Overview of the Microenvironment of Vasculature in Vascular Tone Regulation


Abstract
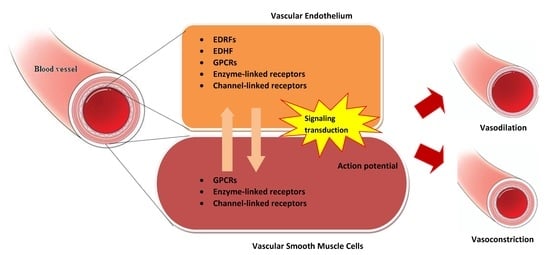
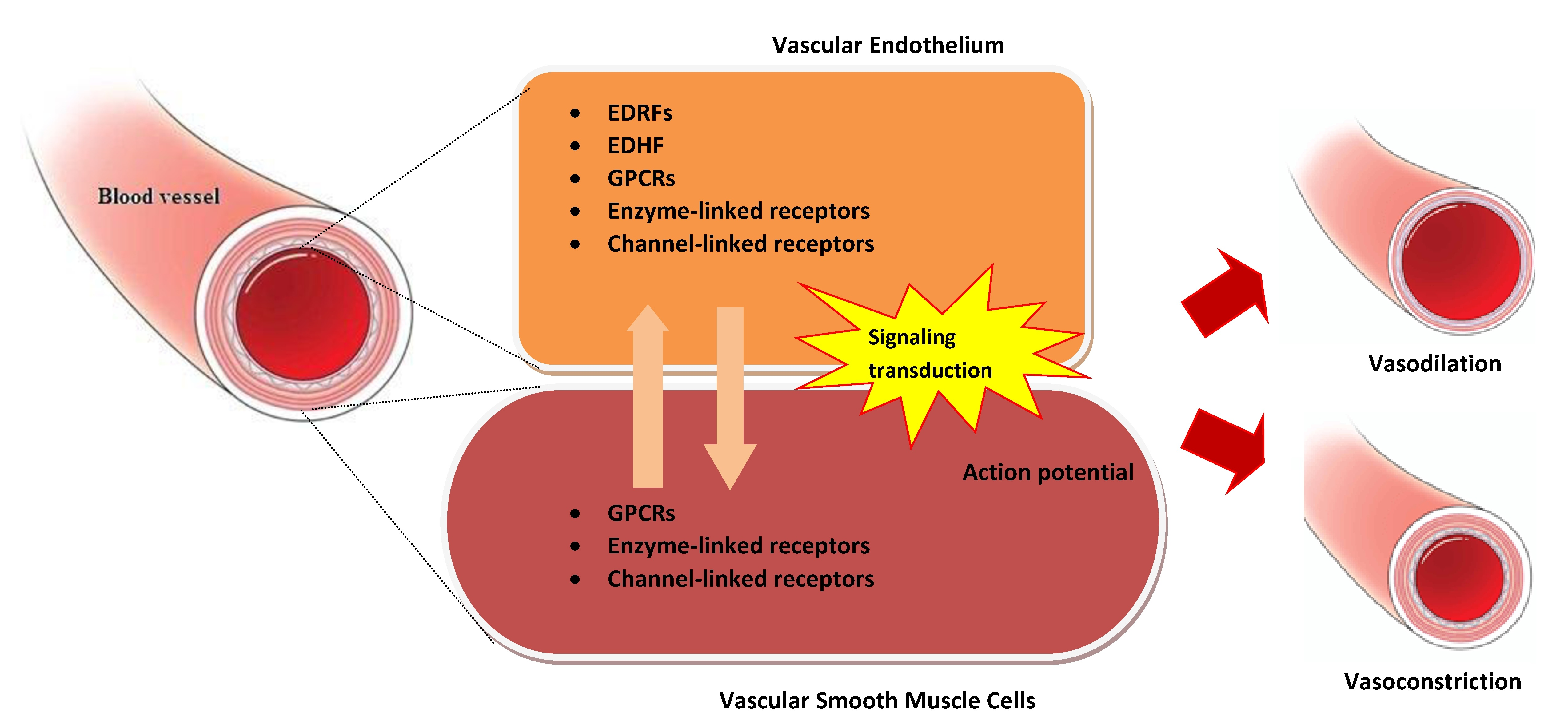{kind=link}
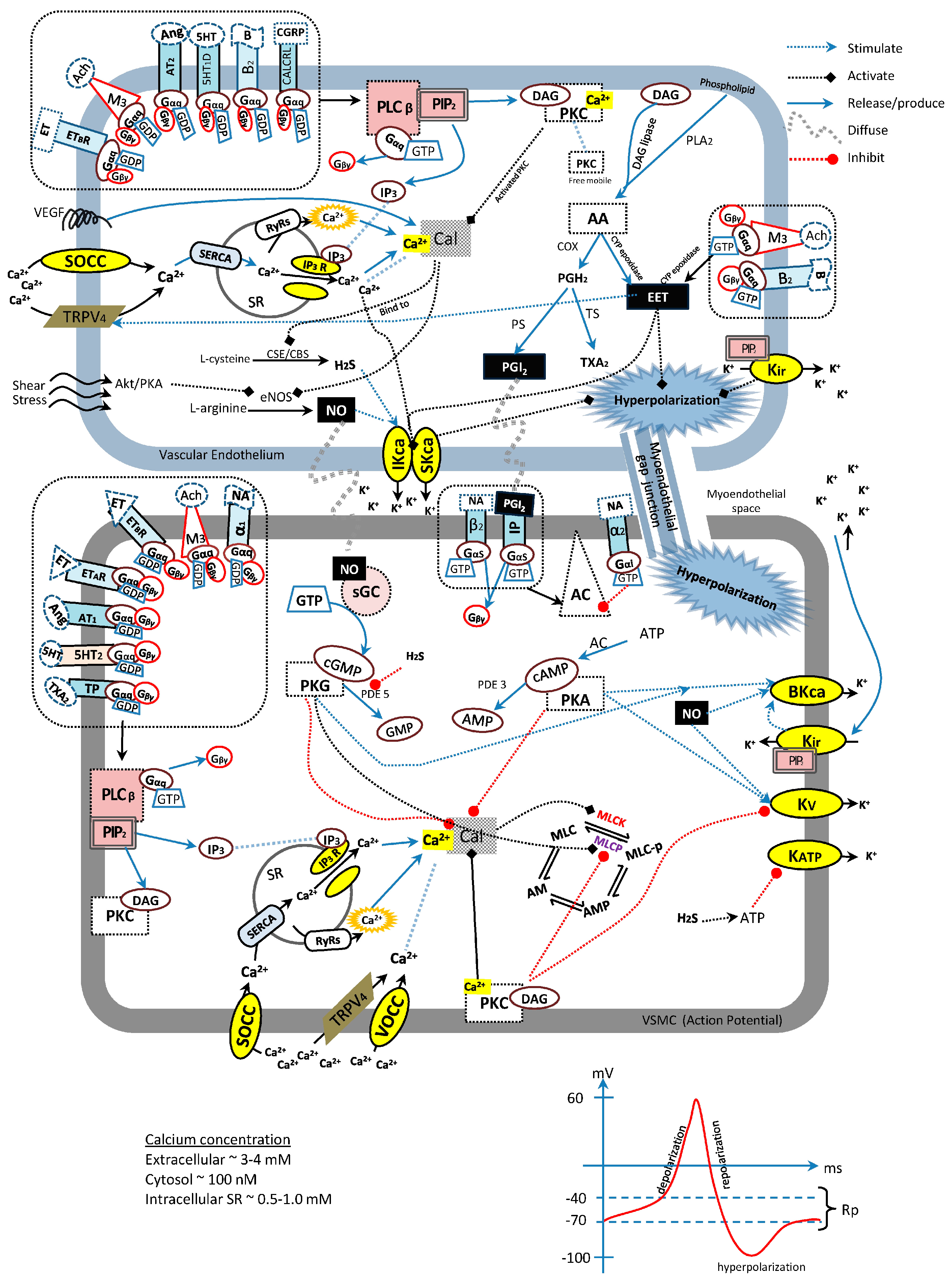{kind=link}
1. Introduction
2. Blood Vessel
3. Endothelium-Derived Relaxing Factors (EDRFs)
3.1. Nitric Oxide
3.2. Prostacyclin (PGI2)
3.3. Endothelium-Derived Hyperpolarizing Factors
3.4. Hydrogen Sulfide
4. Enzyme-Linked Receptors
4.1. Soluble Guanylyl Cyclase
4.2. Serine-Threonine Protein Kinases
5. G-Protein-Coupled Receptors (GPCRs)
5.1. Gqα-Protein-Coupled Receptors
5.2. Giα-Protein-Coupled Receptors
5.3. Gsα-Protein-Coupled Receptors
6. Channel-Linked Receptors
6.1. Potassium Channels
6.2. Calcium Channels
7. General Integration of Vasodilative Receptors
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| α | α-adrenergic receptor |
| β | β-adrenergic receptor |
| 2-APB | 2-Aminoethoxydiphenyl borate |
| 4-AP | 4-Aminopyridine |
| 5-HT | 5-Hydroxytryptamine |
| AA | Arachidonic acid |
