VEGF Signaling in Neurological Disorders


Abstract
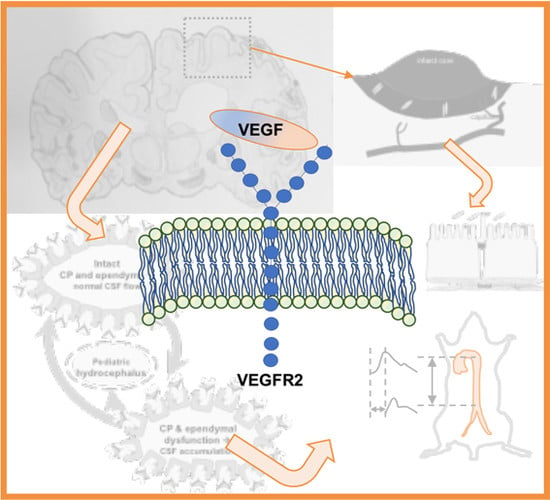
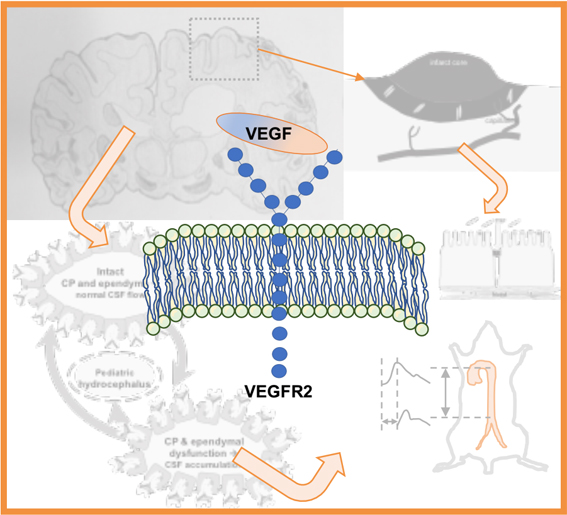
1. Introduction
2. VEGF Signaling in Cerebrovascular Diseases
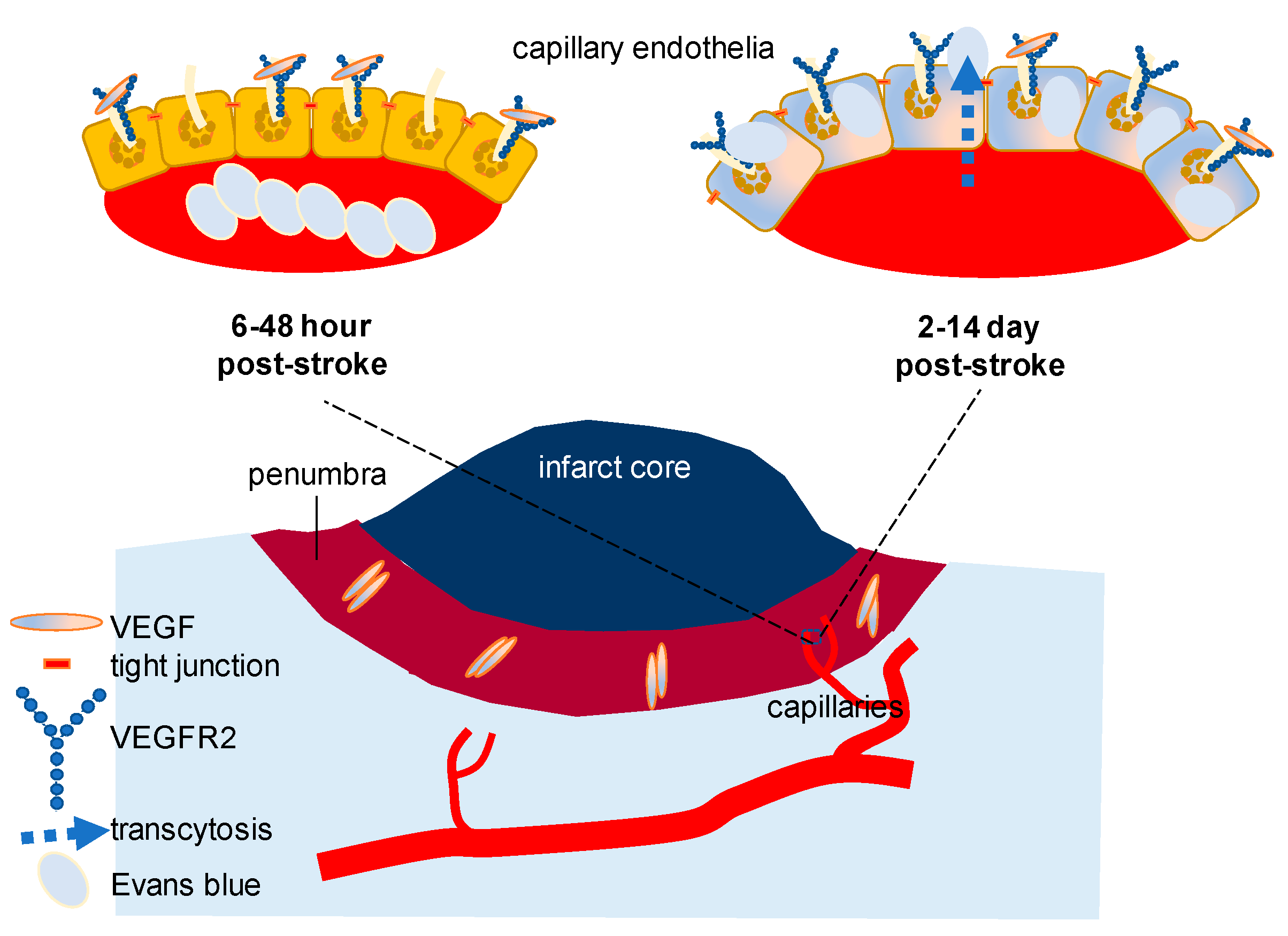
2.1. VEGF Signaling in Stroke
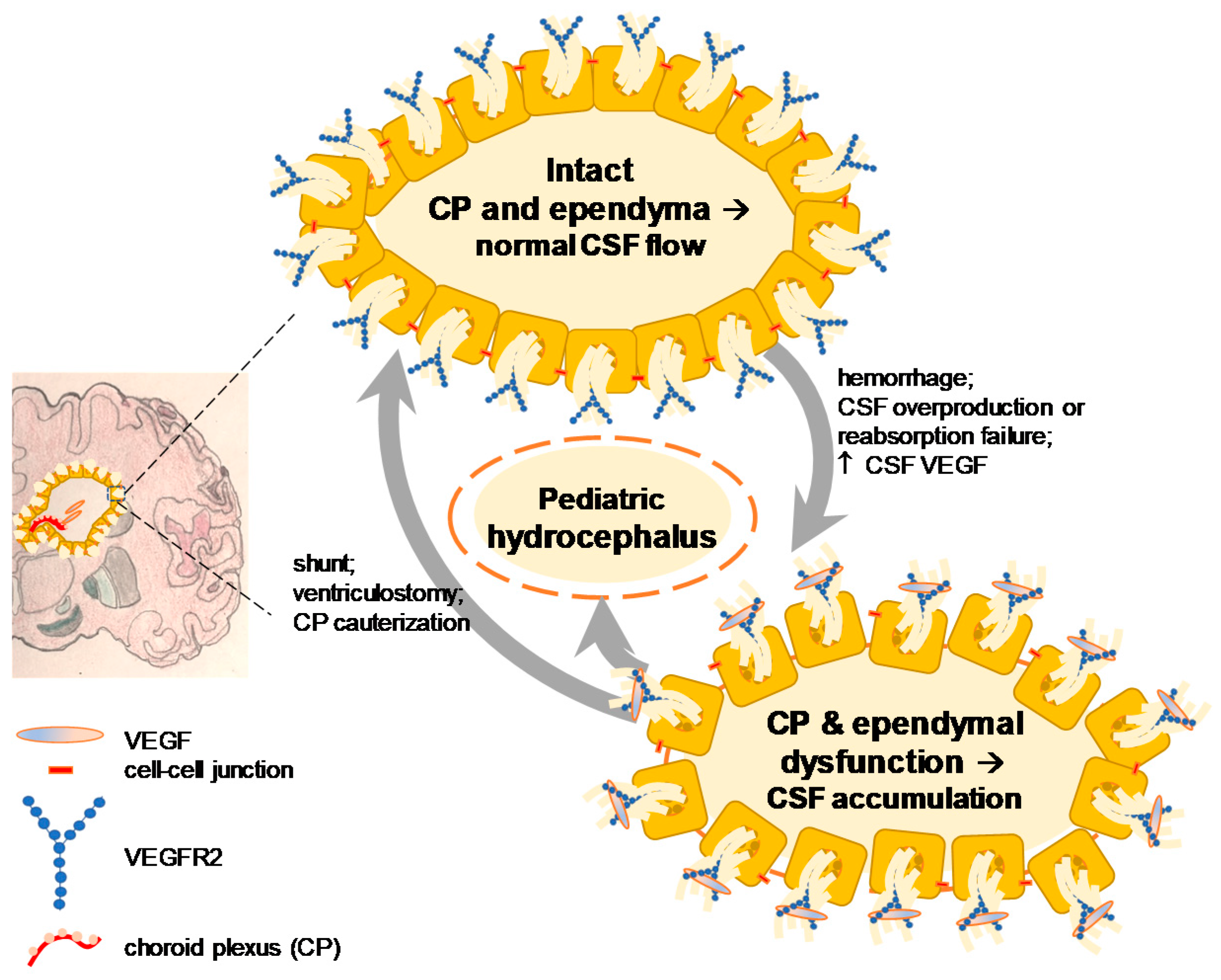
2.2. VEGF Signaling in Hydrocephalus: Lessons from Stroke
2.3. An Alternative (EGFR/ErbB) Signaling in Stroke
2.4. Clinical Implication
3. VEGF Signaling in Age-Related Neurological Disorders
3.1. VEGF Levels in AD
3.2. VEGF Levels in PD
3.3. VEGF and Other Vascular Risk
3.4. Vascular Risk Factors in Neurological Disorders
4. Materials and Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Storkebaum, E.; de Almodóvar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Mirzadeh, Z.; Merkle, F.T.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 2008, 3, 265–278. [Google Scholar] [CrossRef] [PubMed]
- De Almodovar, C.R.; Lambrechts, D.; Mazzone, M.; Carmeliet, P. Role and therapeutic potential of VEGF in the nervous system. Physiol. Rev. 2009, 89, 607–648. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.A.; Zeichner, S.B.; Bartnik, C.M.; Neustadter, E.; Flowers, C.R. Metastatic Colorectal Cancer: A Systematic Review of the Value of Current Therapies. Clin. Colorectal Cancer 2016, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, M.R.; Ennis, S.R.; Masada, T.; Keep, R.F. Intraventricular infusion of vascular endothelial growth factor promotes cerebral angiogenesis with minimal brain edema. Neurosurgery 2002, 50, 589–598. [Google Scholar] [PubMed]
- Harrigan, M.R.; Ennis, S.R.; Sullivan, S.E.; Keep, R.F. Effects of intraventricular infusion of vascular endothelial growth factor on cerebral blood flow, edema, and infarct volume. Acta Neurochir. (Wien) 2003, 145, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Xu, H.; Hu, F.; Braun, A.; Smith, K.; Rivera, A.; Lou, N.; Ungvari, Z.; Goldman, S.A.; Csiszar, A.; Nedergaard, M. Angiogenic inhibition reduces germinal matrix hemorrhage. Nat. Med. 2007, 13, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shanahan, K.J.; Shriver, L.P.; Luciano, M.G. Exercise-induced changes of cerebrospinal fluid vascular endothelial growth factor in adult chronic hydrocephalus patients. J. Clin. Neurosci. 2016, 24, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Alanko, T.; Kataja, V.; Asola, R.; Utriainen, T.; Kokko, R.; Hemminki, A.; Tarkkanen, M.; et al. Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N. Engl. J. Med. 2006, 354, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Safran, H.; Dipetrillo, T.; Akerman, P.; Ng, T.; Evans, D.; Steinhoff, M.; Benton, D.; Purviance, J.; Goldstein, L.; Tantravahi, U.; et al. Phase I/II study of trastuzumab, paclitaxel, cisplatin and radiation for locally advanced, HER2 overexpressing, esophageal adenocarcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.W.; Sandlund, J.; Hameed, M.Q.; Blazer-Yost, B.; Zhou, F.C.; Klagsbrun, M.; Madsen, J.R. Excess HB-EGF, which promotes VEGF signaling, leads to hydrocephalus. Sci. Rep. 2016, 6, 26794. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Tan, X.; Wan, W.; Dixon, B.J.; Fan, R.; Enkhjargal, B.; Li, Q.; Zhang, J.; Chen, G.; Zhang, J.H. ErbB4 protects against neuronal apoptosis via activation of YAP/PIK3CB signaling pathway in a rat model of subarachnoid hemorrhage. Exp. Neurol. 2017, 297, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Investig. 2003, 111, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Organisation for Economic Co-operation and Development (OECD). Health at a Glance 2017: OECD Indicators; OECD Publishing: Paris, France, 2017. [Google Scholar]
- Shim, J.W.; Sandlund, J.; Han, C.H.; Hameed, M.Q.; Connors, S.; Klagsbrun, M.; Madsen, J.R.; Irwin, N. VEGF, which is elevated in the CSF of patients with hydrocephalus, causes ventriculomegaly and ependymal changes in rats. Exp. Neurol. 2013, 247, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, Nv.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; He, J.; Johnson, D.; Wei, Y.; Liu, Y.; Wang, S.; Lutty, G.A.; Duh, E.J.; Semba, R.D. Deletion of placental growth factor prevents diabetic retinopathy and is associated with Akt activation and HIF1α-VEGF pathway inhibition. Diabetes 2015, 64, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Van de Veire, S.; Stalmans, I.; Heindryckx, F.; Oura, H.; Tijeras-Raballand, A.; Schmidt, T.; Loges, S.; Albrecht, I.; Jonckx, B.; Vinckier, S.; et al. Further pharmacological and genetic evidence for the efficacy of PlGF inhibition in cancer and eye disease. Cell 2010, 141, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Mackey, J.K.; Morrison, J.C.; Haigh, J.J.; Adams, R.H.; Faber, J.E. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ. Res. 2012, 111, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dombrowski, S.M.; Krishnan, C.; Krajcir, N.; Deshpande, A.; El-Khoury, S.; Guruprakash, D.K.; Luciano, M.G. Vascular endothelial growth factor in the CSF of elderly patients with ventriculomegaly: Variability, periodicity and levels in drainage responders and non-responders. Clin. Neurol. Neurosurg. 2013, 115, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Alterations of the blood-spinal cord barrier in sporadic amyotrophic lateral sclerosis. Neuropathology 2015, 35, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kawata, A.; Kato, S.; Hayashi, M.; Takamoto, K.; Hayashi, H.; Hirai, S.; Yamaguchi, S.; Komori, T.; Oda, M. Autonomic failure in ALS with a novel SOD1 gene mutation. Neurology 2000, 54, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Briyal, S.; Nguyen, C.; Leonard, M.; Gulati, A. Stimulation of endothelin B receptors by IRL-1620 decreases the progression of Alzheimer’s disease. Neuroscience 2015, 301, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Spuch, C.; Antequera, D.; Portero, A.; Orive, G.; Hernández, R.M.; Molina, J.A.; Bermejo-Pareja, F.; Pedraz, J.L.; Carro, E. The effect of encapsulated VEGF-secreting cells on brain amyloid load and behavioral impairment in a mouse model of Alzheimer’s disease. Biomaterials 2010, 31, 5608–5618. [Google Scholar] [CrossRef] [PubMed]
- Herran, E.; Perez-Gonzalez, R.; Igartua, M.; Pedraz, J.L.; Carro, E.; Hernandez, R.M. Enhanced Hippocampal Neurogenesis in APP/Ps1 Mouse Model of Alzheimer’s Disease After Implantation of VEGF-loaded PLGA Nanospheres. Curr. Alzheimer Res. 2015, 12, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; Palmer, J.C.; Love, S. Pathophysiology of Hypoperfusion of the Precuneus in Early Alzheimer’s Disease. Brain Pathol. 2016, 26, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Lindqvist, D.; Francardo, V.; Hall, S.; Zetterberg, H.; Blennow, K.; Adler, C.H.; Beach, T.G.; Serrano, G.E.; van Westen, D.; et al. Increased CSF biomarkers of angiogenesis in Parkinson disease. Neurology 2015, 85, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Hao, L.J.; Yang, Z.H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.L.; Zhong, M.L.; Wang, T.; Li, J.Y.; et al. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Povysheva, T.; Shmarov, M.; Logunov, D.; Naroditsky, B.; Shulman, I.; Ogurcov, S.; Kolesnikov, P.; Islamov, R.; Chelyshev, Y. Post-spinal cord injury astrocyte-mediated functional recovery in rats after intraspinal injection of the recombinant adenoviral vectors Ad5-VEGF and Ad5-ANG. J. Neurosurg. Spine 2017, 27, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef] [PubMed]
- Reeson, P.; Tennant, K.A.; Gerrow, K.; Wang, J.; Weiser Novak, S.; Thompson, K.; Lockhart, K.L.; Holmes, A.; Nahirney, P.C.; Brown, C.E. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J. Neurosci. 2015, 35, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Hattiangady, B.; Shetty, A.K. Aging does not alter the number or phenotype of putative stem/progenitor cells in the neurogenic region of the hippocampus. Neurobiol. Aging 2008, 29, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.K.; Hattiangady, B.; Shetty, G.A. Stem/progenitor cell proliferation factors FGF-2, IGF-1, and VEGF exhibit early decline during the course of aging in the hippocampus: Role of astrocytes. Glia 2005, 51, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Jean LeBlanc, N.; Guruswamy, R.; ElAli, A. Vascular Endothelial Growth Factor Isoform-B Stimulates Neurovascular Repair After Ischemic Stroke by Promoting the Function of Pericytes via Vascular Endothelial Growth Factor Receptor-1. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.W.; Sandlund, J.; Madsen, J.R. VEGF: A potential target for hydrocephalus. Cell Tissue Res. 2014, 358, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Del Bigio, M.R.; Di Curzio, D.L. Nonsurgical therapy for hydrocephalus: A comprehensive and critical review. Fluids Barriers CNS 2016, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.; Blázquez, J.L.; Rodríguez, E.M. Blood-brain barrier and foetal-onset hydrocephalus, with a view on potential novel treatments beyond managing CSF flow. Fluids Barriers CNS 2017, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Dombrowski, S.M.; Leichliter, A.; Krajcir, N.; Zingales, N.; Inoue, M.; Schenk, S.; Fukamachi, K.; Luciano, M.G. Dissociation between vascular endothelial growth factor receptor-2 and blood vessel density in the caudate nucleus after chronic hydrocephalus. J. Cereb. Blood Flow Metab. 2009, 29, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dombrowski, S.M.; Deshpande, A.; Krajcir, N.; Luciano, M.G. VEGF/VEGFR-2 changes in frontal cortex, choroid plexus, and CSF after chronic obstructive hydrocephalus. J. Neurol. Sci. 2010, 296, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Heep, A.; Stoffel-Wagner, B.; Bartmann, P.; Benseler, S.; Schaller, C.; Groneck, P.; Obladen, M.; Felderhoff-Mueser, U. Vascular endothelial growth factor and transforming growth factor-beta1 are highly expressed in the cerebrospinal fluid of premature infants with posthemorrhagic hydrocephalus. Pediatr. Res. 2004, 56, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; AlMazroa, M.A. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- GBD 2015 Maternal Mortality Collaborators. Global, regional, and national levels of maternal mortality, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1775–1812. [Google Scholar]
- GBD 2016 Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar]
- Ostrowski, R.P.; Colohan, A.R.; Zhang, J.H. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2005, 25, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Gaál, E.I.; Tammela, T.; Anisimov, A.; Marbacher, S.; Honkanen, P.; Zarkada, G.; Leppänen, V.M.; Tatlisumak, T.; Hernesniemi, J.; Niemelä, M.; et al. Comparison of vascular growth factors in the murine brain reveals placenta growth factor as prime candidate for CNS revascularization. Blood 2013, 122, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, I.; Park, E.; Liu, E.; Spratt, S.K.; Surosky, R.; Lee, G.; Ando, D.; Giedlin, M.; Hare, G.M.; Fehlings, M.G.; et al. Treatment of traumatic brain injury using zinc-finger protein gene therapy targeting VEGF-A. J. Neurotrauma 2012, 29, 2647–2659. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Ziaco, B.; Tortiglione, A.; Gala, R.; Cuomo, O.; Vinciguerra, A.; Lapi, D.; Mastantuono, T.; Anzilotti, S.; D’Andrea, L.D.; et al. Neuroprotective Effect of VEGF-Mimetic Peptide QK in Experimental Brain Ischemia Induced in Rat by Middle Cerebral Artery Occlusion. ACS Chem. Neurosci. 2015, 6, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, L.; Humbert-Claude, M. Glial Cell Line-Derived Neurotrophic Factor Gene Delivery in Parkinson’s Disease: A Delicate Balance between Neuroprotection, Trophic Effects, and Unwanted Compensatory Mechanisms. Front. Neuroanat. 2017, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; Lawlor, P.A.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; During, M.J. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 2007, 369, 2097–2105. [Google Scholar] [CrossRef]
- Muramatsu, S.; Fujimoto, K.; Kato, S.; Mizukami, H.; Asari, S.; Ikeguchi, K.; Kawakami, T.; Urabe, M.; Kume, A.; Sato, T.; et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol. Ther. 2010, 18, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.K.; Lang, A.E. Disease-modifying strategies for Parkinson’s disease. Mov. Disord. 2015, 30, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Boldogköi, Z.; Sík, A.; Dénes, A.; Reichart, A.; Toldi, J.; Gerendai, I.; Kovács, K.J.; Palkovits, M. Novel tracing paradigms—Genetically engineered herpesviruses as tools for mapping functional circuits within the CNS: Present status and future prospects. Prog. Neurobiol. 2004, 72, 417–445. [Google Scholar] [CrossRef] [PubMed]
- Mountain, A. Gene therapy: The first decade. Trends Biotechnol. 2000, 18, 119–128. [Google Scholar] [CrossRef]
- Oh, M.S.; Hong, S.J.; Huh, Y.; Kim, K.S. Expression of transgenes in midbrain dopamine neurons using the tyrosine hydroxylase promoter. Gene Ther. 2009, 16, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Volm, M.; Rittgen, W.; Drings, P. Prognostic value of ERBB-1, VEGF, cyclin A, FOS, JUN and MYC in patients with squamous cell lung carcinomas. Br. J. Cancer 1998, 77, 663–669. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yano, S.; Nakataki, E.; Ohtsuka, S.; Inayama, M.; Tomimoto, H.; Edakuni, N.; Kakiuchi, S.; Nishikubo, N.; Muguruma, H.; Sone, S. Retreatment of lung adenocarcinoma patients with gefitinib who had experienced favorable results from their initial treatment with this selective epidermal growth factor receptor inhibitor: A report of three cases. Oncol. Res. 2005, 15, 107–111. [Google Scholar] [PubMed]
