Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors

 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Result and Discussion
2.1. Antibacterial Activity
2.1.1. Determination of the Inhibition Zone Diameters
2.1.2. Determination of MIC and MBC Values
2.2. Virtual Screening
2.2.1. ADMET Profiling
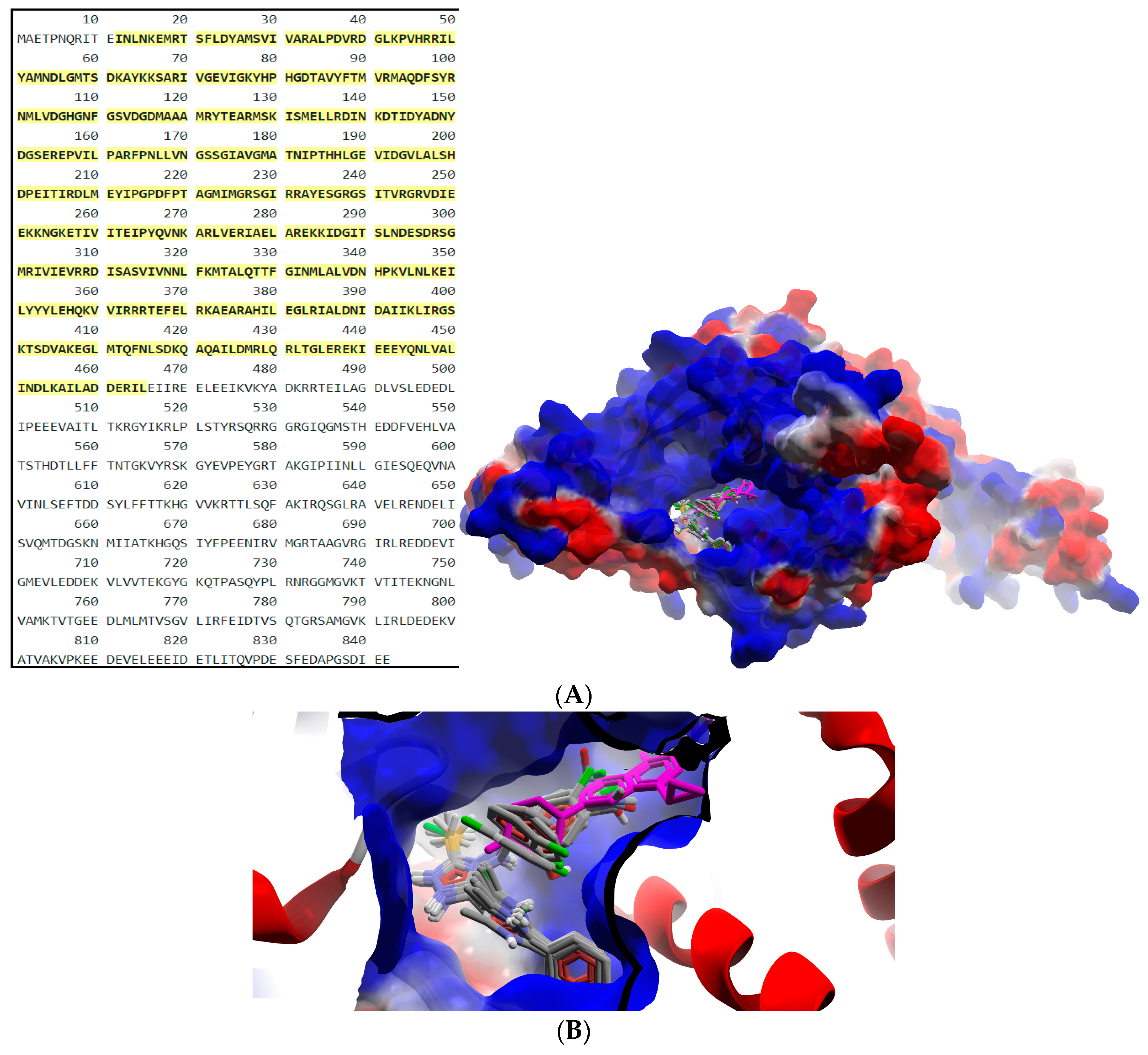
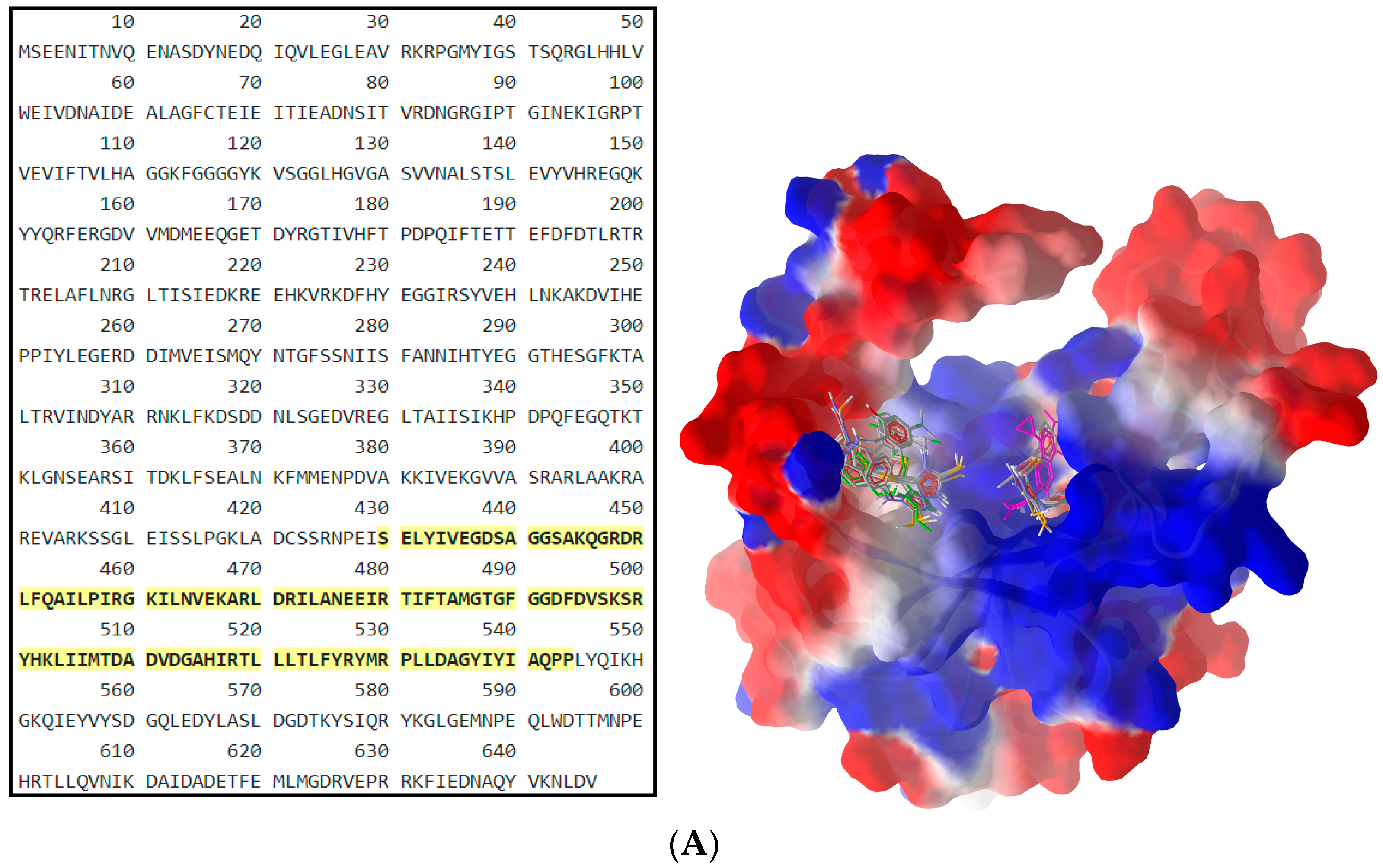
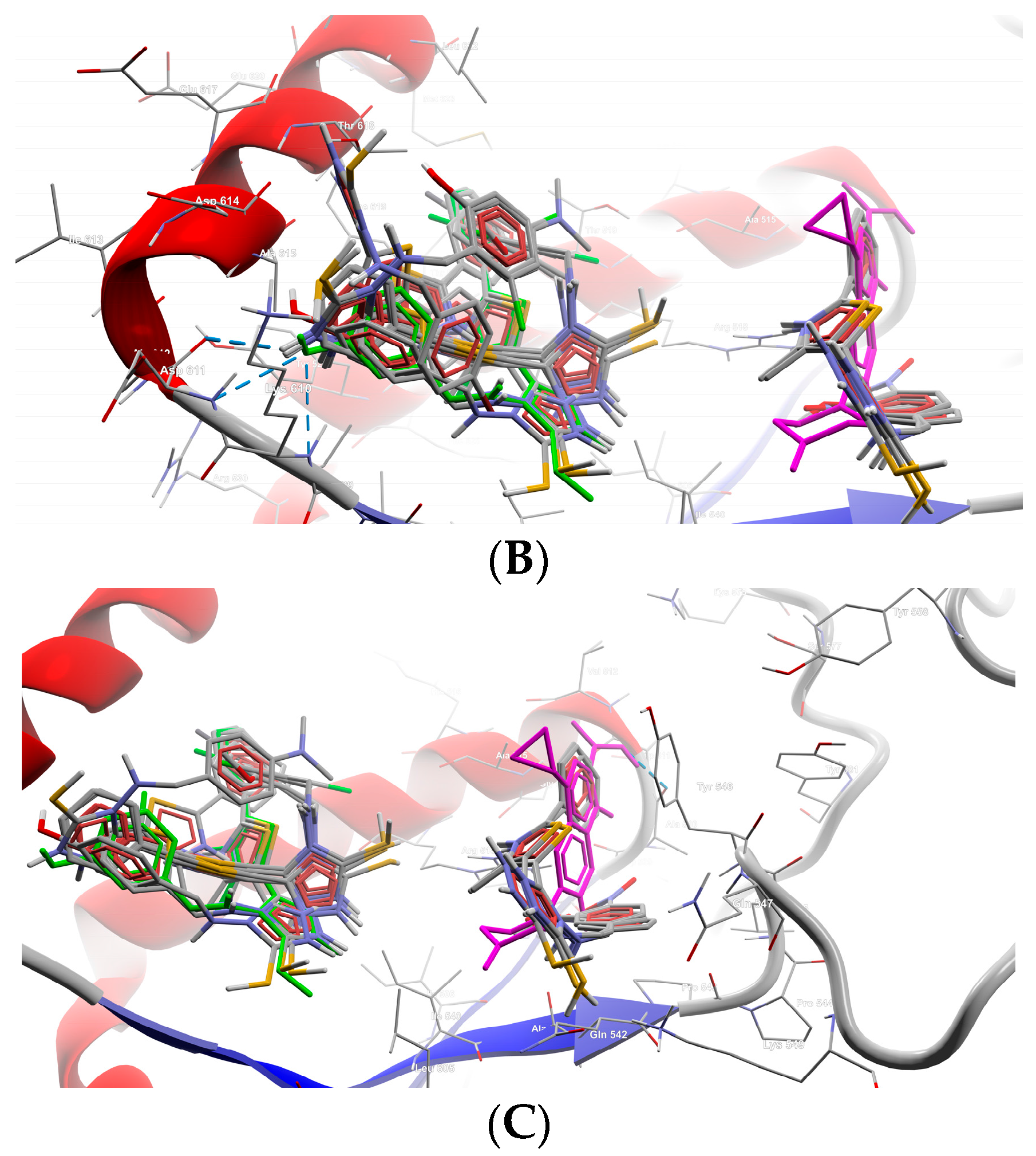
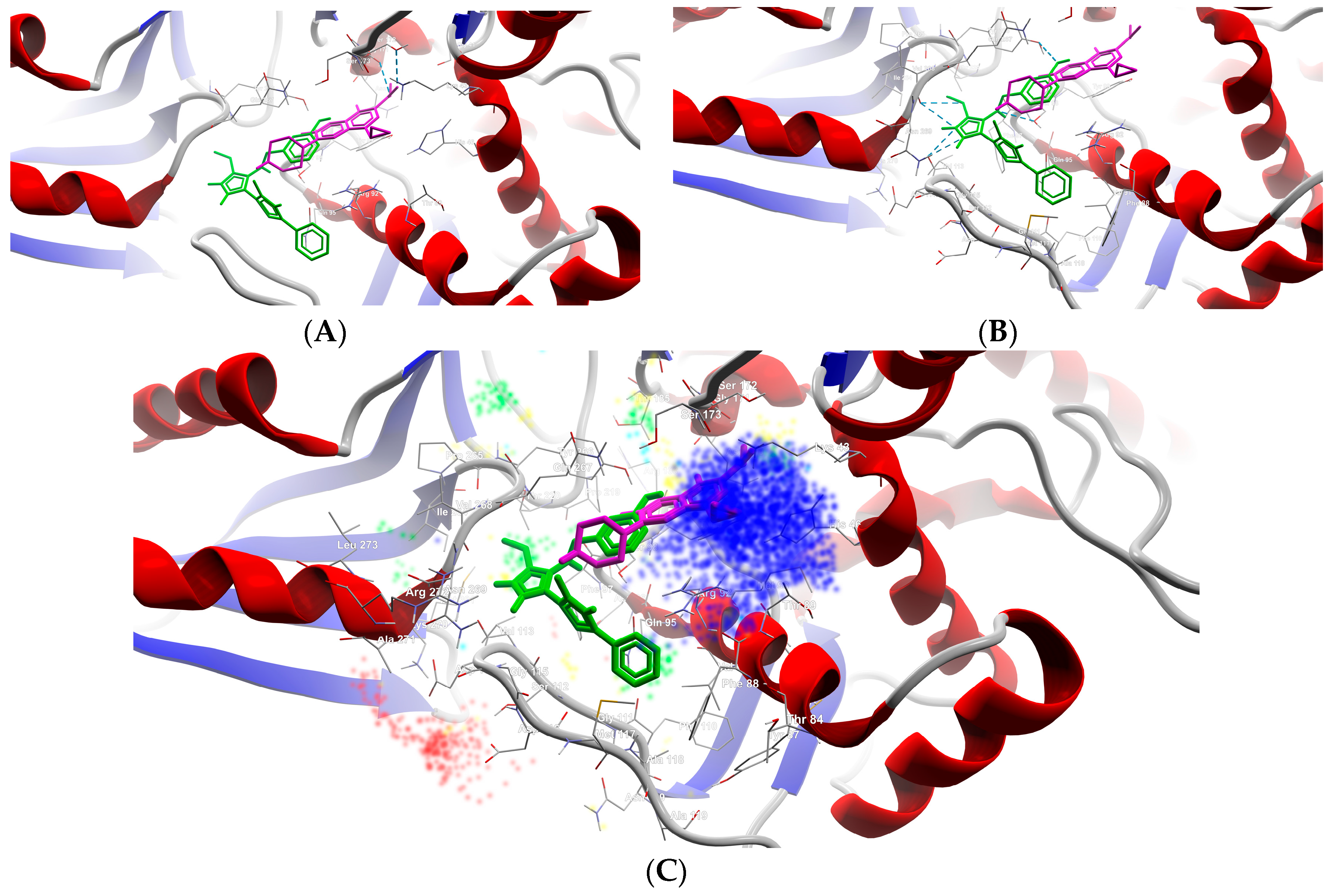
2.2.2. Molecular Docking
3. Materials and Methods
3.1. Antibacterial Activity Assay
3.1.1. Determination of the Inhibition Zone Diameters
3.1.2. Calculation of the Percentage Activity Index (% AI)
3.1.3. Determination of MIC and MBC Values
3.2. Virtual Screening
3.2.1. ADMET Predictions
3.2.2. Molecular Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tang, S.S.; Apisarnthanarak, A.; Hsu, L.Y. Mechanisms of β-lactam antimicrobial resistance and epidemiology of major community- and healthcare-associated multidrug-resistant bacteria. Adv. Drug Deliv. Rev. 2014, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Dodds, D.R. Antibiotic resistance: A current epilogue. Biochem. Pharmacol. 2017, 134, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Johani, K.; Abualsaud, D.; Costa, D.M.; Hu, H.; Whiteley, G.; Deva, A.; Vickery, K. Characterization of microbial community composition, antimicrobial resistance and biofilm on intensive care surfaces. J. Infect. Public Health. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zawadzki, P.; Stracy, M.; Ginda, K.; Zawadzka, K.; Lesterlin, C.; Kapanidis, A.N.; Sherratt, D.J. The Localization and Action of Topoisomerase IV in Escherichia coli Chromosome Segregation Is Coordinated by the SMC Complex, MukBEF. Cell Rep. 2015, 13, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Schoeffler, A.J.; Thomsen, N.D.; Berger, J.M. The