In Silico Analysis of the Association Relationship between Neuroprotection and Flavors of Traditional Chinese Medicine Based on the mGluRs

 ,
,
Abstract
1. Introduction
2. Results
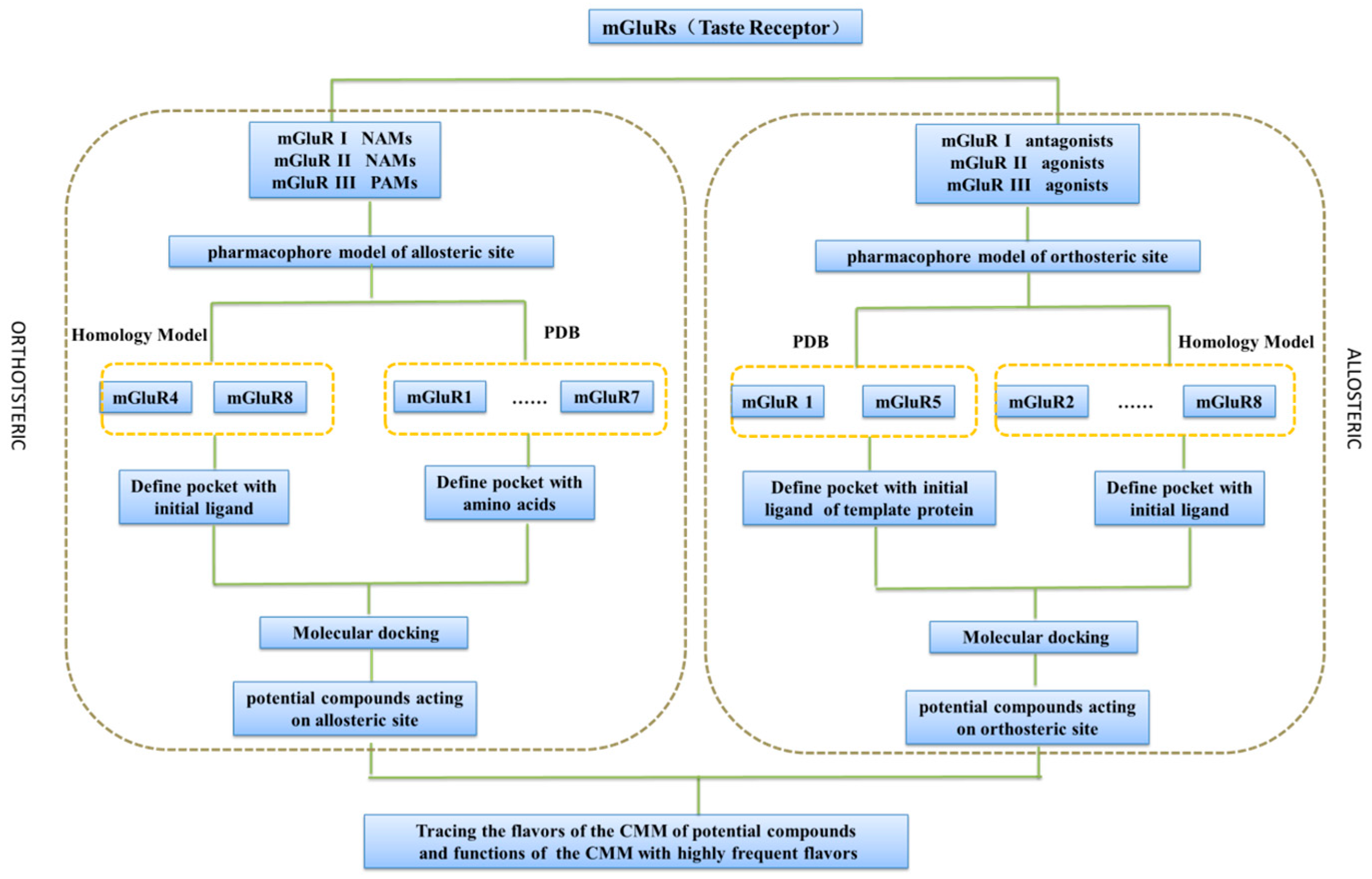
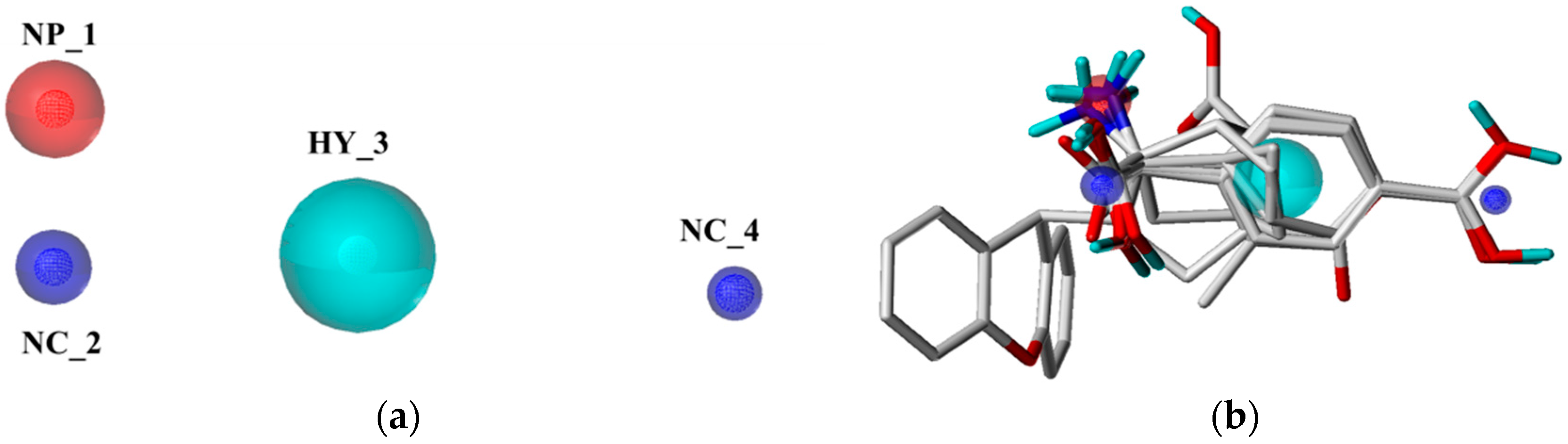
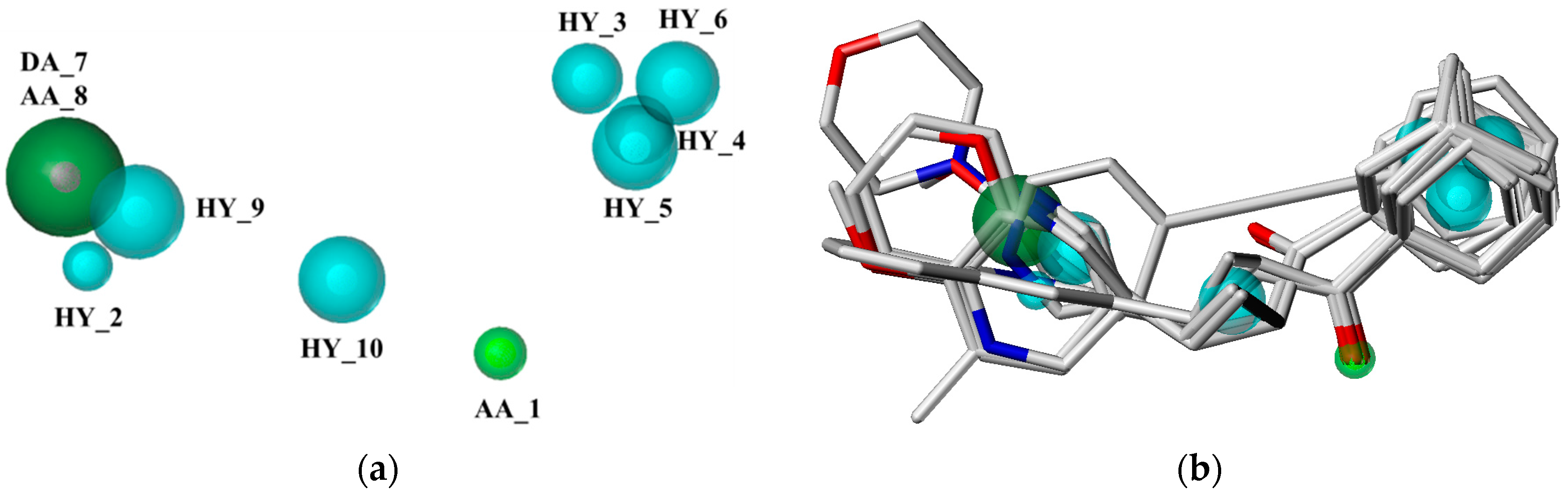
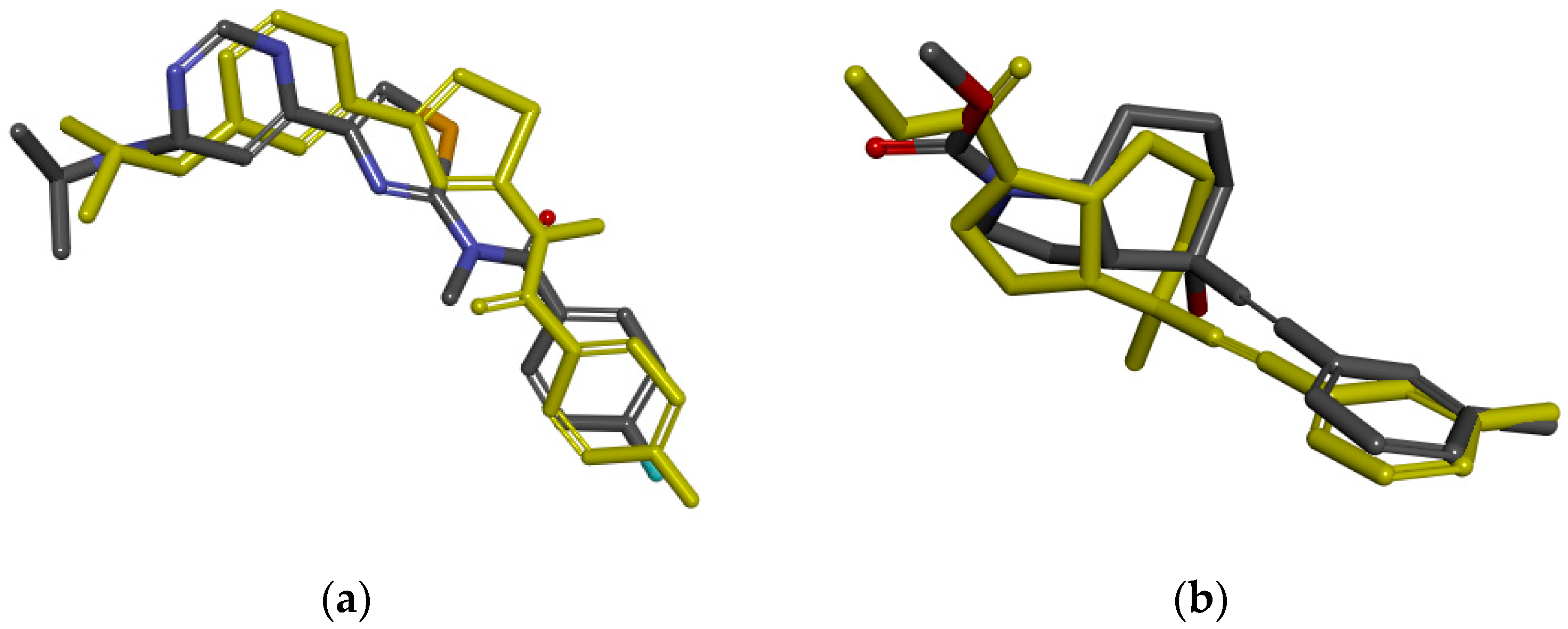
2.1. Pharmacophore Model Studies
2.2. Database Search
2.3. Homology Modeling Studies
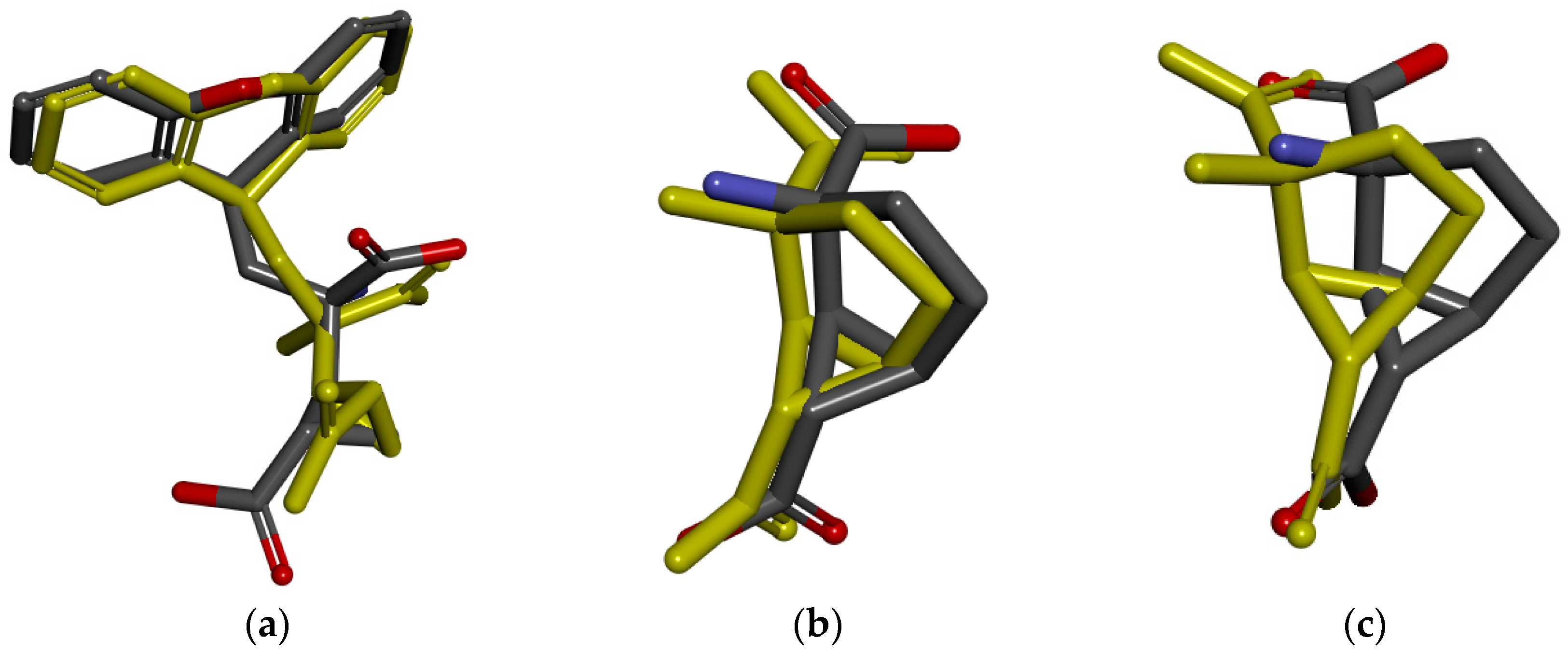
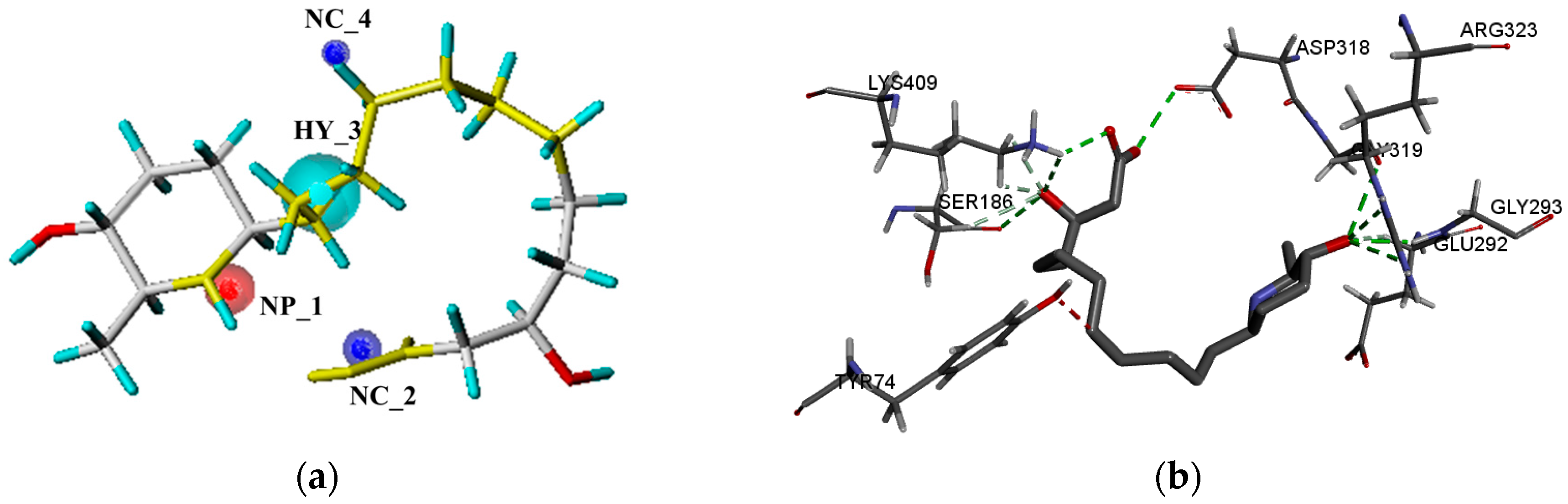
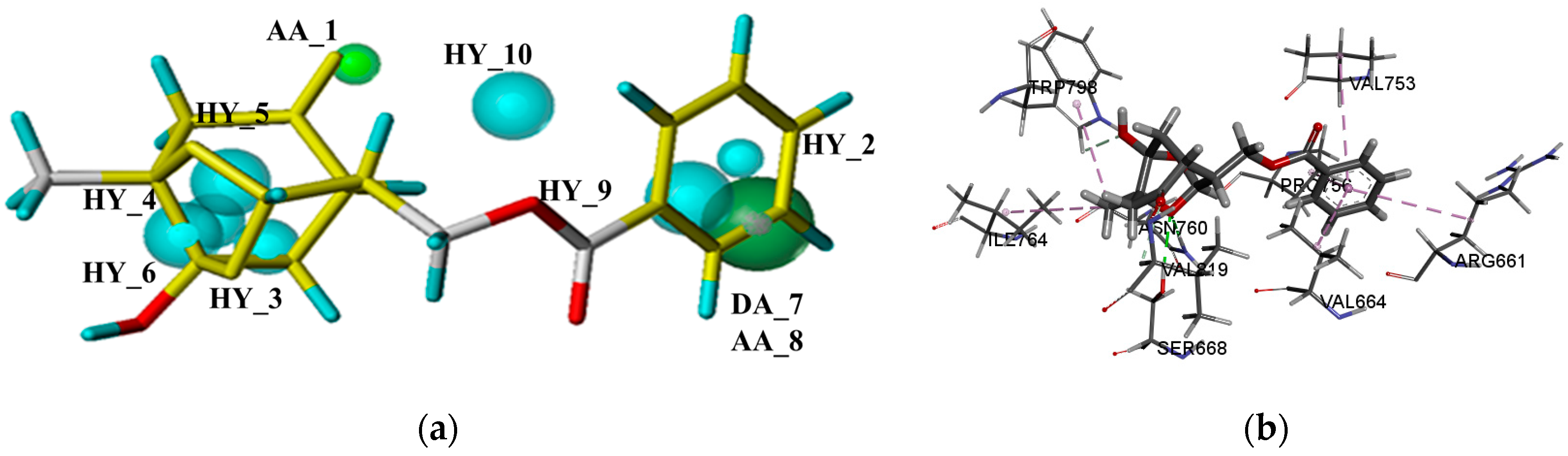
2.4. Molecular Docking Studies
2.4.1. The mGluRs Orthosteric Site
2.4.2. The mGluRs Allosteric Site
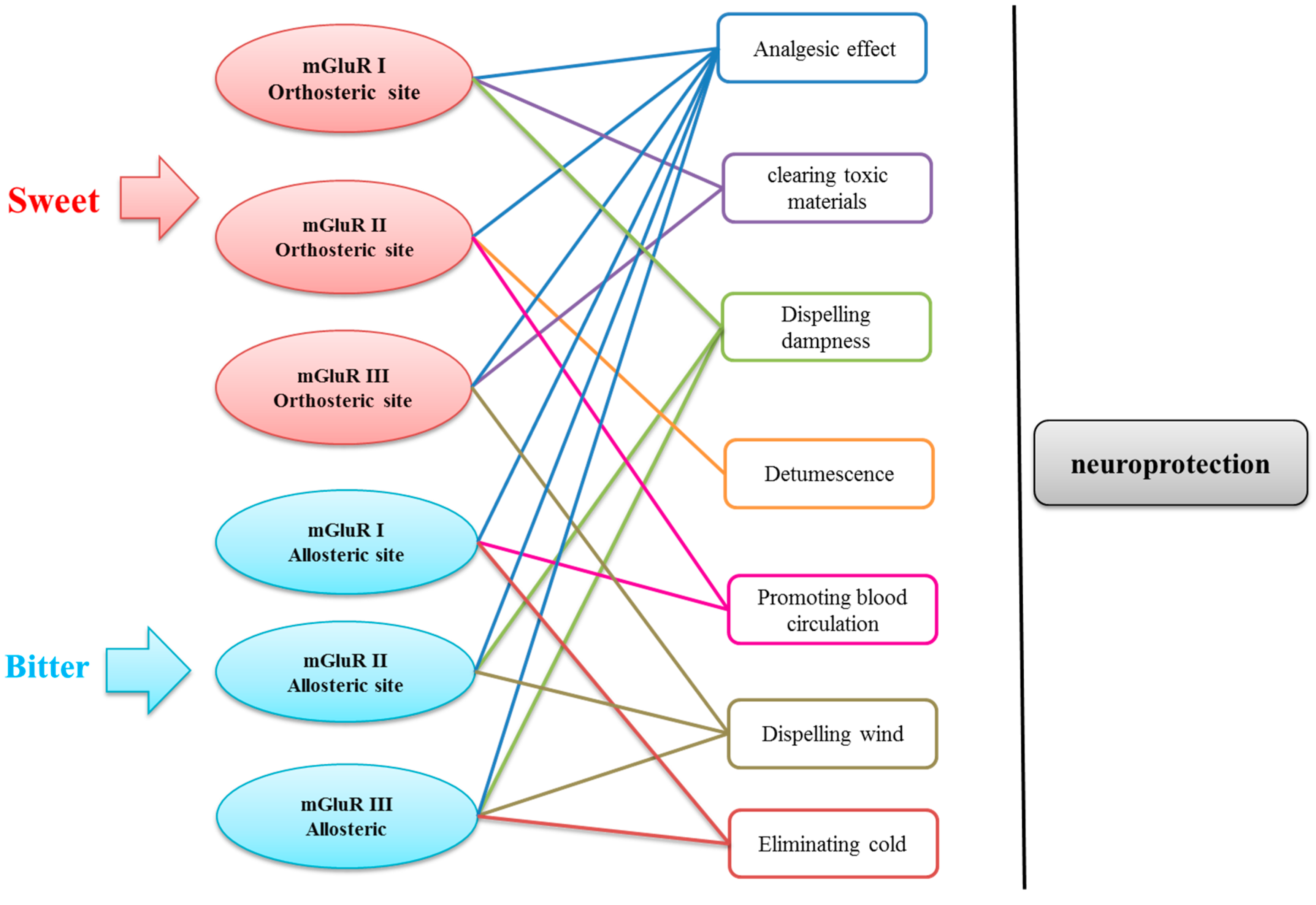
2.5. Data Analysis
3. Materials and Methods
3.1. GALAHAD Pharmacophore Hypotheses Generation
3.2. Homology Modeling Studies
3.3. Molecular Docking
3.4. Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Molares, S.; Ladio, A. Chemosensory perception and medicinal plants for digestive ailments in a Mapuche community in NW Patagonia, Argentina. J. Ethnopharmacol. 2009, 123, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Gilca, M.; Barbulescu, A. Taste of medicinal plants: A potential tool in predicting ethnopharmacological activities? J. Ethnopharmacol. 2015, 174, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, P.M.D.; Pinto, B.L.S.; Nascimento, V.T.D. Can organoleptic properties explain the differential use of medicinal plants? Evidence from Northeastern Brazil. J. Ethnopharmacol. 2015, 159, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Guo, J.; Pang, B.; Zhao, L.; Tong, X. Application of Herbal Medicines with Bitter Flavor and Cold Property on Treating Diabetes Mellitus. Evid.-Based Complement. Altern. Med. 2015, 2015, 529491. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zheng, X.; Sit, C.; Loo, W.T. Using association rules mining to explore pattern of Chinese medicinal formulae (prescription) in treating and preventing breast cancer recurrence and metastasis. J. Transl. Med. 2012, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Yoshida, R.; Ninomiya, Y. Taste transductions in taste receptor cells: Basic tastes and moreover. Curr. Pharm. Des. 2014, 20, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Landin, A.M.; Roper, S.D. A metabotropic glutamate receptor variant functions as a taste receptor. Nat. Neurosci. 2000, 3, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.J.; Pin, J.P. Pharmacology and functions of metabotropic glutamate receptors. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, Y.; Masu, M.; Ishii, T.; Shigemoto, R.; Nakanishi, S. A family of metabotropic glutamate receptors. Neuron 1992, 8, 169–179. [Google Scholar] [CrossRef]
- Chun, L.; Zhang, W.H.; Liu, J.F. Structure and ligand recognition of class C GPCRs. Acta Pharmacol. Sin. 2012, 33, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, X.; Chen, X.; He, Y.; Qiao, L.; Zhang, Y.; Li, G.; Xiang, Y. Virtual Screening and Molecular Dynamics Study of Potential Negative Allosteric Modulators of mGluR1 from Chinese Herbs. Molecules 2015, 20, 12769–12786. [Google Scholar] [CrossRef] [PubMed]
- Celanire, S.; Sebhat, I.; Wichmann, J.; Mayer, S.; Schann, S.; Gatti, S. Novel metabotropic glutamate receptor 2/3 antagonists and their therapeutic applications: A patent review (2005–present). Expert Opin. Ther. Pat. 2014, 25, 1–22. [Google Scholar]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Luneia, R.; Costantino, G.; Marinozzi, M.; Natalini, B.; Jakobsen, P.; Kanstrup, A.; Lombardi, G.; Moroni, F.; Thomsen, C. 1-Aminoindan-1,5-dicarboxylic Acid: A Novel Antagonist at Phospholipase C-Linked Metabotropic Glutamate Receptors. J. Med. Chem. 1995, 38, 3717–3719. [Google Scholar] [CrossRef] [PubMed]
