Telomere Homeostasis: Interplay with Magnesium

 , ,
, ,
Abstract
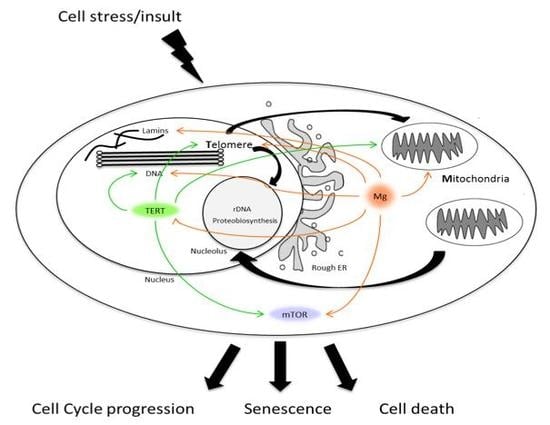
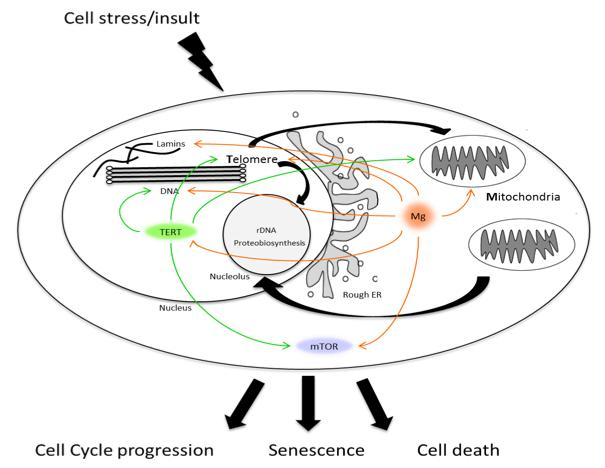{kind=link}
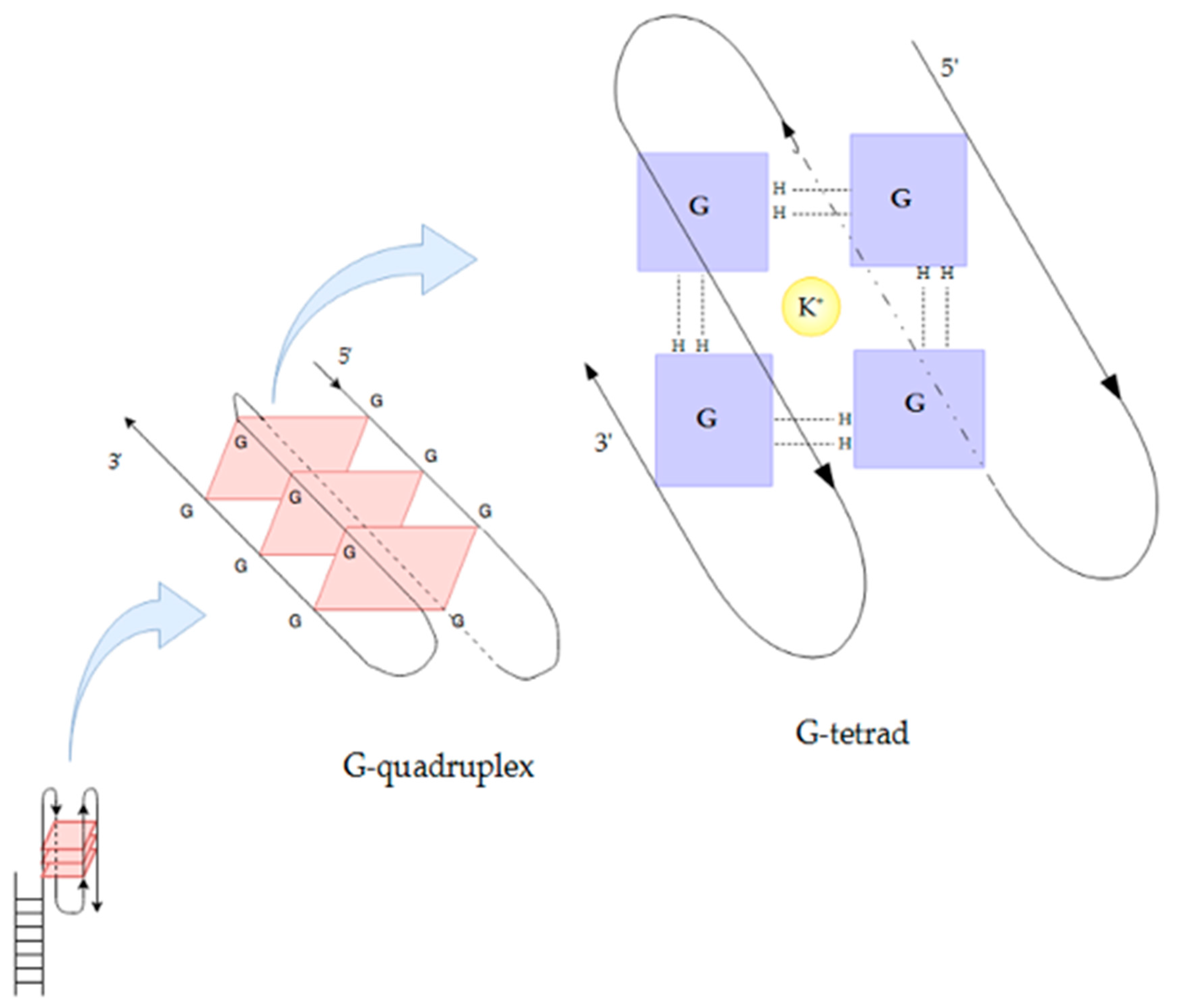{kind=link}
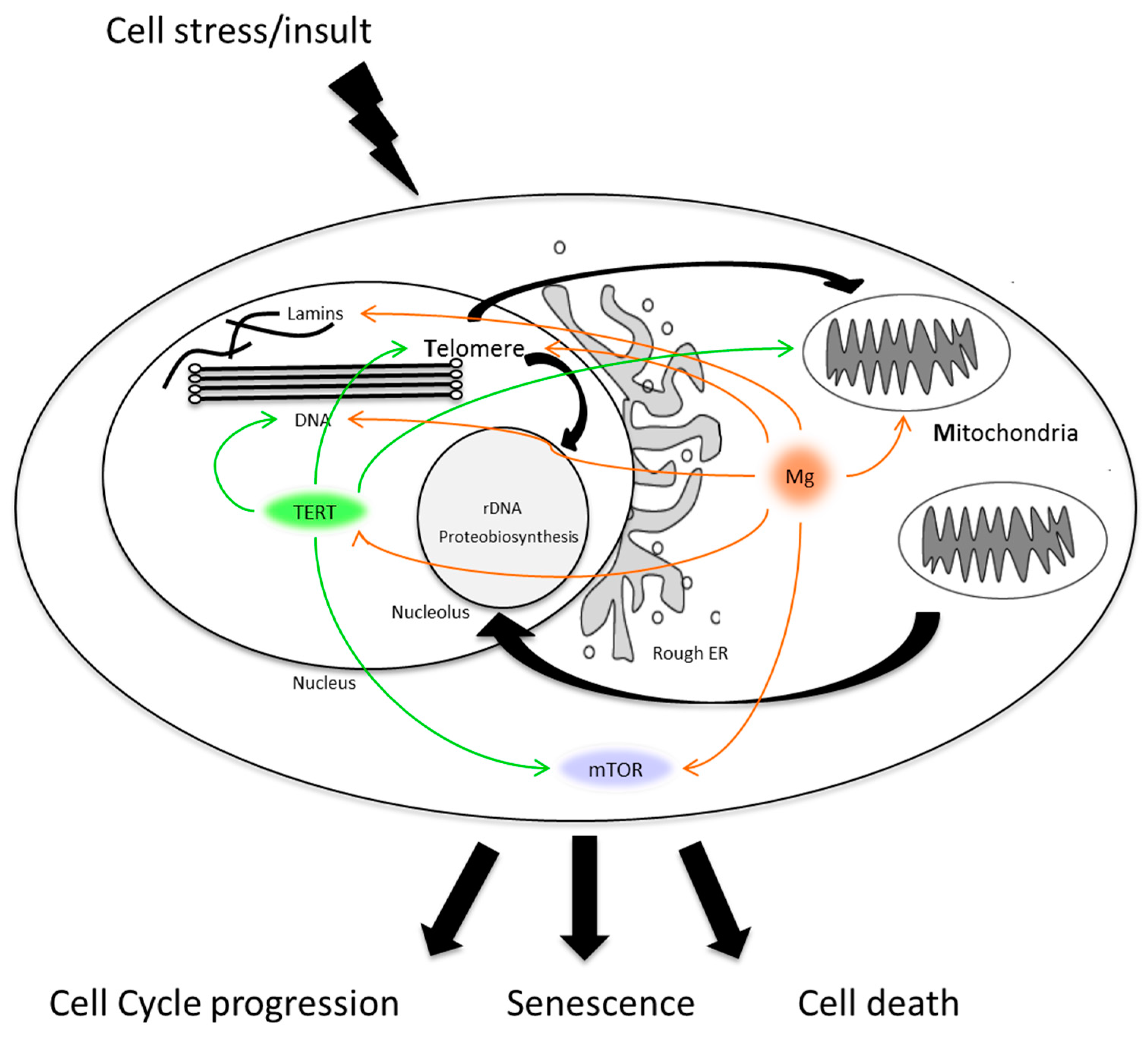{kind=link}
1. Introduction
2. Magnesium Homeostasis and Disease
3. Magnesium and Telomeres
3.1. Telomere Attrition
3.2. Magnesium and Telomeric Structure
3.3. The Role of Telomeric Repeat-Containing RNA (TERRA)
3.4. Telomerase Activity in the Nucleus
3.5. Non-Canonical TERT Functions in the Nucleus
3.6. Non-Canonical TERT Functions in the Mitochondria
3.7. Telomeres and mTOR
4. Mitochondrial Health, Ageing and Cancer
5. Telomere Length and Disease
6. Telomeres, Metabolism, and Circadian Rhythms
7. Conclusions
Author Contributions
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. Principle of marginotomy in template synthesis of polynucleotides. Dokl. Akad. Nauk SSSR 1971, 201, 1496–1499. [Google Scholar] [PubMed]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Ridout, S.J.; Ridout, K.K.; Kao, H.-T.; Carpenter, L.L.; Philip, N.S.; Tyrka, A.R.; Price, L.H. Telomeres, Early-Life Stress and Mental Illness. In Clinical Challenges in the Biopsychosocial Interface; Karger Publishers: Basel, Switzerland, 2015; pp. 92–108. [Google Scholar]
- Blackburn, E.H.; Collins, K. Telomerase: An RNP Enzyme Synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, a003558. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yuan, X.; Xu, D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes 2016, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G. Extra-telomeric functions of human telomerase: Cancer, mitochondria and oxidative stress. Curr. Pharm. Des. 2014, 20, 6386–6403. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Von Zglinicki, T. DNA damage in telomeres and mitochondria during cellular senescence: Is there a connection? Nucleic Acids Res. 2007, 35, 7505–7513. [Google Scholar] [CrossRef] [PubMed]
- Pilchova, I.; Klacanova, K.; Tatarkova, Z.; Kaplan, P.; Racay, P. The Involvement of Mg2+ in Regulation of Cellular and Mitochondrial Functions. Oxid. Med. Cell. Longev. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2017, 35, 265–290. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.M.P. Magnesium in Health and Disease. In Interrelations between Essential Metal Ions and Human Diseases; Springer: Dordrecht, The Netherlands, 2013; pp. 49–79. [Google Scholar]
- Jahnen-Dechent, W.; Ketteler, M. Magnesium basics. Clin. Kidney J. 2012, 5, i3–i14. [Google Scholar] [CrossRef] [PubMed]
- Bertinato, J.; Wang, K.C.; Hayward, S. Serum Magnesium Concentrations in the Canadian Population and Associations with Diabetes, Glycemic Regulation, and Insulin Resistance. Nutrients 2017, 9, 296. [Google Scholar] [CrossRef] [PubMed]
- Veronese, N.; Zurlo, A.; Solmi, M.; Luchini, C.; Trevisan, C.; Bano, G.; Manzato, E.; Sergi, G.; Rylander, R. Magnesium Status in Alzheimer’s Disease: A Systematic Review. Am. J. Alzheimers Dis. Other Dement. 2016, 31, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Vormann, J. Magnesium and Kidney Health—More on the “Forgotten Electrolyte”. Am. J. Nephrol. 2016, 44, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M. Magnesium and type 2 diabetes. World J. Diabetes 2015, 6, 1152. [Google Scholar] [CrossRef] [PubMed]