Structural Basis for Substrate Specificity in DNA Topoisomerase IV. J. Mol. Biol. 2005, 351, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Watt, P.M.; Hickson, I.D. Structure and function of type II DNA topoisomerases. Biochem. J. 1994, 303, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Mun Huang, W. Bacterial diversity based on type II DNA topoisomerase genes. Annu. Rev. Genet. 1996, 30, 79–107. [Google Scholar] [CrossRef] [PubMed]
- Morais Cabral, J.H.; Jackson, A.P.; Smith, C.V.; Shikotra, N.; Maxwell, A.; Liddington, R.C. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature 1997, 388, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Fluit, A.C.; Schmitz, F.J. Fluoroquinolones: Structure and target sites. Curr. Drug Targets 2003, 4, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Nakada, N.; Shimada, H.; Hirata, T.; Aoki, Y.; Kamiyama, T.; Watanabe, J.; Arisawa, M. Biological characterization of cyclothialidine, a new DNA gyrase inhibitor. Antimicrob. Agents Chemother. 1993, 37, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; O'Dea, M.H.; Itoh, T.; Tomizawa, J. Novobiocin and coumeromycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl. Acad. Sci. USA 1976, 73, 4474–4478. [Google Scholar] [CrossRef] [PubMed]
- Pinar, A.; Yurdakul, P.; Yildiz, I.; Temiz-Arpaci, O.; Acan, N.L.; Aki-Sener, E.; Yalcin, I. Some fused heterocyclic compounds as eukaryotic topoisomerase II inhibitors. Biochem. Biophys. Res. Commun. 2004, 317, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Madhavapeddi, P.; Tucker, J.A.; Murugan, K.; Patil, V.; Basavarajappa, H.; Raichurkar, A.V.; Humnabadkar, V.; Hussein, S.; Sharma, S.; et al. Aminopyrazinamides: Novel and Specific GyrB Inhibitors that Kill Replicating and