- Folkman, J. Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 2007, 6, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Johnson, D.H.; Mininberg, E.; Carbone, D.P.; Henderson, T.; Kim, E.S.; Blumenschein, G., Jr.; Lee, J.J.; Liu, D.D.; Truong, M.T.; et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 2544–2555. [Google Scholar] [PubMed]
- Meng, X.; Zhao, R.; Shen, G.; Dong, D.; Ding, L.; Wu, S. Efficacy and safety of bevacizumab treatment for refractory brain edema: Case report. Medicine 2017, 96, e8280. [Google Scholar] [CrossRef] [PubMed]
- Abbassy, M.; Missios, S.; Barnett, G.H.; Brewer, C.; Peereboom, D.M.; Ahluwalia, M.; Neyman, G.; Chao, S.T.; Suh, J.H.; Vogelbaum, M.A. Phase I Trial of Radiosurgery Dose Escalation Plus Bevacizumab in Patients With Recurrent/Progressive Glioblastoma. Neurosurgery 2017. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, M.; Hoppenreijs, S.; van Rensburg, A.J.; Ruyken, M.; Kolk, A.H.; Springer, P.; Hoepelman, A.I.; Geelen, S.P.; Kimpen, J.L.; Schoeman, J.F. Vascular endothelial growth factor and blood-brain barrier disruption in tuberculous meningitis. Pediatr. Infect. Dis. J. 2004, 23, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Koehne, P.; Hochhaus, F.; Felderhoff-Mueser, U.; Ring-Mrozik, E.; Obladen, M.; Bührer, C. Vascular endothelial growth factor and erythropoietin concentrations in cerebrospinal fluid of children with hydrocephalus. Childs Nerv. Syst. 2002, 18, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jiang, J.; Ford, G.; Ford, B.D. Neuregulin-1 is neuroprotective and attenuates inflammatory responses induced by ischemic stroke. Biochem. Biophys. Res. Commun. 2004, 322, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.F.; Wu, C.Y.; Fang, Y.Y.; Zeng, Y.N.; Luo, Z.Y.; Li, S.J.; Li, X.W.; Zhu, X.H.; Mei, L.; Gao, T.M. Neuregulin 1 protects against ischemic brain injury via ErbB4 receptors by increasing GABAergic transmission. Neuroscience 2015, 307, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Crespo, M.; Gleason, D.; Sistos, A.; Toosky, T.; Solaroglu, I.; Zhang, J.H.; Bryant, P.J.; Fallon, J.H. Transforming growth factor-alpha induces neurogenesis and behavioral improvement in a chronic stroke model. Neuroscience 2009, 160, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Sun, Y.; Xie, L.; Childs, J.; Mao, X.O.; Greenberg, D.A. Post-ischemic administration of heparin-binding epidermal growth factor-like growth factor (HB-EGF) reduces infarct size and modifies neurogenesis after focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 2004, 24, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Ander, B.P.; Stamova, B.; Zhan, X.; Liu, D.; Rothstein, L.; Verro, P.; Khoury, J.; Jauch, E.C.; Pancioli, A.M.; et al. RNA in blood is altered prior to hemorrhagic transformation in ischemic stroke. Ann. Neurol. 2013, 74, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, R.; Ago, T.; Kamouchi, M.; Kuroda, J.; Kuwashiro, T.; Hata, J.; Sugimori, H.; Fukuda, K.; Gotoh, S.; Makihara, N.; et al. Clinical significance of plasma VEGF value in ischemic stroke—Research for biomarkers in ischemic stroke (REBIOS) study. BMC Neurol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.; Ropper, A.H. Ischaemic brain oedema. J. Clin. Neurosci. 2002, 9, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243. [Google Scholar] [CrossRef]
