Investigation of New Morpholino Oligomers to Increase Survival Motor Neuron Protein Levels in Spinal Muscular Atrophy



Abstract
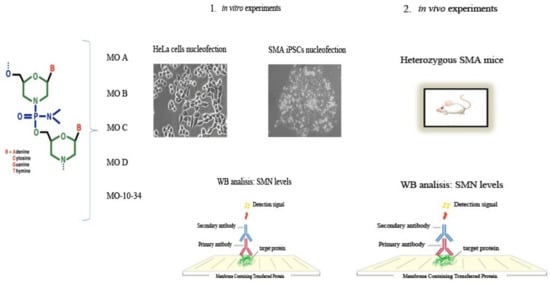
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
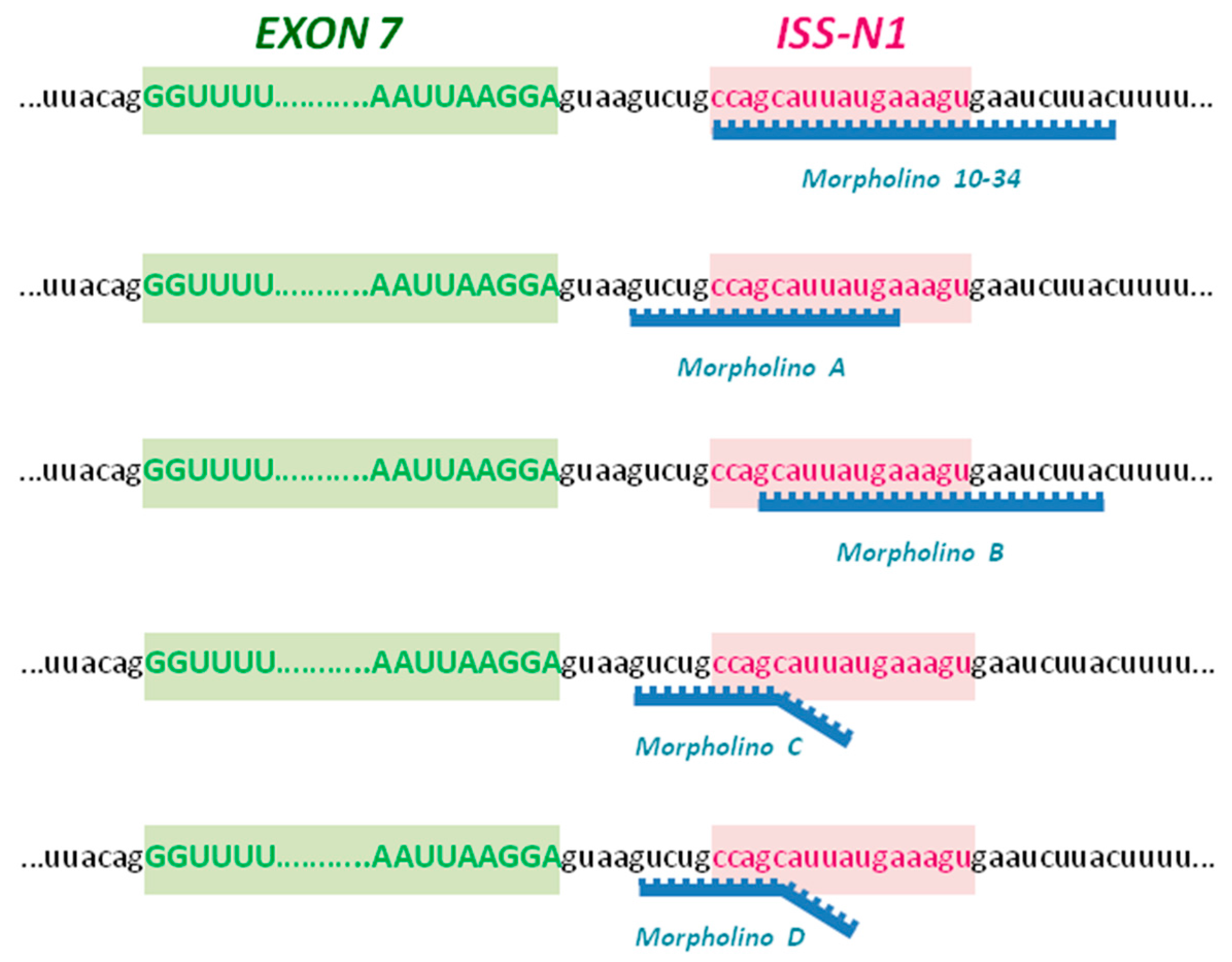
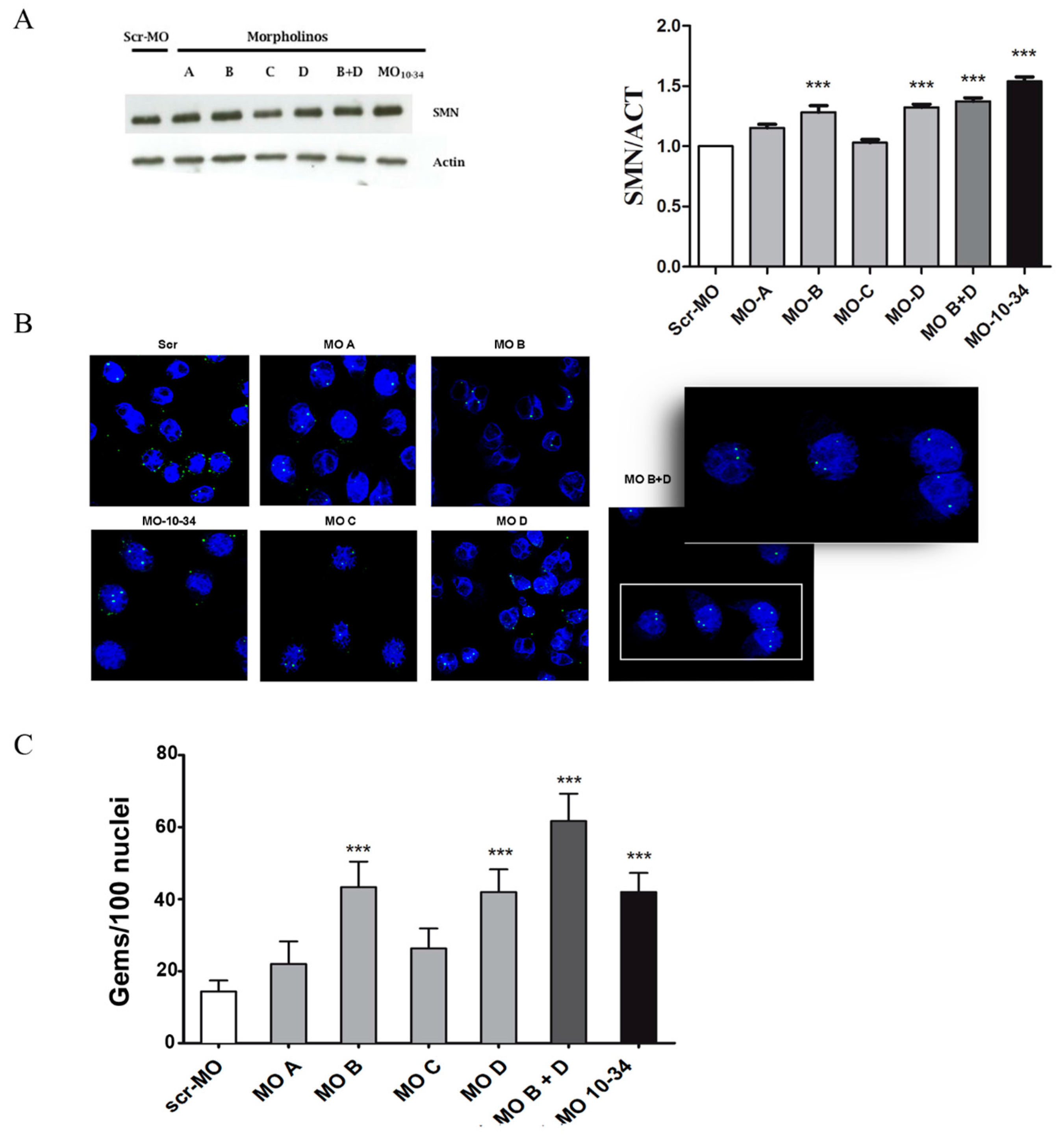
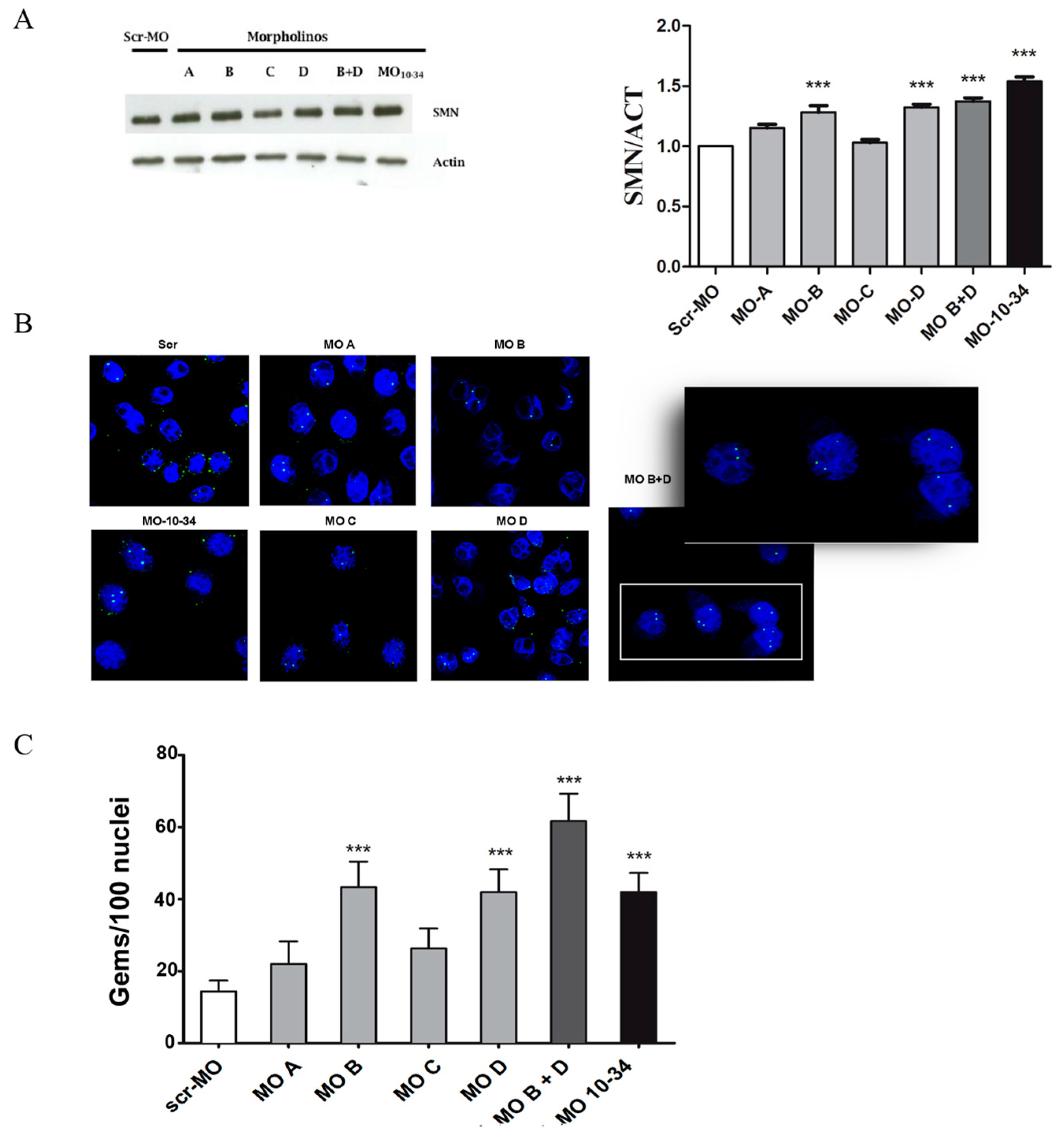
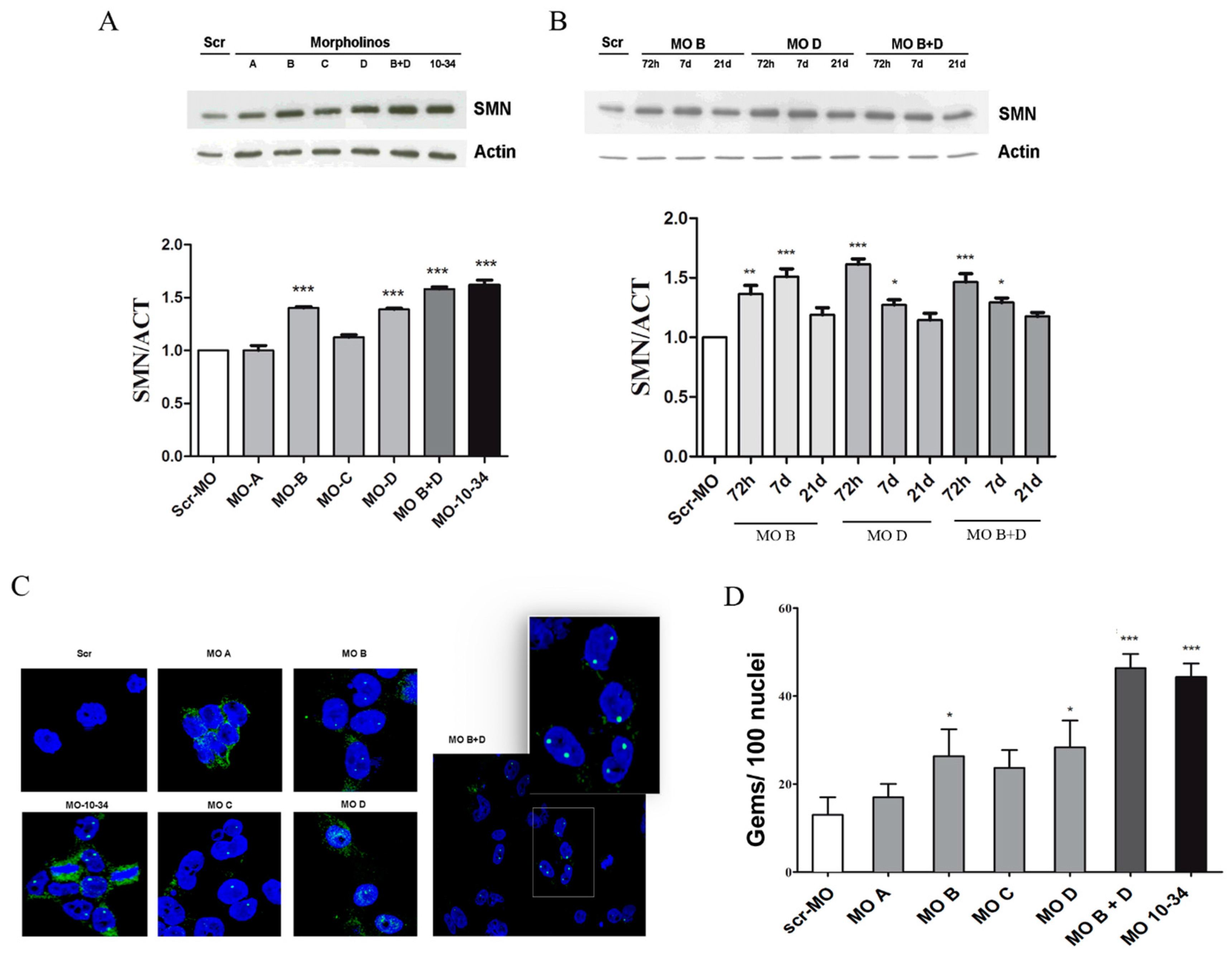
2.1. Upregulation of SMN in Wild-Type Cells
2.2. Upregulation of SMN in SMA iPSCs
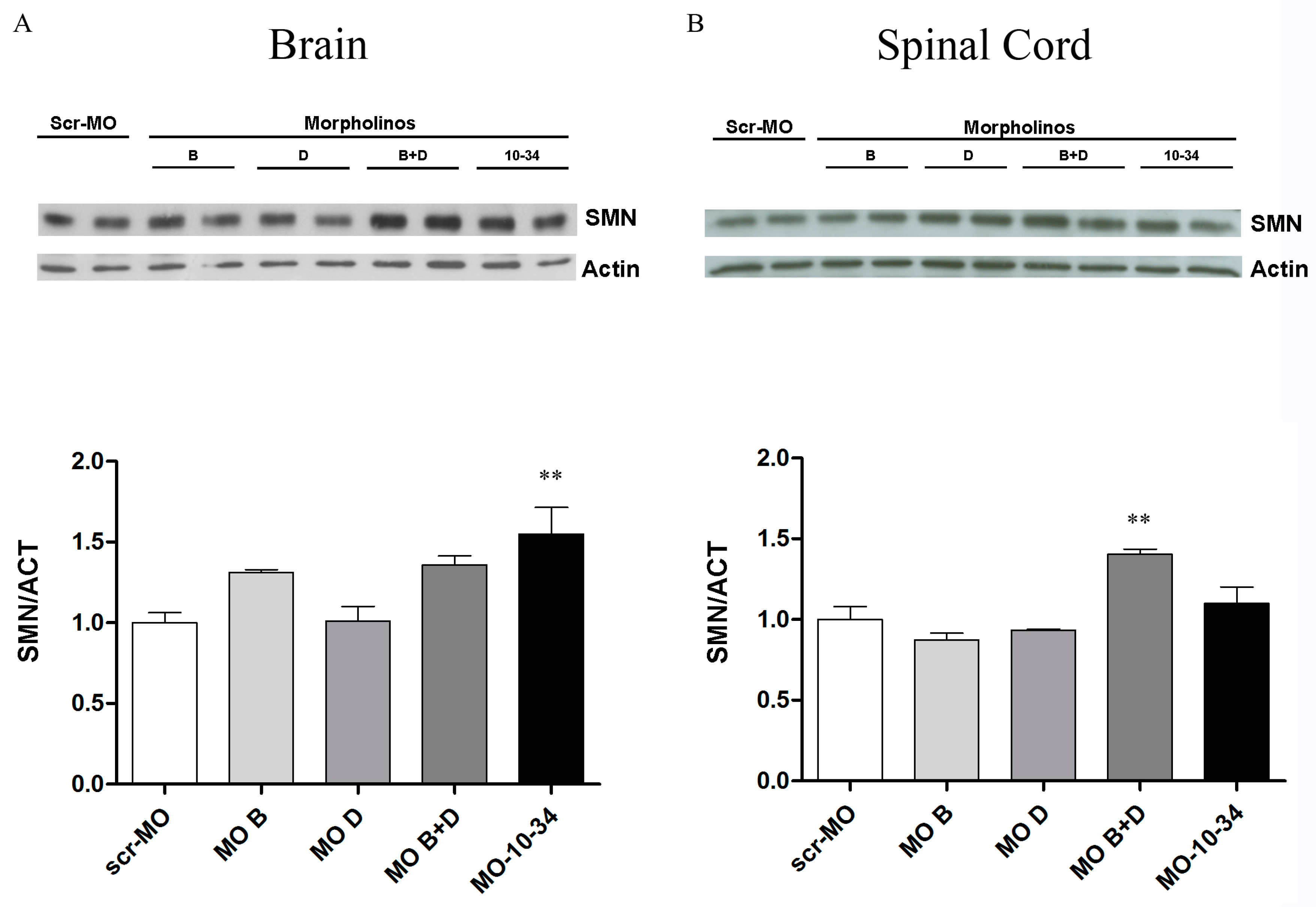
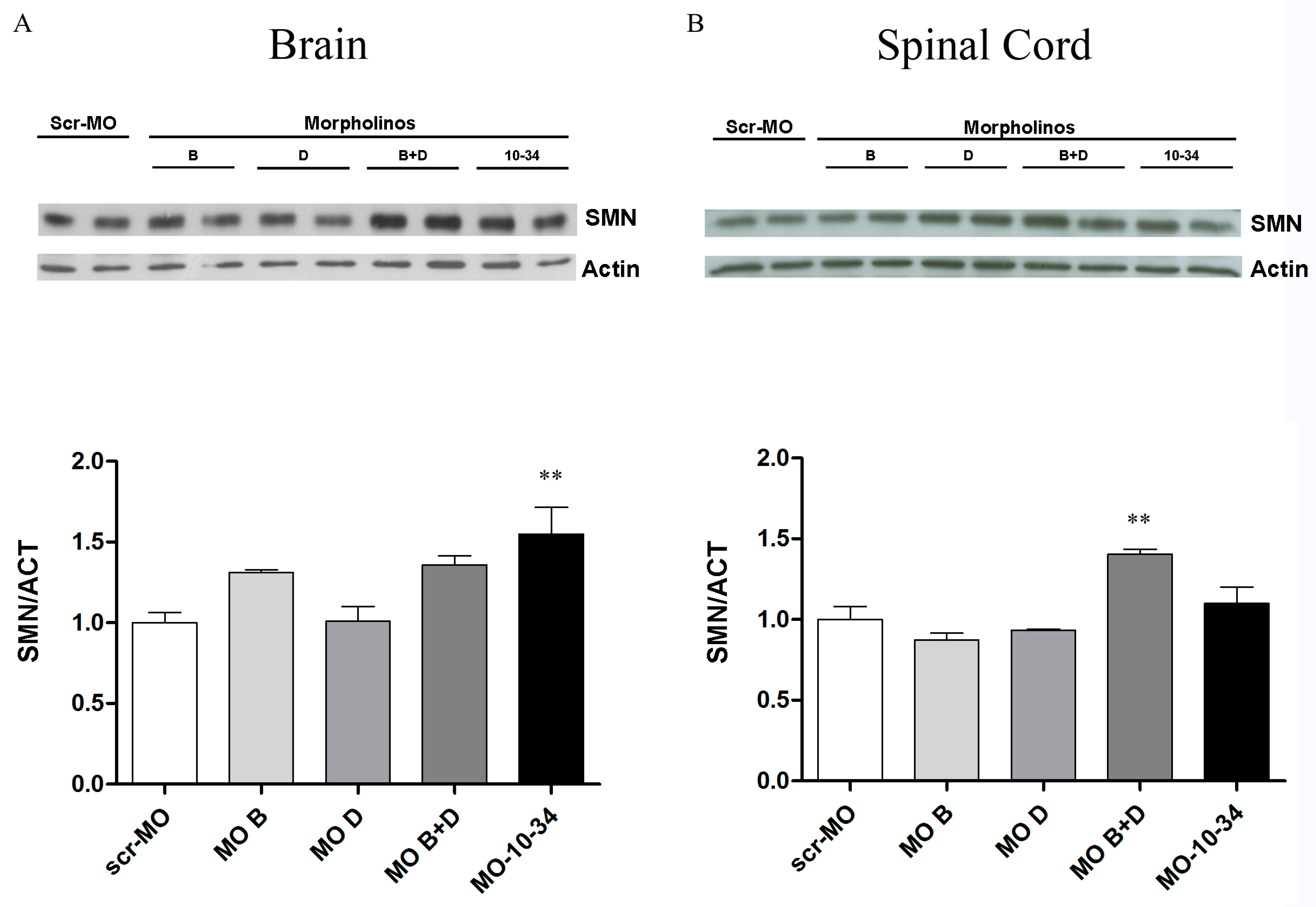
2.3. Effect of Mos on SMN Levels in the Central Nervous System of SMA Mice