Nonreplicating Mycobacterium tuberculosis. ACS Chem. Biol. 2013, 8, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Jeankumar, V.U.; Renuka, J.; Santosh, P.; Soni, V.; Sridevi, J.P.; Suryadevara, P.; Yogeeswari, P.; Sriram, D. Thiazole–aminopiperidine hybrid analogues: Design and synthesis of novel Mycobacterium tuberculosis GyrB inhibitors. Eur. J. Med. Chem. 2013, 70, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Tomašič, T.; Katsamakas, S.; Hodnik, Ž.; Ilaš, J.; Brvar, M.; Solmajer, T.; Montalvão, S.; Tammela, P.; Banjanac, M.; Ergović, G.; et al. Discovery of 4,5,6,7-Tetrahydrobenzo[1,2-d]thiazoles as Novel DNA Gyrase Inhibitors Targeting the ATP-Binding Site. J. Med. Chem. 2015, 58, 5501–5521. [Google Scholar] [CrossRef] [PubMed]
- Brvar, M.; Perdih, A.; Oblak, M.; Mašič, L.P.; Solmajer, T. In silico discovery of 2-amino-4-(2,4-dihydroxyphenyl)thiazoles as novel inhibitors of DNA gyrase B. Bioorg. Med. Chem. Lett. 2010, 20, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Plech, T.; Kaproń, B.; Paneth, A.; Kosikowska, U.; Malm, A.; Strzelczyk, A.; Stączek, P.; Świątek, L.; Rajtar, B.; Polz-Dacewicz, M. Determination of the Primary Molecular Target of 1,2,4-Triazole-Ciprofloxacin Hybrids. Molecules 2015, 20, 6254–6272. [Google Scholar] [CrossRef] [PubMed]
- East, S.P.; White, C.B.; Barker, O.; Barker, S.; Bennett, J.; Brown, D.; Boyd, E.A.; Brennan, C.; Chowdhury, C.; Collins, I.; et al. DNA gyrase (GyrB)/topoisomerase IV (ParE) inhibitors: Synthesis and antibacterial activity. Bioorg. Med. Chem. Lett. 2009, 19, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.P.; Kumar, V.; Gupta, G.K. Synthesis, characterization, and antibacterial activity of a novel heterocyclic Schiff’s base and its metal complexes of first transition series. Med. Chem. Res. 2014, 23, 690–698. [Google Scholar] [CrossRef]