- Guo, L.H.; Alexopoulos, P.; Perneczky, R. Heart-type fatty acid binding protein and vascular endothelial growth factor: Cerebrospinal fluid biomarker candidates for Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Ghezzi, S.; Scarpini, E.; Bresolin, N.; Comi, G.P. VEGF genetic variability is associated with increased risk of developing Alzheimer’s disease. J. Neurol. Sci. 2009, 283, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, R.; Scarlato, M.; Ghezzi, S.; Martinelli Boneschi, F.; Fenoglio, C.; Galbiati, S.; Virgilio, R.; Galimberti, D.; Galimberti, G.; Crimi, M.; et al. Vascular endothelial growth factor gene variability is associated with increased risk for AD. Ann. Neurol. 2005, 57, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Mateo, I.; Llorca, J.; Infante, J.; Rodríguez-Rodríguez, E.; Fernández-Viadero, C.; Peña, N.; Berciano, J.; Combarros, O. Low serum VEGF levels are associated with Alzheimer’s disease. Acta Neurol. Scand. 2007, 116, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jia, J.; Liu, R. Decreased serum levels of the angiogenic factors VEGF and TGF-β1 in Alzheimer’s disease and amnestic mild cognitive impairment. Neurosci. Lett. 2013, 550, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Herrán, E.; Pérez-González, R.; Igartua, M.; Pedraz, J.L.; Carro, E.; Hernández, R.M. VEGF-releasing biodegradable nanospheres administered by craniotomy: A novel therapeutic approach in the APP/Ps1 mouse model of Alzheimer’s disease. J. Control. Release 2013, 170, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Rocha de Paula, M.; Gómez Ravetti, M.; Berretta, R.; Moscato, P. Differences in abundances of cell-signalling proteins in blood reveal novel biomarkers for early detection of clinical Alzheimer’s disease. PLoS ONE 2011, 6, e17481. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Sousa-Ribeiro, D.; Rodriguez-Pallares, J.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Aging and sedentarism decrease vascularization and VEGF levels in the rat substantia nigra. Implications for Parkinson’s disease. J. Cereb. Blood Flow Metab. 2009, 29, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Teema, AM.; Zaitone, S.A.; Moustafa, Y.M. Ibuprofen or piroxicam protects nigral neurons and delays the development of l-dopa induced dyskinesia in rats with experimental Parkinsonism: Influence on angiogenesis. Neuropharmacology 2016, 107, 432–450. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Arai, H.; Takanashi, M.; Fukae, J.; Oizumi, H.; Yasuda, T.; Mizuno, Y.; Mochizuki, H. Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport 2006, 17, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Langston, R.G.; Rudenko, I.N.; Cookson, M.R. The function of orthologues of the human Parkinson’s disease gene LRRK2 across species: Implications for disease modelling in preclinical research. Biochem. J. 2016, 473, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.L.; Stowe, R.; Patel, S.; Rick, C.; Gray, R.; Clarke, C.E. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov. Disord. 2010, 25, 2649–2653. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bezard, E. New animal models of Parkinson’s disease. Mov. Disord. 2011, 26, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Nagy, J.A.; Pal, S.; Vasile, E.; Eckelhoefer, I.A.; Bliss, V.S.; Manseau, E.J.; Dasgupta, P.S.; Dvorak, H.F.; Mukhopadhyay, D. The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 2001, 7, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Zhang, Z.; Huang, J.; Chen, C.; Zhang, Z.; Jia, M.; Xiong, J.; Liu, X.; Wang, F.; Cao, X.; et al. VEGF-expressing human umbilical cord mesenchymal stem cells, an improved therapy strategy for Parkinson’s disease. Gene Ther. 2011, 18, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Shingo, T.; Kobayashi, K.; Takeuchi, A.; Yano, A.; Muraoka, K.; Matsui, T.; Miyoshi, Y.; Hamada, H.; Date, I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur. J. Neurosci. 2004, 19, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Shingo, T.; Muraoka, K.; Kameda, M.; Agari, T.; Ji, Y.W.; Hayase, H.; Hamada, H.; Borlongan, C.V.; Date, I. Neurorescue effects of VEGF on a rat model of Parkinson’s disease. Brain Res. 2005, 1053, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Falk, T.; Zhang, S.; Sherman, S.J. Vascular endothelial growth factor B (VEGF-B) is up-regulated and exogenous VEGF-B is neuroprotective in a culture model of Parkinson’s disease. Mol. Neurodegener. 2009, 4, 49. [Google Scholar] [CrossRef] [PubMed]
- Mihci, E.; Ozkaynak, S.S.; Sallakci, N.; Kizilay, F.; Yavuzer, U. VEGF polymorphisms and serum VEGF levels in Parkinson’s disease. Neurosci. Lett. 2011, 494, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.; Mateo, I.; Rodríguez-Rodríguez, E.; Berciano, J.; Combarros, O. VEGF serum levels are not associated with Parkinson’s disease. Eur. J. Neurol. 2007, 14, e6. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Shingo, T.; Muraoka, K.; Ji, Y.W.; Kameda, M.; Takeuchi, A.; Yano, A.; Nishio, S.; Matsui, T.; Miyoshi, Y.; et al. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson’s disease model. Brain Res. 2005, 1038, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, K.S.; Lee, Y.S.; Park, S.H.; Choe, J.Y. Arterial stiffness and proinflammatory cytokines in fibromyalgia syndrome. Clin. Exp. Rheumatol. 2010, 28, S71–S77. [Google Scholar] [PubMed]