- Osikowicz, M.; Mika, J.; Przewlocka, B. The glutamatergic system as a target for neuropathic pain relief. Exp. Physiol. 2013, 98, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Mercier, M.S.; Lodge, D. Group III Metabotropic Glutamate Receptors: Pharmacology, Physiology and Therapeutic Potential. Neurochem. Res. 2014, 39, 1876–1894. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Chen, L.; Zhang, J.; Xie, X.; Qiu, K.; Fu, J. A Combined Pharmacophore Modeling, 3D QSAR and Virtual Screening Studies on Imidazopyridines as B-Raf Inhibitors. Int. J. Mol. Sci. 2015, 16, 12307–12323. [Google Scholar] [CrossRef] [PubMed]
- Athri, P.; Wenzler, T.; Tidwell, R.; Bakunova, S.M.; Wilson, W.D. Pharmacophore model for pentamidine analogs active against Plasmodium falciparum. Eur. J. Med. Chem. 2010, 45, 6147–6151. [Google Scholar] [CrossRef] [PubMed]
- He, Y.S.; Jiang, L.D.; Yang, Z.; Qiao, Y.J.; Zhang, Y.L. A combination of pharmacophore modeling, molecular docking, and virtual screening for P2Y(12) receptor antagonists from Chinese herbs. Can. J. Chem. 2015, 93, 311–316. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.X.; Liu, Q.; Ai, Z.X.; Zhang, Y.L.; Xiang, Y.H.; Qiao, Y.J. Discovery of Dual ETA/ETB Receptor Antagonists from Traditional Chinese Herbs through in Silico and in Vitro Screening. Int. J. Mol. Sci. 2016, 17, 389. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput.-Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Bräunerosborne, H.; Egebjerg, J.; Nielsen, E.O.; Madsen, U.; Krogsgaardlarsen, P. Ligands for glutamate receptors: Design and therapeutic prospects. J. Med. Chem. 2000, 43, 2609–2645. [Google Scholar] [CrossRef]
- And, T.A.P.; Jain, A.N. Parameter Estimation for Scoring Protein—Ligand Interactions Using Negative Training Data. J. Med. Chem. 2006, 49, 5856–5868. [Google Scholar]
- Babu, S.; Nagarajan, S.K.; Lee, S.H.; Madhavan, T. Structural characterization of human CRTh2: A combined homology modeling, molecular docking and 3D-QSAR-based in silico approach. Med. Chem. Res. 2016, 25, 653–671. [Google Scholar] [CrossRef]