- Khan, K.M.; Ambreen, N.; Karim, A.; Saied, S.; Amyn, A.; Ahmed, A.; Perveen, S. Schiff Bases of Thiazole as Antibacterial and Antifungal Agents. J. Pharm. Res. 2012, 5, 651–656. [Google Scholar]
- Kajal, A.; Bala, S.; Kamboj, S.; Sharma, N.; Shaini, V. Schiff bases: A versatile pharmacophore. J. Catal. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Hameed, A.; al-Rashida, M.; Uroos, M.; Ali, S.A.; Khan, K.M. Schiff bases in medicinal chemistry: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.-F.; Addla, D.; Zhou, C.-H. Novel 3-Aminothiazolquinolones: Design, Synthesis, Bioactive Evaluation, SARs and Preliminary Antibacterial Mechanism. J. Med. Chem. 2016, 59, 4488–4510. [Google Scholar] [CrossRef] [PubMed]
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-based discovery of substituted 4,5′-bithiazoles as novel DNA gyrase inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Damu, G.L.V.; Lv, J.S.; Geng, R.X.; Yang, D.C.; Zhou, C.H. Design, synthesis and evaluation of clinafloxacin triazole hybrids as a new type of antibacterial and antifungal agents. Bioorg. Med. Chem. Lett. 2012, 22, 5363–5366. [Google Scholar] [CrossRef] [PubMed]
- Nastasă, C.; Tiperciuc, B.; Duma, M.; Benedec, D.; Oniga, O. New Hydrazones Bearing Thiazole Scaffold: Synthesis, Characterization, Antimicrobial, and Antioxidant Investigation. Molecules 2015, 20, 17325–17338. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Enache, A.; Vodnar, D.C.; Nastasă, C.; Benedec, D.; Ionuț, I.; Login, C.; Marc, G.; Oniga, O.; Tiperciuc, B. New Thiazolyl-triazole Schiff Bases: Synthesis and Evaluation of the Anti-Candida Potential. Molecules 2016, 21, 1595. [Google Scholar] [CrossRef] [PubMed]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [PubMed]
