factor from fibrin scaffolds and bone marrow transplantation in critical limb ischemia. Tissue Eng. A 2011, 17, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Tasciotti, E.; Kusmic, C.; Arsic, N.; Sorace, O.; Marini, C.; Marzullo, P.; Pardini, S.; Petroni, D.; Pattarini, L.; et al. In vivo imaging shows abnormal function of vascular endothelial growth factor-induced vasculature. Hum. Gene Ther. 2007, 18, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.P.; Peters, M.C.; Ennett, A.B.; Mooney, D.J. Polymeric system for dual growth factor delivery. Nat. Biotechnol. 2001, 19, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Bauters, C.; Zheng, L.P.; Takeshita, S.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation 1995, 92, II365–II371. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caicedo, D.; Díaz, O.; Devesa, P.; Devesa, J. Growth Hormone (GH) and Cardiovascular System. Int. J. Mol. Sci. 2018, 19, 290. https://doi.org/10.3390/ijms19010290
Caicedo D, Díaz O, Devesa P, Devesa J. Growth Hormone (GH) and Cardiovascular System. International Journal of Molecular Sciences. 2018; 19(1):290. https://doi.org/10.3390/ijms19010290
Chicago/Turabian StyleCaicedo, Diego, Oscar Díaz, Pablo Devesa, and Jesús Devesa. 2018. "Growth Hormone (GH) and Cardiovascular System" International Journal of Molecular Sciences 19, no. 1: 290. https://doi.org/10.3390/ijms19010290
APA StyleCaicedo, D., Díaz, O., Devesa, P., & Devesa, J. (2018). Growth Hormone (GH) and Cardiovascular System. International Journal of Molecular Sciences, 19(1), 290. https://doi.org/10.3390/ijms19010290