- Feng, Z.; Ma, S.; Hu, G.; Xie, X.Q. Allosteric Binding Site and Activation Mechanism of Class C G-Protein Coupled Receptors: Metabotropic Glutamate Receptor Family. AAPS J. 2015, 17, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.S.; Yuen, W.H.; So, K.F.; Chang, R. Neuroprotective effects of extracts from anti-aging Chinese medicine Lycium barbarum (Gou-Qi-Zi). J. Neurochem. 2004, 88 (Suppl. 1), 69. [Google Scholar]
- Saleem, S.; Zhuang, H.; Biswal, S.; Christen, Y.; Doré, S. Ginkgo Biloba Extract Neuroprotective Action Is Dependent on Heme Oxygenase 1 in Ischemic Reperfusion Brain Injury. Stroke 2008, 39, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.K.; Jin, Y.H.; Kim, H.B.; Ryu, J.H.; Sun, Y.K. Neuroprotective effects of the cyanidin-3-O-β-d-glucopyranoside isolated from mulberry fruit against cerebral ischemia. Neurosci. Lett. 2006, 391, 122–126. [Google Scholar]
- Rodrigues, M.J.; Gangadhar, K.N.; Zengin, G.; Mollica, A.; Varela, J.; Barreira, L.; Custódio, L. Juncaceae species as sources of innovative bioactive compounds for the food industry: In vitro antioxidant activity, neuroprotective properties and in silico studies. Food Chem. Toxicol. 2017, 107, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Prema, A.; Justin, T.A.; Manivasagam, T.; Mohamed, E.M.; Guillemin, G.J. Fenugreek Seed Powder Attenuated Aluminum Chloride-Induced Tau Pathology, Oxidative Stress, and Inflammation in a Rat Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, S209–S220. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Sun, H.; Shen, J.; Fan, Y.; Zhang, L.; Zhang, J. Radix Angelica Sinensis Promotes Synaptic Plasticity During Cognitive Recovery in Chronically Stressed Rats. Curr. Neurovasc. Res. 2015, 12, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, J.; Yu, C.; Tang, Y.; Liu, J.; Chen, H.; Jin, B.; Mei, Q.; Cao, S.; Qin, D. Lychee Seed Saponins Improve Cognitive Function and Prevent Neuronal Injury via Inhibiting Neuronal Apoptosis in a Rat Model of Alzheimer’s Disease. Nutrients 2017, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- CİHAN, Y.B.; Arsav, V.; GÖCEN, E. Royal Jelly in the Prevention of Radiation-Induced Brain Damages. J. Neurol. Sci. 2011, 28, 475–486. [Google Scholar]