- Hafidh, R.R.; Abdulamir, A.S.; Vern, L.S.; Abu Bakar, F.; Abas, F.; Jahanshiri, F.; Sekawi, Z. Inhibition of Growth of Highly Resistant Bacterial and Fungal Pathogens by a Natural Product. Open Microbiol. J. 2011, 5, 96–106. [Google Scholar] [CrossRef] [PubMed]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D. Computational Methods Applied to Rational Drug Design. Open Med. Chem. J. 2016, 10, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Beesu, M.; Caruso, G.; Salyer, A.C.D.; Khetani, K.K.; Sil, D.; Weerasinghe, M.; Tanji, H.; Ohto, U.; Shimizu, T.; David, S.A. Structure-Based Design of Human TLR8-Specific Agonists with Augmented Potency and Adjuvanticity. J. Med. Chem. 2015, 58, 7833–7849. [Google Scholar] [CrossRef] [PubMed]
- Beesu, M.; Caruso, G.; Salyer, A.C.D.; Shukla, N.M.; Khetani, K.K.; Smith, L.J.; Fox, L.M.; Tanji, H.; Ohto, U.; Shimizu, T.; et al. Identification of a Human Toll-Like Receptor (TLR) 8-Specific Agonist and a Functional Pan-TLR Inhibitor in 2-Aminoimidazoles. J. Med. Chem. 2016, 59, 3311–3330. [Google Scholar] [CrossRef] [PubMed]
- Varnek, A.; Tropsha, A. Chemoinformatics Approaches to Virtual Screening; Royal Society of Chemistry: Cambridge, UK, 2008; pp. 1–338. ISBN 978-0-85404-144-2. [Google Scholar]
- Trosset, J.-Y.; Carbonell, P. Synthetic biology for pharmaceutical drug discovery. Drug Des. Dev. Ther. 2015, 9, 6285–6302. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Matter, H. Integrating virtual screening in lead discovery. Curr. Opin. Chem. Biol. 2004, 8, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, H.; Najafi, A.; Eslami, H.; Negahdari, B.; Moghaddam, M.M. Identification of novel bacterial DNA gyrase inhibitors: An in silico study. Res. Pharm. Sci. 2016, 11, 250–258. [Google Scholar] [PubMed]
- ChemAxon. Available online: https://chemaxon.com.