- De Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J.M. Magnesium in Man: Implications for Health and Disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Kolisek, M.; Sponder, G.; Mastrototaro, L.; Smorodchenko, A.; Launay, P.; Vormann, J.; Schweigel-Röntgen, M. Substitution p.A350V in Na+/Mg2+ Exchanger SLC41A1, Potentially Associated with Parkinson’s Disease, Is a Gain-of-Function Mutation. PLoS ONE 2013, 8, e71096. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Magnesium intake and risk of type 2 diabetes: A meta-analysis. J. Intern. Med. 2007, 262, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, K.; Han, D.; He, X.; Wei, J.; Zhao, L.; Imam, M.U.; Ping, Z.; Li, Y.; Xu, Y.; et al. Dietary magnesium intake and the risk of cardiovascular disease, type 2 diabetes, and all-cause mortality: A dose–response meta-analysis of prospective cohort studies. BMC Med. 2016, 14, 210. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Hamano, T.; Isaka, Y. Effects of Magnesium on the Phosphate Toxicity in Chronic Kidney Disease: Time for Intervention Studies. Nutrients 2017, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Ferrè, S.; Mazur, A.; Maier, J.A.M. Low-magnesium induces senescent features in cultured human endothelial cells. Magnes. Res. 2007, 20, 66–71. [Google Scholar] [PubMed]
- Killilea, D.W.; Ames, B.N. Magnesium deficiency accelerates cellular senescence in cultured human fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 5768–5773. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.I.; Torsello, A.; Fasanella, S.; Cittadini, A. Cell physiology of magnesium. Mol. Asp. Med. 2003, 24, 11–26. [Google Scholar] [CrossRef]
- Kooman, J.P.; Kotanko, P.; Schols, A.M.W.J.; Shiels, P.G.; Stenvinkel, P. Chronic kidney disease and premature ageing. Nat. Rev. Nephrol. 2014, 10, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; McGuinness, D.; Eriksson, M.; Kooman, J.P.; Stenvinkel, P. The role of epigenetics in renal ageing. Nat. Rev. Nephrol. 2017, 13, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ter Braake, A.D.; Shanahan, C.M.; de Baaij, J.H.F.F. Magnesium Counteracts Vascular Calcification: Passive Interference or Active Modulation? Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A.; Farese, S.; Graber, S.; Wald, J.; Richtering, W.; Floege, J.; Jahnen-Dechent, W. Nanoparticle-Based Test Measures Overall Propensity for Calcification in Serum. J. Am. Soc. Nephrol. 2012, 23, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Kuro-O, M.; Ishikawa, S.-E.; Funazaki, S.; Kusaka, I.; Kakei, M.; Hara, K. Daily variability in serum levels of calciprotein particles and their association with mineral metabolism parameters: A cross-sectional pilot study. Nephrology 2017. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Romani, A.M.P. Cellular magnesium homeostasis. Arch. Biochem. Biophys. 2011, 512, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sahni, J.; Scharenberg, A.M. The SLC41 family of MgtE-like magnesium transporters. Mol. Asp. Med. 2013, 34, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Mastrototaro, L.; Smorodchenko, A.; Aschenbach, J.R.; Kolisek, M.; Sponder, G. Solute carrier 41A3 encodes for a mitochondrial Mg2+ efflux system. Sci. Rep. 2016, 6, 27999. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Tabata, S.; Shindo, Y.; Hotta, K.; Suzuki, K.; Soga, T.; Oka, K. Mitochondrial Mg2+ homeostasis decides cellular energy metabolism and vulnerability to stress. Sci. Rep. 2016, 6, 30027. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.J. Correcting magnesium deficiencies may prolong life. Clin. Interv. Aging 2012, 7, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Sun, L.; Tan, R.; Xu, J.; LaFace, J.; Gao, Y.; Xiao, Y.; Attar, M.; Neumann, C.; Li, G.-M.; Su, B.; et al. Targeted DNA damage at individual telomeres disrupts their integrity and triggers cell death. Nucleic Acids Res. 2015, 43, 6334–6347. [Google Scholar] [CrossRef] [PubMed]