- Kim, Y.J.; Lim, H.S.; Kim, Y.; Lee, J.; Kim, B.Y.; Jeong, S.J. Neuroprotective Effect of Corydalis ternata Extract and Its Phytochemical Quantitative Analysis. Chem. Pharm. Bull. 2017, 65, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Sang, H.S.; Hong, R.A.; Sang, H.J. Protective effects of the compounds isolated from the seed of Psoralea corylifolia on oxidative stress-induced retinal damage. Toxicol. Appl. Pharmacol. 2013, 269, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Q.; Zhang, W.Y.; Luo, X.T.; Ye, Y.; Zhu, X.Z. Paeoniflorin attenuates neuroinflammation and dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease by activation of adenosine A1 receptor. Br. J. Pharmacol. 2006, 148, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model. 2010, 29, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kothandan, G.; Madhavan, T.; Gadhe, C.G.; Cho, S.J. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: An in silico approach. Med. Chem. Res. 2012, 22, 1773–1787. [Google Scholar] [CrossRef]
- Wu, Y.; He, C.; Gao, Y.; He, S.; Liu, Y.; Lai, L. Dynamic modeling of human 5-lipoxygenase-inhibitor interactions helps to discover novel inhibitors. J. Med. Chem. 2012, 55, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- Nandy, S.K.; Bhuyan, R.; Seal, A. Modelling family 2 cystatins and their interaction with papain. J. Biomol. Struct. Dyn. 2013, 31, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; He, Y.; Luo, G.; Yang, Y.; Li, G.; Zhang, Y. Discovery of potential novel microsomal triglyceride transfer protein inhibitors via virtual screening of pharmacophore modelling and molecular docking. Mol. Simul. 2016, 42, 1223–1232. [Google Scholar] [CrossRef]