- Suzuki, J.; Sakakibara, R.; Tateno, F.; Tsuyusaki, Y.; Kishi, M.; Ogata, T.; Tomaru, T.; Shirai, K.; Kurosu, T. Parkinson’s disease and the cardio-ankle vascular stiffness index. Intern. Med. 2014, 53, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Park, K.W.; Seo, W.K.; Park, M.H.; Han, C.; Jo, I.; Ahn Jo, S. Carotid intima-media thickness in Parkinson’s disease. Mov. Disord. 2007, 22, 2446–2449. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ryu, D.W.; Oh, J.H.; Lee, Y.H.; Park, S.J.; Jeon, K.; Lee, J.Y.; Ho, S.H.; So, J.; Im, J.H.; et al. Cardiovascular Autonomic Dysfunction in Patients with Drug-Induced Parkinsonism. J. Clin. Neurol. 2017, 13, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Han, S.W.; Baik, J.S. Comparative Study of Central Hemodynamics in Parkinson’s Disease. J. Mov. Disord. 2017, 10, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Hariri, D.J.; Caballero, B.; Zhang, S.; Bartlett, M.J.; Kaut, O.; Mount, D.W.; Wüllner, U.; Sherman, S.J.; Falk, T. Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson’s disease. Neuroscience 2014, 258, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P. VEGF kinase inhibitors: How do they cause hypertension? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1–R5. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Osborne, L.D.; Guilluy, C.; Chen, Z.; O’Brien, E.T., 3rd; Reader, J.S.; Burridge, K.; Superfine, R.; Tzima, E. Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat. Commun. 2014, 5, 3984. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Fry, J.L.; Al Sayah, L.; Weisbrod, R.M.; Van Roy, I.; Weng, X.; Cohen, R.A.; Bachschmid, M.M.; Seta, F. Vascular Smooth Muscle Sirtuin-1 Protects Against Diet-Induced Aortic Stiffness. Hypertension 2016, 68, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Komaroff, A.L. Ask the doctor. Is it true that B vitamins can reduce my risk of Alzheimer’s disease and other types of dementia? Harv. Health Lett. 2014, 39, 2. [Google Scholar] [PubMed]
- Sehgel, N.L.; Vatner, S.F.; Meininger, G.A. “Smooth Muscle Cell Stiffness Syndrome”-Revisiting the Structural Basis of Arterial Stiffness. Front. Physiol. 2015, 6, 335. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.Z.; Saphirstein, R.J.; Yamin, R.; Suki, B.; Morgan, K.G. Aging impairs smooth muscle-mediated regulation of aortic stiffness: A defect in shock absorption function? Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1252–H1261. [Google Scholar] [CrossRef] [PubMed]
- McVeigh, G.E.; Bratteli, C.W.; Morgan, D.J.; Alinder, C.M.; Glasser, S.P.; Finkelstein, S.M.; Cohn, J.N. Age-related abnormalities in arterial compliance identified by pressure pulse contour analysis: Aging and arterial compliance. Hypertension 1999, 33, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Veronese, M.L.; Mosenkis, A.; Flaherty, K.T.; Gallagher, M.; Stevenson, J.P.; Townsend, R.R.; O’Dwyer, P.J. Mechanisms of hypertension associated with BAY 43–9006. J. Clin. Oncol. 2006, 24, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Wagshul, M.E.; Eide, P.K.; Madsen, J.R. The pulsating brain: A review of experimental and clinical studies of intracranial pulsatility. Fluids Barriers CNS 2011, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.R.; Egnor, M.; Zou, R. Cerebrospinal fluid pulsatility and hydrocephalus: The fourth circulation. Clin. Neurosurg. 2006, 53, 48. [Google Scholar] [PubMed]
- Meaume, S.; Benetos, A.; Henry, O.F.; Rudnichi, A.; Safar, M.E. Aortic pulse wave velocity predicts cardiovascular mortality in subjects >70 years of age. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Scott, D.E.; Shandas, R.; Stenmark, K.R.; Tan, W. High pulsatility flow induces adhesion molecule and cytokine mRNA expression in distal pulmonary artery endothelial cells. Ann. Biomed. Eng. 2009, 37, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Talwalkar, J.A.; Glaser, K.J.; Manduca, A.; Grimm, R.C.; Rossman, P.J.; Fidler, J.L.; Ehman, R.L. Assessment of hepatic fibrosis with magnetic resonance elastography. Clin. Gastroenterol. Hepatol. 