- Neeley, W.L.; Essigmann, J.M. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 2006, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Burrows, C.J.; Muller, J.G. Oxidative Nucleobase Modifications Leading to Strand Scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef] [PubMed]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M.; et al. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Henle, E.S.; Han, Z.; Tang, N.; Rai, P.; Luo, Y.; Linn, S. Sequence-specific DNA Cleavage by Fe2+-mediated Fenton Reactions Has Possible Biological Implications. J. Biol. Chem. 1999, 274, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Tada-Oikawa, S.; Kawanishi, S. Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 2001, 40, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Oikawa, S. Mechanism of Telomere Shortening by Oxidative Stress. Ann. N. Y. Acad. Sci. 2004, 1019, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.M.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.J.; Wu, Y.; Zakian, V.A. DNA Repair at Telomeres: Keeping the Ends Intact. Cold Spring Harb. Perspect. Biol. 2013, 5, a012666. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; et al. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Moye, A.L.; Porter, K.C.; Cohen, S.B.; Phan, T.; Zyner, K.G.; Sasaki, N.; Lovrecz, G.O.; Beck, J.L.; Bryan, T.M. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 2015, 6, 7643. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Luo, H.Q.; Li, N.B. A highly sensitive resonance Rayleigh scattering method to discriminate a parallel-stranded G-quadruplex from DNA with other topologies and structures. Chem. Commun. 2013, 49, 6209. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, G.; D’Errico, S.; Pinto, B.; Nici, F.; Dardano, P.; Rea, I.; De Stefano, L.; Mayol, L.; Piccialli, G.; Borbone, N. Self-Assembly of G-Rich Oligonucleotides Incorporating a 3′-3′ Inversion of Polarity Site: A New Route Towards G-Wire DNA Nanostructures. ChemistryOpen 2017, 6, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kosman, J.; Juskowiak, B. Hemin/G-quadruplex structure and activity alteration induced by magnesium cations. Int. J. Biol. Macromol. 2016, 85, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Rico, D.; Herrera, L.A. Regulated expression of the lncRNA TERRA and its impact on telomere biology. Mech. Ageing Dev. 2017, 167, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.Y.; Lin, J.; Ou, T.M.; Tan, J.H.; Li, D.; Gu, L.Q.; Huang, Z.S. Selective recognition of oncogene promoter G-quadruplexes by Mg2+. Biochem. Biophys. Res. Commun. 2010, 402, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhao, Y.; Li, N. Genome-wide colonization of gene regulatory elements by G4 DNA motifs. Nucleic Acids Res. 2009, 37, 6784–6798. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Martínez-Fernández, L.; Balty, C.; Perron, M.; Douki, T.; Improta, R.; Markovitsi, D. Absorption of Low-Energy UV Radiation by Human Telomere G-Quadruplexes Generates Long-Lived Guanine Radical Cations. J. Am. Chem. Soc. 2017, 139, 10561–10568. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Horvath, K.; Kark, J.D.; Susser, E.; Tishkoff, S.A.; Aviv, A. Telomere Length and the Cancer–Atherosclerosis Trade-Off. PLoS Genet. 2016, 12, e1006144. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.E.B.; Hunt, S.C.; Stone, R.C.; Horvath, K.; Herbig, U.; Ranciaro, A.; Hirbo, J.; Beggs, W.; Reiner, A.P.; Wilson, J.G.; et al. Shorter telomere length in Europeans than in Africans due to polygenetic adaptation. Hum. Mol. Genet. 2016, 25, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Qin, Z.; Spagnol, S.T.; Biegler, M.T.; Coffey, K.A.; Kalinowski, A.; Buehler, M.J.; Dahl, K.N. The tail domain of lamin B1 is more strongly modulated by divalent cations than lamin A. Nucleus 2015, 6, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Romero, C.; Chartrand, P. Telomeric Noncoding RNA TERRA Is Induced by Telomere Shortening to Nucleate Telomerase Molecules at Short Telomeres. Mol. Cell 2013, 51, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takahama, K.; Takada, A.; Tada, S.; Shimizu, M.; Sayama, K.; Kurokawa, R.; Oyoshi, T. Regulation of telomere length by G-quadruplex telomere DNA- and TERRA-binding protein TLS/FUS. Chem. Biol. 2013, 20, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.; Li, Y.; Tergaonkar, V. Current Insights to Regulation and Role of Telomerase in Human Diseases. Antioxidants 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.A.; Tomita, K. Fundamental mechanisms of telomerase action in yeasts and mammals: Understanding telomeres and telomerase in cancer cells. Open Biol. 2017, 7, 160338. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Wright, W.E.; Shay, J.W. Alternative splicing regulation of telomerase: A new paradigm? Trends Genet. 2014, 30, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Maida, Y.; Masutomi, K. Telomerase reverse transcriptase moonlights: Therapeutic targets beyond telomerase. Cancer Sci. 2015, 106, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012, 40, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Czapiewski, R.; Wan, T.; Bell, A.; Hill, K.N.; von Zglinicki, T.; Saretzki, G. Decreased mTOR signalling reduces mitochondrial ROS in brain via accumulation of the telomerase protein TERT within mitochondria. Aging 2016, 8, 2551–2567. [Google Scholar] [CrossRef] [PubMed]
- Saretzki, G. Telomerase, mitochondria and oxidative stress. Exp. Gerontol. 2009, 44, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Devkota, S.; Roh, J.-I.; Lee, J.; Lee, H.-W. Telomerase reverse transcriptase induces basal and amino acid starvation-induced autophagy through mTORC1. Biochem. Biophys. Res. Commun. 2016, 478, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Sundin, T.; Hentosh, P. InTERTesting association between telomerase, mTOR and phytochemicals. Expert Rev. Mol. Med. 2012, 14, e8. [Google Scholar] [CrossRef] [PubMed]
- Feeney, K.A.; Hansen, L.L.; Putker, M.; Olivares-Yañez, C.; Day, J.; Eades, L.J.; Larrondo, L.F.; Hoyle, N.P.; O’Neill, J.S.; van Ooijen, G. Daily magnesium fluxes regulate cellular timekeeping and energy balance. Nature 2016, 532, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, G.; O’Neill, J.S. Intracellular magnesium and the rhythms of life. Cell Cycle 2016, 15, 2997–2998. [Google Scholar] [CrossRef] [PubMed]
- Killilea, D.W.; Maier, J.A.M. A connection between magnesium deficiency and aging: New insights from cellular studies. Magnes. Res. 2008, 21, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Nair, K.S. Mitochondrial function as a determinant of life span. Pfluger Arch. 2010, 459, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Shinohara, A.; Matsukawa, T.; Chiba, M.; Wu, J.; Iesaki, T.; Okada, T. Chronic magnesium deficiency decreases tolerance to hypoxia/reoxygenation injury in mouse heart. Life Sci. 2011, 88, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Kobayashi, K.; Takahashi, M.; Katahira, K.; Goryo, K.; Matsushita, N.; Yasumoto, K.; Fujii-Kuriyama, Y.; Sogawa, K. Magnesium Deficiency Causes Loss of Response to Intermittent Hypoxia in Paraganglion Cells. J. Biol. Chem. 2009, 284, 19077–19089. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, T.J.; Trinh, T.; Kogelschatz, C.; Fujimoto, E.; Lush, M.E.; Piotrowski, T.; Brimley, C.J.; Bonkowsky, J.L. Hypoxia Disruption of Vertebrate CNS Pathfinding through EphrinB2 Is Rescued by Magnesium. PLoS Genet. 2012, 8, e1002638. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Crowther, C.A.; Middleton, P.; Marret, S.; Rouse, D. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. In Cochrane Database of Systematic Reviews; Doyle, L.W., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 2001, 11, 293–299. [Google Scholar] [CrossRef]
- Jacobs, T.L.; Epel, E.S.; Lin, J.; Blackburn, E.H.; Wolkowitz, O.M.; Bridwell, D.A.; Zanesco, A.P.; Aichele, S.R.; Sahdra, B.K.; MacLean, K.A.; et al. Intensive meditation training, immune cell telomerase activity, and psychological mediators. Psychoneuroendocrinology 2011, 36, 664–681. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Elmofty, M.; Castillo, E.; DeRouen, M.; Shariff-Marco, S.; Allen, L.; Gomez, S.L.; Nápoles, A.M.; Márquez-Magaña, L. Evaluation of cortisol and telomere length measurements in ethnically diverse women with breast cancer using culturally sensitive methods. J. Community Genet. 2017, 8, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sundquist, K.; Hedelius, A.; Palmér, K.; Memon, A.A.; Sundquist, J. Leukocyte telomere length and depression, anxiety and stress and adjustment disorders in primary health care patients. BMC Psychiatry 2017, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Ridout, K.K.; Ridout, S.J.; Price, L.H.; Sen, S.; Tyrka, A.R. Depression and telomere length: A meta-analysis. J. Affect. Disord. 2016, 191, 237–247. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, D.; McGlynn, L.M.; Johnson, P.C.; MacIntyre, A.; Batty, G.D.; Burns, H.; Cavanagh, J.; Deans, K.A.; Ford, I.; McConnachie, A.; et al. Socio-economic status is associated with epigenetic differences in the pSoBid cohort. Int. J. Epidemiol. 2012, 41, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Ridout, K.K.; Levandowski, M.; Ridout, S.J.; Gantz, L.; Goonan, K.; Palermo, D.; Price, L.H.; Tyrka, A.R. Early life adversity and telomere length: A meta-analysis. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, B.M.; Carvalho, C.M.; Moretti, P.N.; Mello, M.F.; Belangero, S.I. Stress-related telomere length in children: A systematic review. J. Psychiatr. Res. 2017, 92, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; McGlynn, L.M.; MacIntyre, A.; Johnson, P.C.D.; da Batty, G.D.; Burns, H.; Cavanagh, J.; Deans, K.A.; Ford, I.; McConnachie, A.; et al. Accelerated telomere attrition is associated with relative household income, diet and inflammation in the pSoBid Cohort. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Bakunina, N.; Pariante, C.M.; Zunszain, P.A. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.-J.; Ratka, A. Telomere shortening and Alzheimer’s disease. Neuromol. Med. 2013, 15, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Saijonmaa, O. Telomere length and cardiovascular aging. Ann. Med. 2012, 44, S138–S142. [Google Scholar] [CrossRef] [PubMed]