- Holt, P.A.; Chaires, J.B.; Trent, J.O. Molecular docking of intercalators and groove-binders to nucleic acids using Autodock and Surflex. J. Chem. Inf. Model. 2008, 48, 1602–1615. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cai, S.; Mei, H.; Li, J.; Yan, N.; Wang, Y. Docking and 3D QSAR study of thiourea analogs as potent inhibitors of influenza virus neuraminidase. J. Mol. Model. 2010, 16, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, F.; Chen, Y.K.; Luo, G.G.; Jiang, L.D.; Qiao, L.S.; Zhang, Y.L.; Xiang, Y.H. Discovery of Potential Orthosteric and Allosteric Antagonists of P2Y1R from Chinese Herbs by Molecular Simulation Methods. Evid.-Based Complement. Altern. Med. 2016, 2016, 4320201. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Specificity | N_HITS | PARETP | Energy | Sterics | HBOND | MOL-QRY |
|---|---|---|---|---|---|---|---|
| model 01 | 3.458 | 6 | 0 | 10.79 | 203.10 | 107.90 | 2.29 |
| model 02 | 3.456 | 6 | 0 | 4.52 | 193.60 | 101.10 | 2.90 |
| model 03 a | 3.458 | 6 | 0 | 13.70 | 203.10 | 107.90 | 2.29 |
| model 11 | 3.458 | 6 | 0 | 14.14 | 200.50 | 108.90 | 2.29 |
| model 13 | 3.454 | 6 | 0 | 16.61 | 203.30 | 107.40 | 2.15 |
| model 15 | 3.451 | 6 | 0 | 3.73 | 187.10 | 107.70 | 0.66 |
| model 16 | 3.452 | 6 | 0 | 6.98 | 188.50 | 102.70 | 2.29 |
| model 17 | 3.452 | 6 | 0 | 8.95 | 195.10 | 99.40 | 2.29 |
| model 18 | 3.452 | 6 | 0 | 10.72 | 187.40 | 104.70 | 2.29 |
| Model | Specificity | Energy | Sterics | HBOND | MOL-QRY | ∑Ranking |
|---|---|---|---|---|---|---|
| model 01 | 1 | 7 | 2 | 2 | 2 | 15 |
| model 02 | 4 | 2 | 6 | 8 | 1 | 22 |
| model 03 a | 1 | 6 | 2 | 2 | 2 | 14 |
| model 11 | 1 | 8 | 4 | 1 | 2 | 17 |
| model 13 | 4 | 9 | 1 | 5 | 6 | 26 |
| model 15 | 4 | 1 | 9 | 4 | 7 | 26 |
| model 16 | 7 | 3 | 7 | 7 | 2 | 27 |
| model 17 | 7 | 4 | 5 | 9 | 2 | 28 |
| model 18 | 7 | 5 | 8 | 6 | 2 | 29 |
| Number of Compounds | Pharmacophore Screening | Drug-Like Compounds | Blood–Brain Barrier Permeability | |
|---|---|---|---|---|
| Crystal Structure | ||||