- Potashman, M.H.; Duggan, M.E. Covalent Modifiers: An Orthogonal Approach to Drug Design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W.; Natsch, A. High Throughput Kinetic Profiling Approach for Covalent Binding to Peptides: Application to Skin Sensitization Potency of Michael Acceptor Electrophiles. Chem. Res. Toxicol. 2009, 22, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P. Generation of a Set of Simple, Interpretable ADMET Rules of Thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Bruns, R.F.; Ian, A.W. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical Drug Properties Associated with In Vivo Toxicological Outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Drugbank. Available online: https://www.drugbank.ca/drugs/DB00537.
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- National Committee for Clinical Laboratory Standards/Clinical and Laboratory Standards Institute (NCCLS/CLSI). Methods for Dilution Antibacterial Susceptibility Test for Bacteria That Grow Aerobically: Approved Standard, 8th ed.; CLSI Document M07-A8; Clinical and Laboratory standards Institute: Wayne, PA, USA, 2009. [Google Scholar]
- Ahmed, M.; Qadir, M.A.; Shafiq, M.I.; Muddassar, M.; Samra, Z.Q.; Hameed, A. Synthesis, characterization, biological activities and molecular modeling of Schiff bases of benzene sulfonamides bearing curcumin scaffold. Arab. J. Chem. 2016. [Google Scholar] [CrossRef]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A Web Server for Compound Property Calculation and Chemical Library Design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational Analysis of Calculated Physicochemical and ADMET Properties of Protein-Protein Interaction Inhibitors. Sci. Rep. 2017, 7, 46277. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, S.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2004, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Pihan, E.; Colliandre, L.; Guichou, J.-F.; Douguet, D. E-Drug3D: 3D Structure Collections Dedicated to Drug Repurposing and Fragment-Based Drug Design. Bioinformatics 2012, 28, 1540–1541. [Google Scholar] [CrossRef] [PubMed]
- Reynès, C.; Host, H.; Camproux, A.-C.; Laconde, G.; Leroux, F.; Mazars, A.; Deprez, B.; Fahraeus, R.; Villoutreix, B.O.; Sperandio, O. Designing Focused Chemical Libraries Enriched in Protein-Protein Interaction Inhibitors Using Machine-Learning Methods. PLoS Comput. Biol. 2010, 6, e1000695. [Google Scholar]
- Horvath, D.; Lisurek, M.; Rupp, B.; Kühne, R.; Specker, E.; Von Kries, J.; Rognan, D.; Andersson, C.D.; Almgvist, C.; Elofsson, M.; et al. Design of a General-Purpose European Compound Screening Library for EU-OPENSCREEN. ChemMedChem 2014, 9, 2309–2326. [Google Scholar] [CrossRef] [PubMed]
- Cumming, J.G.; Davis, A.M.; Muresan, S.; Haeberlein, M.; Chen, H. Chemical Predictive Modelling to Improve Compound Quality. Nat. Rev. Drug Discov. 2013, 12, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Mok, N.Y.; Maxe, S.; Brenk, R. Locating Sweet Spots for Screening Hits and Evaluating Pan-Assay Interference Filters from the Performance Analysis of Two Lead-like Libraries. J. Chem. Inf. Model. 2013, 53, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Galons, H.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs2: Free ADME/tox Filtering Tool to Assist Drug Discovery and Chemical Biology Projects. BMC Bioinform. 2008, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Przybylak, K.R.; Alzahrani, A.R.; Cronin, M.T.D. How Does the Quality of Phospholipidosis Data Influence the Predictivity of Structural Alerts? J. Chem. Inf. Model. 2014, 54, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Uniprot. Available online: http://www.uniprot.org.
- RCSB Protein Data Bank. Available online: http://www.rcsb.org.
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- SWISS-MODEL. Available online: https://swissmodel.expasy.org.