2007, 5, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Bae, J.M.; Joo, S.K.; Woo, H.; Lee, D.H.; Jung, Y.J.; Kim, B.G.; Lee, K.L.; Kim, W. Prospective comparison among transient elastography, supersonic shear imaging, and ARFI imaging for predicting fibrosis in nonalcoholic fatty liver disease. PLoS ONE 2017, 12, e0188321. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Friedman, S.L.; Aloman, C. Hepatic fibrosis. Curr. Opin. Gastroenterol. 2009, 25, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Pong, A.C.; Jugé, L.; Bilston, L.E.; Cheng, S. Development of acute hydrocephalus does not change brain tissue mechanical properties in adult rats, but in juvenile rats. PLoS ONE 2017, 12, e0182808. [Google Scholar] [CrossRef] [PubMed]
- McEniery, C.M.; Yasmin Hall, I.R.; Qasem, A.; Wilkinson, I.B.; Cockcroft, J.R.; ACCT Investigators. Normal vascular aging: Differential effects on wave reflection and aortic pulse wave velocity: The Anglo-Cardiff Collaborative Trial (ACCT). J. Am. Coll. Cardiol. 2005, 46, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Heijl, A.; Leske, M.C.; Bengtsson, B.; Hyman, L.; Bengtsson, B.; Hussein, M.; Early Manifest Glaucoma Trial Group. Reduction of intraocular pressure and glaucoma progression: Results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002, 120, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- MacMahon, S.; Peto, R.; Cutler, J.; Collins, R.; Sorlie, P.; Neaton, J.; Abbott, R.; Godwin, J.; Dyer, A.; Stamler, J. Blood pressure, stroke, and coronary heart disease. Part 1, Prolonged differences in blood pressure: Prospective observational studies corrected for the regression dilution bias. Lancet 1990, 335, 765–774. [Google Scholar] [CrossRef]
- Ringstad, G.; Emblem, K.E.; Geier, O.; Alperin, N.; Eide, P.K. Aqueductal Stroke Volume: Comparisons with Intracranial Pressure Scores in Idiopathic Normal Pressure Hydrocephalus. Am. J. Neuroradiol. 2015, 36, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Hayashi, H.; Kato, S.; Hayashi, M.; Tanabe, H.; Oda, M. Circulatory collapse and sudden death in respirator-dependent amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 124, 45–55. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Waldstein, S.R.; Rice, S.C.; Thayer, J.F.; Najjar, S.S.; Scuteri, A.; Zonderman, A.B. Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension 2008, 51, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Godwin-Austen, R.B.; Lee, P.N.; Marmot, M.G.; Stern, G.M. Smoking and Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1982, 45, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Hernán, M.A.; Takkouche, B.; Caamaño-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Grandinetti, A.; Morens, D.M.; Reed, D.; MacEachern, D. Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson’s disease. Am. J. Epidemiol. 1994, 139, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Lee, P.C.; Lassen, C.F.; Arah, O.A. Parkinson disease and smoking revisited: Ease of quitting is an early sign of the disease. Neurology 2014, 83, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Morris, C.M.; Court, J.A.; Cheng, A.; Fairbairn, A.F.; McKeith, I.G.; Irving, D.; Brown, A.; Perry, R.H. Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: Possible index of early neuropathology. Neuroscience 1995, 64, 385–395. [Google Scholar] [CrossRef]
- Villafane, G.; Cesaro, P.; Rialland, A.; Baloul, S.; Azimi, S.; Bourdet, C.; Le Houezec, J.; Macquin-Mavier, I.; Maison, P. Chronic high dose transdermal nicotine in Parkinson’s disease: An open trial. Eur. J. Neurol. 2007, 14, 1313–1316. [Google Scholar] [CrossRef] [PubMed]
- Conklin, B.S.; Zhao, W.; Zhong, D.S.; Chen, C. Nicotine and cotinine up-regulate vascular endothelial growth factor expression in endothelial cells. Am. J. Pathol. 2002, 160, 413–418. [Google Scholar] [CrossRef]
- Ansari, K.A.; Johnson, A. Olfactory function in patients with Parkinson’s disease. J. Chronic Dis. 1975, 28, 493–497. [Google Scholar] [CrossRef]
- Doty, R.L. Olfactory dysfunction in Parkinson disease. Nat. Rev. Neurol. 2012, 8, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Martínez, J.; Gorostidi, A.; Goyenechea, E.; Alzualde, A.; Poza, J.J.; Rodríguez, F.; Bergareche, A.; Moreno, F.; López de Munain, A.; Martí Massó, J.F. Olfactory deficits and cardiac 123I-MIBG in Parkinson’s disease related to the LRRK2 R1441G and G2019S mutations. Mov. Disord. 2011, 26, 2026–2031. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