| mGluR I | Orthoteric site | 123 | 89 | 43 |
| Allosteric site | 320 | 221 | 94 | |
| mGluR II | Orthoteric site | 178 | 103 | 30 |
| Allosteric site | 532 | 287 | 105 | |
| mGluR III | Orthoteric site | 110 | 80 | 3 |
| Allosteric site | 1714 | 392 | 23 |
| Domain | Target | Templates | Identity Value | Ramachandran Plot | ERRAT |
|---|---|---|---|---|---|
| Extracellular Domain | mGluR4 | 3MQ4 | 99% | 93.79% | 80.000 |
| mGluR8 | 3MQ4 | 72% | 94.26% | 83.559 | |
| 7TMD | mGluR2 | 4OR2 | 52% | 99.60% | 89.879 |
| mGluR3 | 4OO9 | 47% | 95.70% | 87.391 | |
| mGluR4 | 4OR2 | 43% | 98.43% | 94.400 | |
| mGluR7 | 4OO9 | 43% | 98.11% | 89.453 | |
| mGluR8 | 5CGC | 44% | 100% | 88.462 |
| No_Compounds | Ki | Rank of Ki | Total Score | Rank of Total Score |
|---|---|---|---|---|
| CHEMBL33567 | 910 | 1 | 7.2647 | 2 |
| CHEMBL277475 | 5200 | 2 | 7.4445 | 1 |
| CHEMBL329236 | 8800 | 3 | 6.6985 | 3 |
| CHEMBL89000 | 21,000 | 4 | 6.6781 | 4 |
| CHEMBL90501 | 23,000 | 5 | 6.1632 | 5 |
| CHEMBL88612 | 26,000 | 6 | 4.3929 | 7 |
| CHEMBL41221 | 470,000 | 7 | 5.6421 | 6 |
| Correlation | 0.9286 | |||
| No_Compounds | Ki (nM) | Rank of Ki | Total Score | Rank of Total Score |
|---|---|---|---|---|
| CHEMBL33567 | 175,000 | 1 | 6.8675 | 1 |
| CHEMBL277475 | 185,000 | 2 | 6.4943 | 2 |
| BDBM17657 | 5,400,000 | 3 | 5.215 | 3 |
| Correlation | 1.0000 | |||
| No_Compounds | Ki | Rank of Ki | Total Score | Rank of Total Score |
|---|---|---|---|---|
| CHEMBL33567 | 61 | 1 | 6.3650 | 2 |
| CHEMBL277475 | 210 | 2 | 6.9249 | 1 |
| CHEMBL89000 | 1700 | 3 | 6.0272 | 3 |
| CHEMBL280563 | 3400 | 4 | 5.9511 | 4 |
| CHEMBL88999 | 7300 | 5 | 5.1538 | 9 |
| BDBM17657 | 9500 | 6 | 4.6927 | 5 |
| CHEMBL8759 | 12,000 | 7 | 4.1048 | 6 |
| CHEMBL330097 | 15,000 | 8 | 3.6829 | 7 |
| CHEMBL34453 | 45,000 | 9 | 5.9186 | 8 |
| Correlation | 0.8167 | |||
| Domian | Target | Crystal Structure | Initial Ligand | RMSD/Correlation a |
|---|---|---|---|---|
| Extracellular Domain | mGluR1 | 3KS9 | Z99 | 1.4946 Å |