- Aviv, A.; Anderson, J.J.; Shay, J.W. Mutations, Cancer and the Telomere Length Paradox. Trends Cancer 2017, 3, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Andrew, T.; Gardner, J.P.; Kimura, M.; Oelsner, E.; Cherkas, L.F.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Astuti, Y.; Wardhana, A.; Watkins, J.; Wulaningsih, W. PILAR Research Network Cigarette smoking and telomere length: A systematic review of 84 studies and meta-analysis. Environ. Res. 2017, 158, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Panich, U.; Sittithumcharee, G.; Rathviboon, N.; Jirawatnotai, S. Ultraviolet Radiation-Induced Skin Aging: The Role of DNA Damage and Oxidative Stress in Epidermal Stem Cell Damage Mediated Skin Aging. Stem Cells Int. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Blackburn, E.H. Human cancer cells harbor T-stumps, a distinct class of extremely short telomeres. Mol. Cell 2007, 28, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.B.; Balakrishna Pai, S.; Yang, L.; Joshi, H.C. Abundance of a distinct cluster of telomere t-stumps in advanced breast cancer cell line. Oncol. Lett. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Bruderlein, S.; Wegener, S.; Lichtensztejn, D.; Lichtensztejn, Z.; Lemieux, B.; Moller, P.; Mai, S. 3D nuclear organization of telomeres in the Hodgkin cell lines U-HO1 and U-HO1-PTPN1: PTPN1 expression prevents the formation of very short telomeres including “t-stumps”. BMC Cell Biol. 2010, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Righolt, C.; Mai, S. Genomic Instability: The Driving Force behind Refractory/Relapsing Hodgkin’s Lymphoma. Cancers 2013, 5, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sfeir, A.J.; Zou, Y.; Buseman, C.M.; Chow, T.T.; Shay, J.W.; Wright, W.E. Telomere Extension Occurs at Most Chromosome Ends and Is Uncoupled from Fill-In in Human Cancer Cells. Cell 2009, 138, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Major, J.M.; Cawthon, R.; Weinstein, S.J.; Virtamo, J.; Lan, Q.; Rothman, N.; Albanes, D.; Stolzenberg-Solomon, R.Z. A prospective analysis of telomere length and pancreatic cancer in the α-tocopherol β-carotene cancer (ATBC) prevention study. Int. J. Cancer 2013, 133, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Julin, B.; Shui, I.; Heaphy, C.M.; Joshu, C.E.; Meeker, A.K.; Giovannucci, E.; De Vivo, I.; Platz, E.A. Circulating leukocyte telomere length and risk of overall and aggressive prostate cancer. Br. J. Cancer 2015, 112, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhou, Z.; Wei, S.; Liu, Z.; Pooley, K.A.; Dunning, A.M.; Svenson, U.; Roos, G.; Hosgood, H.D.; Shen, M.; et al. Shortened Telomere length is associated with increased risk of cancer: A meta-analysis. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, I.M.; Mirabello, L.; Pfeiffer, R.M.; Savage, S.A. The association of telomere length and cancer: A meta-analysis. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, X.; Li, L.; Zhou, Y.; Wang, C.; Hou, S. The Association between Telomere Length and Cancer Prognosis: Evidence from a Meta-Analysis. PLoS ONE 2015, 10, e0133174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Han, W.; Xue, W.; Zou, Y.; Xie, C.; Du, J.; Jin, G. The association between telomere length and cancer risk in population studies. Sci. Rep. 2016, 6, 22243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Q.; Zhu, W.; Liu, T.; Xie, S.-H.; Zhong, L.-X.; Cai, Y.-Y.; Li, X.-N.; Liang, M.; Chen, W.; et al. The Association of Telomere Length in Peripheral Blood Cells with Cancer Risk: A Systematic Review and Meta-analysis of Prospective Studies. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.; Díez-González, L.; Ocaña, A.; Šeruga, B.; Amir, E.; Templeton, A.J. Prognostic role of telomere length in malignancies: A meta-analysis and meta-regression. Exp. Mol. Pathol. 