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminform. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cp. | Gram-Positive Bacteria | Gram-Negative Bacteria | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Staphylococcus aureus ATCC 25923 | Listeria monocytogenes ATCC 35152 | Escherichia coli ATCC 25922 | Salmonella typhimurium ATCC 13311 | P. aeruginosa ATCC 27853 | ||||||
| Diameter (mm) | %AI | Diameter (mm) | %AI | Diameter (mm) | %AI | Diameter (mm) | %AI | Diameter (mm) | %AI | |
| B1 | 14 | 50 | 18 | 100 | 14 | 51.8 | 16 | 72.7 | 19 | 73 |
| B2 | 14 | 50 | 18 | 100 | 14 | 51.8 | 18 | 81.8 | 19 | 73 |
| B3 | 14 | 50 | 16 | 88.8 | 14 | 51.8 | 18 | 81.8 | 16 | 61.5 |
| B4 | 14 | 50 | 14 | 77.7 | 14 | 51.8 | 18 | 81.8 | 18 | 69.2 |
| B5 | 14 | 50 | 14 | 77.7 | 14 | 51.8 | 18 | 81.8 | 21 | 80.7 |
| B6 | 14 | 50 | 14 | 77.7 | 14 | 51.8 | 16 | 72.7 | 21 | 80.7 |
| B7 | 16 | 57.1 | 12 | 66.6 | 14 | 51.8 | 16 | 72.7 | 18 | 69.2 |
| B8 | 12 | 42.8 | 12 | 66.6 | 14 | 51.8 | 16 | 72.7 | 18 | 69.2 |
| B9 | 14 | 50 | 18 | 100 | 16 | 59.2 | 16 | 72.7 | 20 | 76.9 |
| B10 | 18 | 64.2 | 20 | 111.1 | 16 | 59.2 | 18 | 81.8 | 18 | 69.2 |
| B11 | 12 | 42.8 | 8 | 44.4 | 16 | 59.2 | 18 | 81.8 | 21 | 80.7 |
| B12 | 12 | 42.8 | 14 | 77.7 | 14 | 51.8 | 18 | 81.8 | 21 | 80.7 |
| B13 | 12 | 42.8 | 12 | 66.6 | 14 | 51.8 | 18 | 81.8 | 21 | 80.7 |
| B14 | 12 | 42.8 | 16 | 88.8 | 16 | 59.2 | 18 | 81.8 | 21 | 80.7 |
| B15 | 16 | 57.1 | 10 | 55.5 | 14 | 51.8 | 18 | 81.8 | 21 | 80.7 |
| CIP | 28 | 100 | 18 | 100 | 27 | 100 | 22 | 100 | 26 | 100 |
| Cp. | S. aureus ATCC 49444 | L. monocytogenes ATCC 19115 | P. aeruginosa ATCC 27853 | S. typhimurium ATCC 14028 | ||||
|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| B1 | 31.25 | 31.25 | 1.95 | 3.9 | 7.81 | 15.62 | 62.5 | 125 |
| B2 | 31.25 | 31.25 | 3.9 | 7.8 | 7.81 | 15.62 | 62.5 | 62.5 |
| B3 | 62.5 | 62.5 | 3.9 | 7.8 | 15.62 | 31.25 | 62.5 | 62.5 |
| B4 | 31.25 | 62.5 | 3.9 | 7.8 | 7.81 | 15.62 | 62.5 | 62.5 |
| B5 | 31.25 | 31.25 | 1.95 | 3.9 | 1.95 | 3.9 | 62.5 | 62.5 |
| B6 | 31.25 | 62.5 | 1.95 | 3.9 | 1.95 | 3.9 | 62.5 | 62.5 |
| B7 | 31.25 | 31.25 | 1.95 | 3.9 | 7.81 | 15.62 | 62.5 | 125 |
| B8 | 62.5 | 62.5 | 3.9 | 3.9 | 7.81 | 15.62 | 62.5 | 125 |
| B9 | 31.25 | 31.25 | 1.95 | 3.9 | 3.9 | 7.8 | 62.5 | 125 |
| B10 | 31.25 | 31.25 | 3.9 | 7.8 | 7.81 | 15.62 | 62.5 | 62.5 |
| B11 | 62.5 | 62.5 | 1.95 | 3.9 | 1.95 | 3.9 | 62.5 | 62.5 |
| B12 | 31.25 | 62.5 | 1.95 | 3.9 | 1.95 | 1.95 | 62.5 | 125 |
| B13 | 31.25 | 62.5 | 1.95 | 3.9 | 1.95 | 3.9 | 31.25 | 62.5 |
| B14 | 15.62 | 31.25 | 1.95 | 3.9 | 1.95 | 3.9 | 62.5 | 125 |
| B15 | 31.25 | 62.5 | 1.95 | 3.9 | 1.95 | 3.9 | 62.5 | 62.5 |
| Ciprofloxacin | 1.95 | 3.9 | 3.9 | 7.8 | 3.9 | 7.8 | 0.97 | 1.95 |
| Inoculum Control | +++ | +++ | +++ | +++ | ||||
| Broth control | No growth | No growth | No growth | No growth | ||||
| Cp. | MW (Da) | logP | tPSA (Å2) | PPIs | UMSs | CIs | PAINS Filters | PPDI | Med Chem | GSK 4/400 | Pfizer 3/75 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | |||||||||||
| B1 | 446.38 | 5.69 | 104.1 | Yes | thiol hal. | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B2 | 446.38 | 5.69 | 104.1 | Yes | thiol hal. | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B3 | 446.38 | 5.69 | 104.10 | Yes | thiol hal. | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B4 | 411.93 | 5.06 | 123.00 | Yes | thiol hal. | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B5 | 456.38 | 5.13 | 123.00 | Yes | hal. | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B6 | 395.48 | 4.53 | 123.00 | Yes | thiol hal. F | thiol | ND | ND | ND | NI | hydr. | good | warn. |
| B7 | 393.49 | 4.08 | 143.23 | Yes | thiol phenol | thiol | I479 I479b | ND | ND | NI | hydr. | good | warn. |
| B8 | 393.49 | 4.08 | 143.23 | Yes | thiol phenol | thiol | ND | ND | ND | NI | hydr. | good | warn. |
| B9 | 393.49 | 4.08 | 143.23 | Yes | thiol phenol | thiol | I215 | ND | ND | NI | hydr. | good | warn. |