| mGluR5 | 3LMK | NAG | / | |
| mGluR2 | 4XAQ | 40F | 1.1403 Å | |
| mGluR3 | 4XAR | 40F | 0.5063 Å | |
| mGluR4 | Homology Model | Z99 | 0.9286 | |
| MGluR7 | 3MQ4 | 1.0000 | ||
| MGluR8 | Homology Model | 0.8167 |
| Domian | Target | Crystal Structure | Define Pocket | RMSD |
|---|---|---|---|---|
| 7TMD | mGluR1 | 4OR2 | FM9 | 1.6314 Å |
| mGluR5 | 4OO9 | 2U8 | 1.0754 Å | |
| mGluR2 | Homology Model | amino acids | / | |
| mGluR3 | Homology Model | amino acids | / | |
| mGluR4 | Homology Model | amino acids | / | |
| mGluR7 | Homology Model | amino acids | / | |
| mGluR8 | Homology Model | amino acids | / |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Qiao, L.; Chen, Y.; Zhao, B.; Gu, Y.; Huo, X.; Zhang, Y.; Li, G. In Silico Analysis of the Association Relationship between Neuroprotection and Flavors of Traditional Chinese Medicine Based on the mGluRs. Int. J. Mol. Sci. 2018, 19, 163. https://doi.org/10.3390/ijms19010163
Zhang X, Qiao L, Chen Y, Zhao B, Gu Y, Huo X, Zhang Y, Li G. In Silico Analysis of the Association Relationship between Neuroprotection and Flavors of Traditional Chinese Medicine Based on the mGluRs. International Journal of Molecular Sciences. 2018; 19(1):163. https://doi.org/10.3390/ijms19010163
Chicago/Turabian StyleZhang, Xu, Liansheng Qiao, Yankun Chen, Bowen Zhao, Yu Gu, Xiaoqian Huo, Yanling Zhang, and Gongyu Li. 2018. "In Silico Analysis of the Association Relationship between Neuroprotection and Flavors of Traditional Chinese Medicine Based on the mGluRs" International Journal of Molecular Sciences 19, no. 1: 163. https://doi.org/10.3390/ijms19010163
APA StyleZhang, X., Qiao, L., Chen, Y., Zhao, B., Gu, Y., Huo, X., Zhang, Y., & Li, G. (2018). In Silico Analysis of the Association Relationship between Neuroprotection and Flavors of Traditional Chinese Medicine Based on the mGluRs. International Journal of Molecular Sciences, 19(1), 163. https://doi.org/10.3390/ijms19010163