3. Discussion
4. Materials and Methods
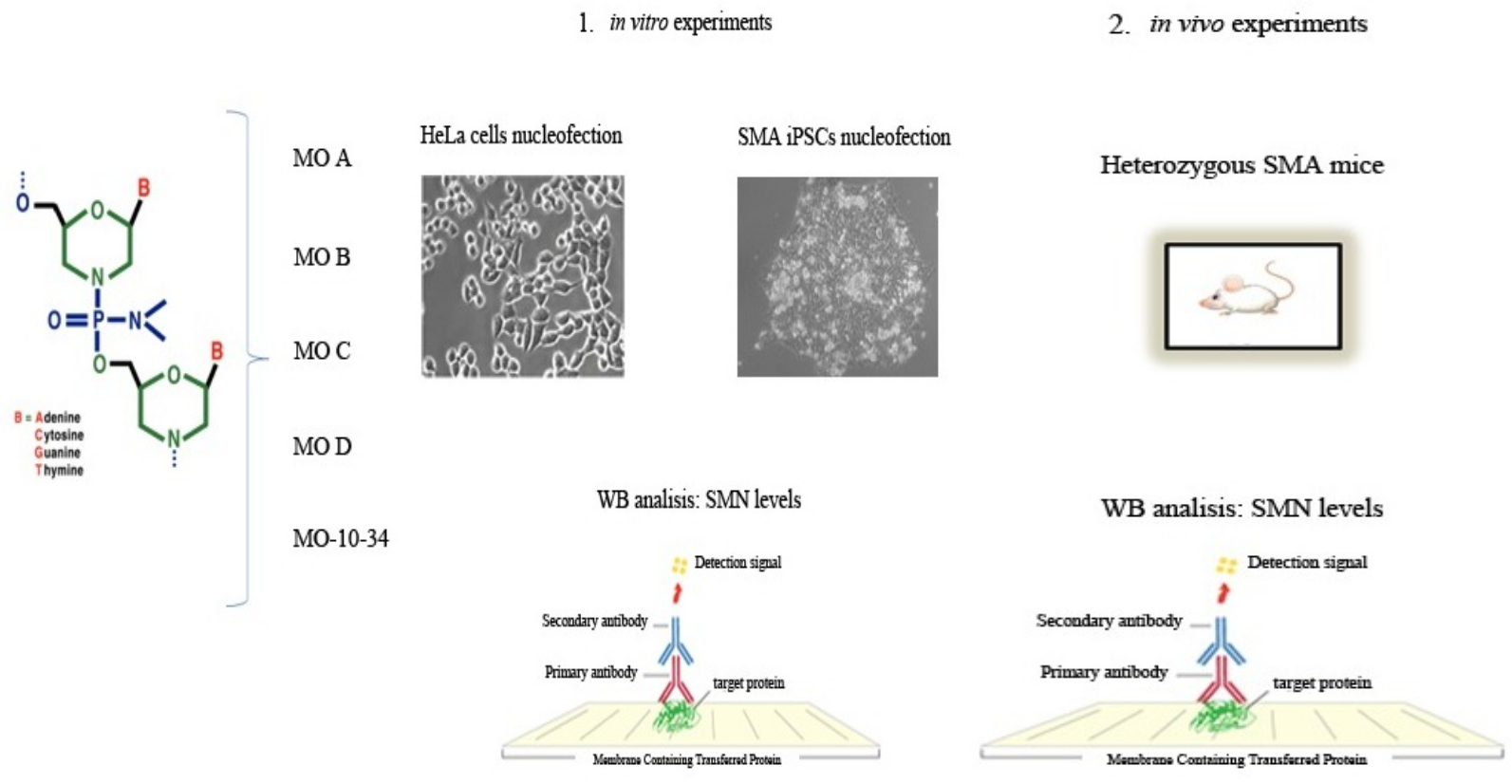
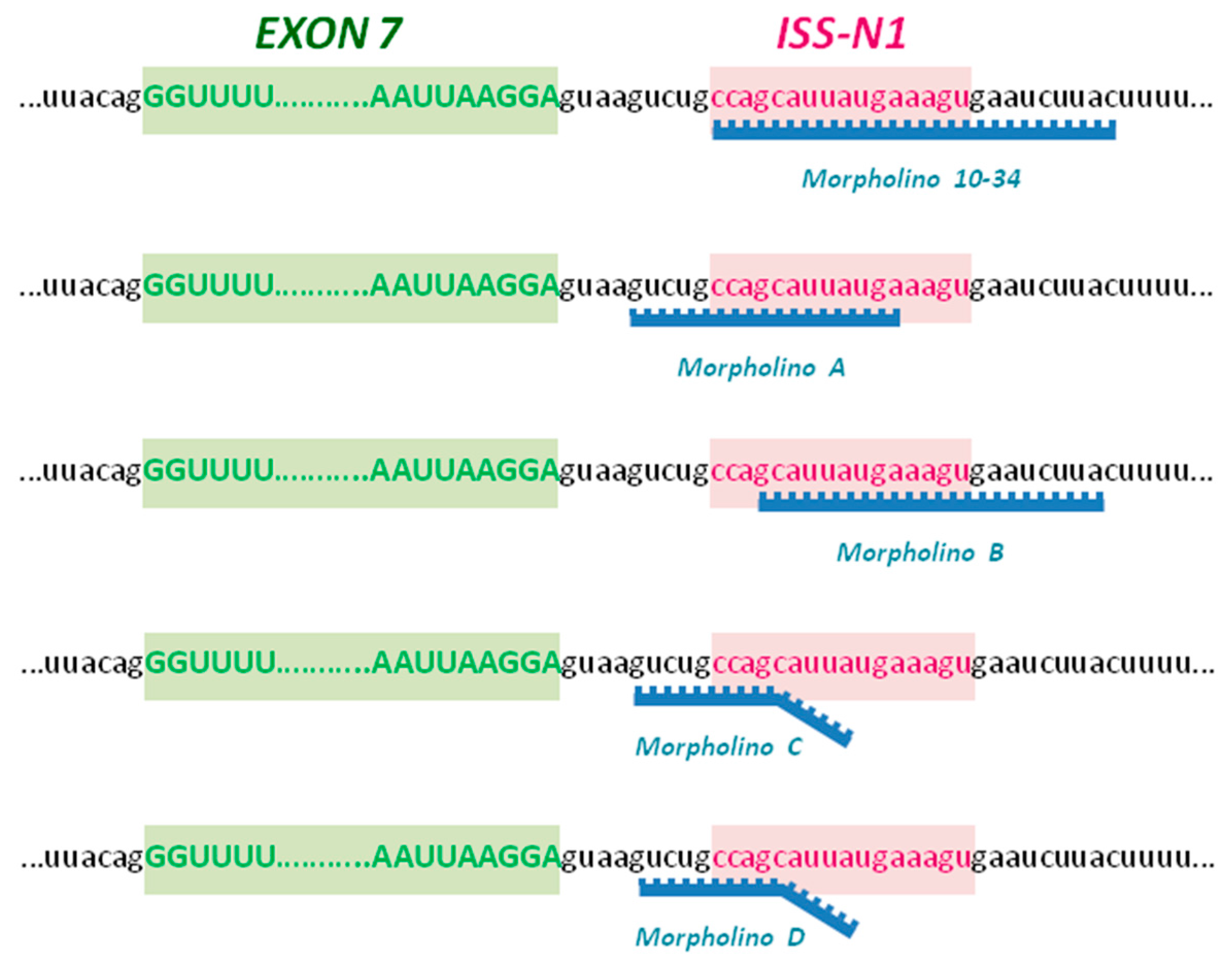
4.1. Morpholino Oligomers
- MO-10-34: GTAAGATTCACTTTCATAATGCTGG
- Scr-MO: GTAACATTGACTTTGATATTCCTGG
- MO A: TCATAATGCTGGCAGAC
- MO B: GTAAGATTCACTTTCATAATGC
- MO C: GATTCACTGTCAGAAGGCTGGCAGAC
- MO D: GATTCACTCTCACAACGCTGGCAGAC
4.2. Cell Cultures
4.3. MO Transfection
4.4. Immunocytochemistry of HeLa Cells and iPSCs
4.5. Western Blot
4.6. Animal Procedures
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| SMA | Spinal Muscular Atrophy |
| SMN | Survival Motor Neuron |
| ASO | Antisense oligonucleotide |