| B10 | 422.48 | 4.26 | 149.92 | Yes | thiol nitro | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B11 | 422.48 | 4.26 | 149.92 | Yes | thiol nitro | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B12 | 407.51 | 4.41 | 132.23 | Yes | thiol | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B13 | 407.51 | 4.41 | 132.23 | Yes | thiol | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| B14 | 383.51 | 4.45 | 151.24 | Yes | thiol thp. | thiol | ND | ND | ND | NI | hydr. | good | warn. |
| B15 | 420.55 | 4.56 | 126.24 | Yes | thiol | thiol | ND | ND | ND | NI | hydr. | bad | warn. |
| CIP | 331.34 | 0.28 | 81.98 | Not | hal. F | ND | I215 | ND | ND | NI | ND | good | bad |
| Backbone of the Compounds B1–15 |  | ||
|---|---|---|---|
| Compound | gyrA BA (kcal/mol) | Atom ID of Ligand | Interacting AA Residue |
| B1 | −8.9 | N (2) | Ser112 |
| N (4) | Val113 | ||
| N (21) | Ser98 | ||
| B2 | −7.6 | N (2) | Ser112, Val113 |
| N (4) | Val113 | ||
| N (21) | Ser98 | ||
| S (32) | Val268 | ||
| B3 | −8.1 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| S (32) | Val268 | ||
| B4 | −8.5 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| B5 | −8.8 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| S (32) | Val268 | ||
| B6 | −8.8 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| B7 | −8.1 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| Phenolic O | Gln95, Ser98 | ||
| B8 | −8.7 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| Phenolic O | Tyr266 | ||
| S (32) | Val268 | ||
| B9 | −8.5 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| S (32) | Val268 | ||
| B10 | −9.1 | N (2) | Val113 |
| N (4) | Val113, Val 268 | ||
| N (21) | Ser98 | ||
| Nitro N | Tyr266 | ||
| Nitro O | Gln267 | ||
| B11 | −8.8 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| S (32) | Val268 | ||
| B12 | −8 | N (4) | Val113, Val268 |
| N (21) | Ser98 | ||
| Methoxy O | Ser98 | ||
| S (32) | Val268 | ||
| B13 | −8.8 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| Methoxy O | Tyr266 | ||
| B14 | −7.9 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| S (32) | Val268 | ||
| B15 | −8.8 | N (2) | Val113 |
| N (4) | Val113, Val268 | ||
| N (21) | Ser98 | ||
| CIP | −7.1 | O (24) | Ser172 |
| O (25) | Gly171 | ||
| Backbone of the compounds B1–15 |  | ||
|---|---|---|---|
| Compound | gyrB BA (kcal/mol) | Atom ID of Ligand | Interacting AA Residue |
| B1 | −6.3 | N (4) | Asp611 |
| S (32) | Asp614 | ||
| B2 | −6.7 | NA | NA |
| B3 | −6.6 | NA | NA |
| B4 | −7.3 | NA | NA |
| B5 | −6.3 | N (2) | Asp614, Thr618 |
| B6 | −7.2 | NA | NA |
| B7 | −6.3 | N (4) | Asp611 |
| Phenolic O (30) | Thr618 | ||
| Thiazole S (16) | Asp614 | ||
| B8 | −7.3 | Phenolic O (30) | Lys610, Asp611 |
| B9 | −7.2 | N (21) | Asn608 |
| Phenolic O (30) | Asp611, Ala615 | ||
| B10 | −6.8 | Nitro O (31,32) | Arg518 |
| B11 | −6.8 | Nitro N (30) | Arg518 |
| Nitro O (32) | Arg518 | ||
| S (33) | Gln542 | ||
| B12 | −6.4 | NA | NA |
| B13 | −6.6 | N (21) | Gln542 |
| Methoxy O (30) | Arg518 | ||
| B14 | −6.5 | NA | NA |
| B15 | −6.2 | N (4) | Asp611 |
| CIP | −6.5 | O (25) | Ala510 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nastasă, C.; Vodnar, D.C.; Ionuţ, I.; Stana, A.; Benedec, D.; Tamaian, R.; Oniga, O.; Tiperciuc, B. Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors. Int. J. Mol. Sci. 2018, 19, 222. https://doi.org/10.3390/ijms19010222
Nastasă C, Vodnar DC, Ionuţ I, Stana A, Benedec D, Tamaian R, Oniga O, Tiperciuc B. Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors. International Journal of Molecular Sciences. 2018; 19(1):222. https://doi.org/10.3390/ijms19010222
Chicago/Turabian StyleNastasă, Cristina, Dan C. Vodnar, Ioana Ionuţ, Anca Stana, Daniela Benedec, Radu Tamaian, Ovidiu Oniga, and Brînduşa Tiperciuc. 2018. "Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors" International Journal of Molecular Sciences 19, no. 1: 222. https://doi.org/10.3390/ijms19010222
APA StyleNastasă, C., Vodnar, D. C., Ionuţ, I., Stana, A., Benedec, D., Tamaian, R., Oniga, O., & Tiperciuc, B. (2018). Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors. International Journal of Molecular Sciences, 19(1), 222. https://doi.org/10.3390/ijms19010222