2017, 102, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qu, K.; Pang, Q.; Wang, Z.; Zhou, Y.; Liu, C. Association between telomere length and survival in cancer patients: A meta-analysis and review of literature. Front. Med. 2016, 10, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.; Du, M.; Wong, J.Y.Y.; Han, J.; De Vivo, I. Paternal age at birth is associated with offspring leukocyte telomere length in the nurses’ health study. Hum. Reprod. 2012, 27, 3622–3631. [Google Scholar] [CrossRef] [PubMed]
- Broer, L.; Codd, V.; Nyholt, D.R.; Deelen, J.; Mangino, M.; Willemsen, G.; Albrecht, E.; Amin, N.; Beekman, M.; de Geus, E.J.C.; et al. Meta-analysis of telomere length in 19,713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur. J. Hum. Genet. 2013, 21, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ruiz, C.M.; Baird, D.; Roger, L.; Boukamp, P.; Krunic, D.; Cawthon, R.; Dokter, M.M.; van der Harst, P.; Bekaert, S.; de Meyer, T.; et al. Reproducibility of telomere length assessment: An international collaborative study. Int. J. Epidemiol. 2015, 44, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Montpetit, A.J.; Alhareeri, A.A.; Montpetit, M.; Starkweather, A.R.; Elmore, L.W.; Filler, K.; Mohanraj, L.; Burton, C.W.; Menzies, V.S.; Lyon, D.E.; et al. Montpetit Telomere Length: A Review of Methods for Measurement. Nurs. Res. 2015, 63, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Abdallah, P.; Luciano, P.; Runge, K.W.; Lisby, M.; Géli, V.; Gilson, E.; Teixeira, M.T. A two-step model for senescence triggered by a single critically short telomere. Nat. Cell Biol. 2009, 11, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cheon, J.; Brown, R.; Coccia, M.; Puterman, E.; Aschbacher, K.; Sinclair, E.; Epel, E.; Blackburn, E.H. Systematic and Cell Type-Specific Telomere Length Changes in Subsets of Lymphocytes. J. Immunol. Res. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Blumenreich, M.S. The White Blood Cell and Differential Count; Butterworths: Boston, MA, USA, 1990; ISBN 040990077X. [Google Scholar]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2015, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, S.; Wei, Q.; Barrera, S.L.; Dong, Y.Q.; Etzel, C.J.; Spitz, M.R.; Forman, M.R. Dietary magnesium and DNA repair capacity as risk factors for lung cancer. Carcinogenesis 2008, 29, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.I.; Trapani, V. Magnesium and its transporters in cancer: A novel paradigm in tumour development. Clin. Sci. 2012, 123, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.I.; Trapani, V.; Cittadini, A.; Maier, J.A.M. Hypomagnesaemia in oncologic patients: To treat or not to treat? Magnes. Res. 2009, 22, 5–9. [Google Scholar] [PubMed]
- Wolf, F.I.; Trapani, V.; Simonacci, M.; Boninsegna, A.; Mazur, A.; Maier, J.A.M. Magnesium deficiency affects mammary epithelial cell proliferation: Involvement of oxidative stress. Nutr. Cancer 2009, 61, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Zastre, J.A.; Sweet, R.L.; Hanberry, B.S.; Ye, S. Linking vitamin B1 with cancer cell metabolism. Cancer Metab. 2013, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Peake, R.W.A.; Godber, I.M.; Maguire, D. The effect of magnesium administration on erythrocyte transketolase activity in alcoholic patients treated with thiamine. Scott. Med. J. 2013, 58, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2016, 18, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.N.; Saafir, T.B.; Tosini, G. The role of retinal photoreceptors in the regulation of circadian rhythms. Rev. Endocr. Metab. Disord. 2009, 10, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Kettner, N.M. The Circadian Clock in Cancer Development and Therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar] [PubMed]