| MO | Morpholino |
| IPSCs | Induced pluripotent stem cells |
| MN | Motor neuron |
| SCR | Scrambled |
| ICV | Intracerebroventricular |
| SC | Subcutaneous |
References
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the sma gene smn1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Burglen, L.; Lefebvre, S.; Clermont, O.; Burlet, P.; Viollet, L.; Cruaud, C.; Munnich, A.; Melki, J. Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996, 32, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Vitte, J.; Fassier, C.; Tiziano, F.D.; Dalard, C.; Soave, S.; Roblot, N.; Brahe, C.; Saugier-Veber, P.; Bonnefont, J.P.; Melki, J. Refined characterization of the expression and stability of the smn gene products. Am. J. Pathol. 2007, 171, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Singh, R.N.; Androphy, E.J. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007, 35, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP a1/a2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Simone, C.; Salani, S.; Ruepp, M.D.; Rizzo, F.; Ruggieri, M.; Zanetta, C.; Brajkovic, S.; Moulton, H.M.; Muehlemann, O.; et al. Effect of combined systemic and local morpholino treatment on the spinal muscular atrophy Δ7 mouse model phenotype. Clin. Ther. 2014, 36, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Simone, C.; Dametti, S.; Salani, S.; Ulzi, G.; Pagliarani, S.; Rizzo, F.; Frattini, E.; Pagani, F.; Bresolin, N.; et al. Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci. Rep. 2015, 5, 11746. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Wood, M.J. Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 2011, 10, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.; Nizzardo, M.; Rizzo, F.; Ruggieri, M.; Riboldi, G.; Salani, S.; Bucchia, M.; Bresolin, N.; Comi, G.P.; Corti, S. Ipsc-derived neural stem cells act via kinase inhibition to exert neuroprotective effects in spinal