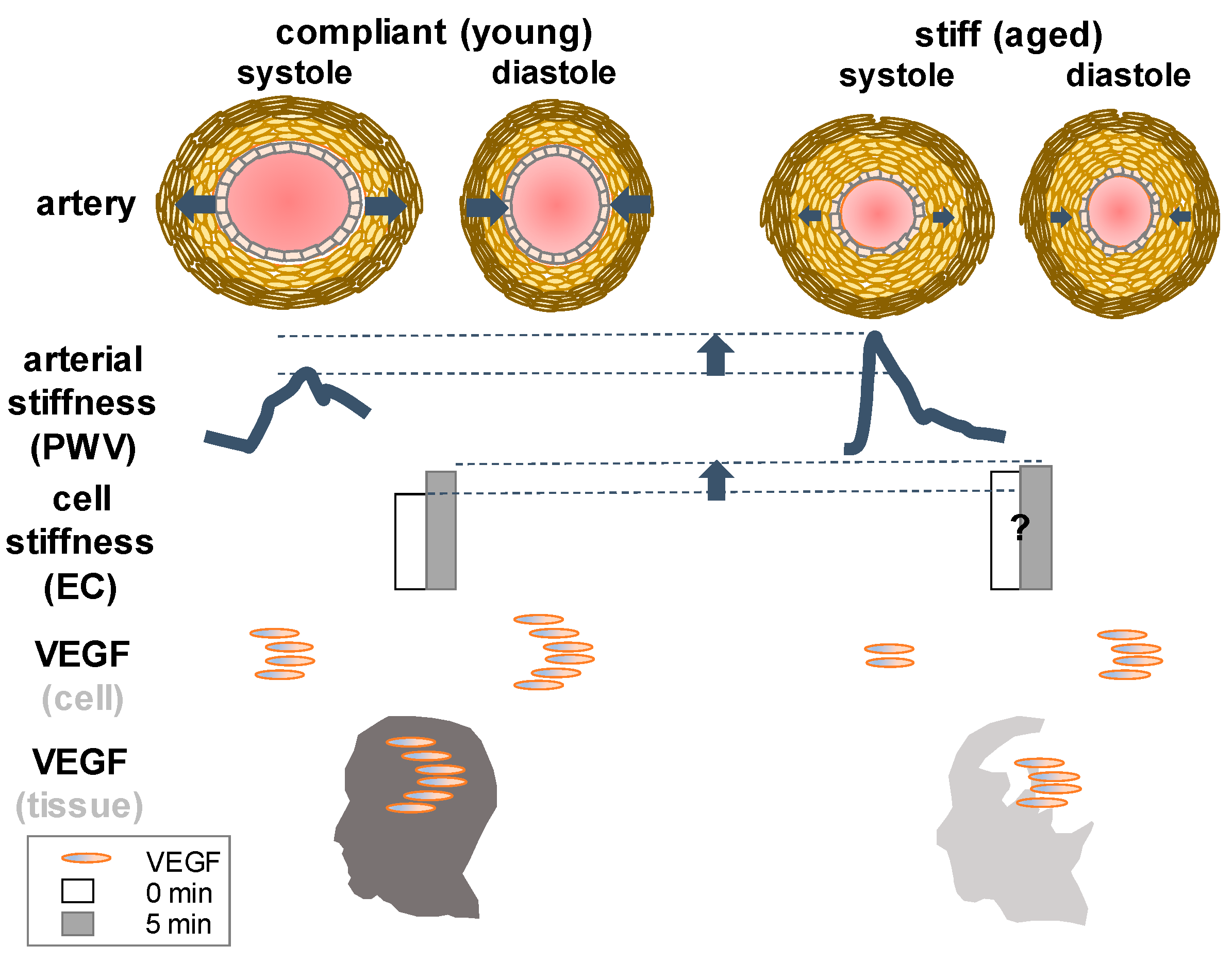{kind=link}
| Disease | VEGF Level (Where) | Concurrent Cerebral Events | Inhibit VEGF? | Promote VEGF? | Reference |
|---|---|---|---|---|---|
| Ocular disease | ↑ VEGF (ocular capillaries) | leaky microvasculature | yes | [18,19] m | |
| Stroke (early phase) | ↓ VEGF; ↑ VEGF (cerebral cortex) | leaky vasculature | yes | [17] r | |
| Stroke (collateral forming phase) | ↓ VEGF (cerebral cortex) | yes | [17] r | ||
| ↑ VEGF (cerebral cortex) | reparative angiogenesis | yes | [20] m | ||
| Hydrocephalus (young) | ↑ VEGF (CSF) | ventriculomegaly and hemorrhage | yes | [12] m, [16] h, [8] rabb,h,* | |
| Hydrocephalus (aged brains) | ↑ VEGF (CSF) | a slight increase in intracranial pressure (NPH) | yes | [9,21] h | |
| ALS | ↓ VEGF (cerebral cortex) | insufficient neuroprotection; motor neuron degeneration; BBB dysfunction | yes | [22,23] h | |
| AD | ↑ VEGF (cerebral cortex) | cerebral hypoperfusion; neural & BBB dysfunction and loss | yes | [24,25,26] m, [27] r | |
| PD | ↑ VEGF (CSF) | leaky vasculature; white matter lesion | yes | [28] h, [29] m |
| Disease | Ligand Level (Where) | Concurrent Cerebral Events | Inhibit ErbB? | Promote ErbB? | Reference |
|---|---|---|---|---|---|
| Stroke (ischemic) | ↑ neuregulin (cerebral cortex) | neuroprotection | Yes (ErbB4) | [71] r | |
| ↑ neuregulin (cerebral cortex) | reduced apoptosis | Yes (ErbB4) | [70] m | ||
| ↑ TGFα (striatal infunsion) | neuroprotection; neural migration | Yes (ErbB1) | [72] r | ||
| ↑ HB-EGF (icv infusion) | neuroprotection; reduced infarct size | Yes (ErbB1; ErbB4) | [73] r | ||
| ↑ amphiregulin (systemic blood) | hemorrhagic transformation | Yes (ErbB1) | [74] h | ||
| Stroke (hemorrhagic) | ↑ neuregulin (cerebral cortex) | neuroprotection after SAH | Yes (ErbB4) | [13] r |
| Condition | VEGF Level (Contributor) | Symptoms | Reference |
|---|---|---|---|
| Pulmonary artery stiffening | ↑ VEGF (elevated pulsatility) | Reduced compliance Pulmonary hypertension | [122] |
| Solid tumor progression | ↑ VEGF (matrix cross-linking) | Increased matrix stiffness Increased matrix metalloproteinase activity | [112] |
| Anti-cancer treatment | ↓ VEGF (VEGF kinase inhibition) | Increased hypertension | [110] |
| Condition | Vascular Factor | Interrelationship | Reference |
|---|---|---|---|
| Aging | vascular stiffness (pulse wave velocity, PWV) | Proportionate ↑ aging → ↑ PWV: yes ↑ PWV → ↑ aging: likely | [127] |
| Ocular disease | Intraocular pressure, IOP | Proportionate ↑ glaucoma → ↑ IOP: yes ↑ IOP → ↑ glaucoma: likely | [128] |
| Stroke (early phase) | blood pressure (hypertension) | Proportionate ↑ blood pressure → ↑ stroke: yes ↑ stroke → ↑ blood pressure: likely | [129] |
| Stroke (collateral forming phase) | blood pressure | Varied | [2] |
| Hydrocephalus (young) | cerebral pulsatility (↑ intracranial pressure) | Proportionate ↑ hydrocephalus → ↑ pulsatility: likely ↑ pulsatility → ↑ hydrocephalus: likely | [119,120] |
| Hydrocephalus (aged brain) | intracranial pressure | No significant association (normal pressure hydrocephalus) | [130] |
| ALS | blood pressure (diurnal variation) | varied with chronic ischemia, lack of neuroprotection, and autonomic failure | [2,23,131,132] |
| AD | vascular stiffness (PWV) | Proportionate ↑ AD → ↑ PWV: yes ↑ PWV → ↑ AD: not always but likely | [133] |
| PD | vascular stiffness (PWV) | Inverse ↑ PD → ↓ PWV: most likely ↓ PWV → ↑ PD: not always but likely | [105,106] |
| Smoking | Inverse ↑ smoking → ↓ PD: yes ↓ smoking → ↑ PD: likely ↑ PD → ↓ smoking: yes (easier to quit) ↓ PD → ↑ smoking: not always but likely | [134,135,136,137,138] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, J.W.; Madsen, J.R. VEGF Signaling in Neurological Disorders. Int. J. Mol. Sci. 2018, 19, 275. https://doi.org/10.3390/ijms19010275
Shim JW, Madsen JR. VEGF Signaling in Neurological Disorders. International Journal of Molecular Sciences. 2018; 19(1):275. https://doi.org/10.3390/ijms19010275
Chicago/Turabian StyleShim, Joon W., and Joseph R. Madsen. 2018. "VEGF Signaling in Neurological Disorders" International Journal of Molecular Sciences 19, no. 1: 275. https://doi.org/10.3390/ijms19010275
APA StyleShim, J. W., & Madsen, J. R. (2018). VEGF Signaling in Neurological Disorders. International Journal of Molecular Sciences, 19(1), 275. https://doi.org/10.3390/ijms19010275