- Chen, W.-D.; Wen, M.-S.; Shie, S.-S.; Lo, Y.-L.; Wo, H.-T.; Wang, C.-C.; Hsieh, I.-C.; Lee, T.-H.; Wang, C.-Y. The circadian rhythm controls telomeres and telomerase activity. Biochem. Biophys. Res. Commun. 2014, 451, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Samulin Erdem, J.; Notø, H.Ø.; Skare, Ø.; Lie, J.-A.S.; Petersen-Øverleir, M.; Reszka, E.; Pepłońska, B.; Zienolddiny, S. Mechanisms of breast cancer risk in shift workers: Association of telomere shortening with the duration and intensity of night work. Cancer Med. 2017, 6, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Kunieda, T.; Minamino, T.; Katsuno, T.; Tateno, K.; Nishi, J.; Miyauchi, H.; Orimo, M.; Okada, S.; Komuro, I. Cellular senescence impairs circadian expression of clock genes in vitro and in vivo. Circ. Res. 2006, 98, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Uetani, N.; Hardy, S.; Gravel, S.-P.; Kiessling, S.; Pietrobon, A.; Wong, N.N.; Chénard, V.; Cermakian, N.; St-Pierre, J.; Tremblay, M.L. PRL2 links magnesium flux and sex-dependent circadian metabolic rhythms. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.P.; Kofuji, P. Stoichiometry of N-methyl-D-aspartate receptors within the suprachiasmatic nucleus. J. Neurophysiol. 2010, 103, 3448–3464. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, N.G.; Siegler Retchless, B.; Johnson, J.W. Molecular bases of NMDA receptor subtype-dependent properties. J. Physiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Voma, C. Low Hepatic Mg2+ Content promotes Liver dysmetabolism: Implications for the Metabolic Syndrome. J. Metab. Syndr. 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Voma, C.; Barfell, A.; Croniger, C.; Romani, A. Reduced cellular Mg2+ content enhances hexose 6-phosphate dehydrogenase activity and expression in HepG2 and HL-60 cells. Arch. Biochem. Biophys. 2014, 548, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Büttner, M.; Jezova, D.; Greene, B.; Konrad, C.; Kircher, T.; Murck, H. Target-based biomarker selection—Mineralocorticoid receptor-related biomarkers and treatment outcome in major depression. J. Psychiatr. Res. 2015, 66–67, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; Davies, R.W. Ageing and the death of neurones. In Molecular Biology of the Neuron; Oxford University Press: Oxford, UK, 2004; pp. 439–468. [Google Scholar]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maguire, D.; Neytchev, O.; Talwar, D.; McMillan, D.; Shiels, P.G. Telomere Homeostasis: Interplay with Magnesium. Int. J. Mol. Sci. 2018, 19, 157. https://doi.org/10.3390/ijms19010157
Maguire D, Neytchev O, Talwar D, McMillan D, Shiels PG. Telomere Homeostasis: Interplay with Magnesium. International Journal of Molecular Sciences. 2018; 19(1):157. https://doi.org/10.3390/ijms19010157
Chicago/Turabian StyleMaguire, Donogh, Ognian Neytchev, Dinesh Talwar, Donald McMillan, and Paul G. Shiels. 2018. "Telomere Homeostasis: Interplay with Magnesium" International Journal of Molecular Sciences 19, no. 1: 157. https://doi.org/10.3390/ijms19010157
APA StyleMaguire, D., Neytchev, O., Talwar, D., McMillan, D., & Shiels, P. G. (2018). Telomere Homeostasis: Interplay with Magnesium. International Journal of Molecular Sciences, 19(1), 157. https://doi.org/10.3390/ijms19010157