muscular atrophy with respiratory distress type 1. Stem Cell Rep. 2014, 3, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Simone, C.; Rizzo, F.; Ruggieri, M.; Salani, S.; Riboldi, G.; Faravelli, I.; Zanetta, C.; Bresolin, N.; Comi, G.P.; et al. Minimally invasive transplantation of ipsc-derived ALDHhiSSCloVLA4+ neural stem cells effectively improves the phenotype of an amyotrophic lateral sclerosis model. Hum. Mol. Genet. 2014, 23, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H. SMNΔ7, the major product of the centromeric survival motor neuron (smn2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez, A.; Crisafulli, S.G.; Rizzuti, M.; Bresolin, N.; Comi, G.P.; Corti, S.; Nizzardo, M. Investigation of New Morpholino Oligomers to Increase Survival Motor Neuron Protein Levels in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2018, 19, 167. https://doi.org/10.3390/ijms19010167
Ramirez A, Crisafulli SG, Rizzuti M, Bresolin N, Comi GP, Corti S, Nizzardo M. Investigation of New Morpholino Oligomers to Increase Survival Motor Neuron Protein Levels in Spinal Muscular Atrophy. International Journal of Molecular Sciences. 2018; 19(1):167. https://doi.org/10.3390/ijms19010167
Chicago/Turabian StyleRamirez, Agnese, Sebastiano G. Crisafulli, Mafalda Rizzuti, Nereo Bresolin, Giacomo P. Comi, Stefania Corti, and Monica Nizzardo. 2018. "Investigation of New Morpholino Oligomers to Increase Survival Motor Neuron Protein Levels in Spinal Muscular Atrophy" International Journal of Molecular Sciences 19, no. 1: 167. https://doi.org/10.3390/ijms19010167
APA StyleRamirez, A., Crisafulli, S. G., Rizzuti, M., Bresolin, N., Comi, G. P., Corti, S., & Nizzardo, M. (2018). Investigation of New Morpholino Oligomers to Increase Survival Motor Neuron Protein Levels in Spinal Muscular Atrophy. International Journal of Molecular Sciences, 19(1), 167. https://doi.org/10.3390/ijms19010167