| AC | Adenylyl cyclase |
| AHA | American Heart Association |
| AMP | Adenosine monophosphate |
| Akt | Protein kinase B |
| ATP | Adenosine triphosphate |
| AT | Angiotensin receptor |
| B2 | Bradykinin receptor B2 |
| BaCl2 | Barium Chloride |
| BKca | Big conductance calcium-activated potassium channel |
| BP | Blood pressure |
| CALCRL | Calcitonin receptor-like |
| cAMP | Cyclic adenosine monophosphate |
| CBS | Cystathionine β-synthase |
| cGMP | Cyclic guanosine monophosphate |
| CICR | Calcium-induced calcium release |
| COX | Cyclooxygenase |
| CSE | Cystathionine γ-lyase |
| CYP | Cytochrome P450 |
| DAG | Diacylglycerol |
| DMSO | Dimethyl sulfoxide |
| eNOS | Endothelium nitric oxide synthase |
| EDHF | Endothelium-derived hyperpolarizing factor |
| EDRFs | Endothelium-derived relaxing factors |
| EETs | Epoxyeicosatrienoic acids |
| ETR | Endothelin receptor |
| Gd3+ | Gadolinium |
| GDP | Guanosine diphosphate |
| GEF | Guanine nucleotide exchange factor |
| GMP | Guanosine monophosphate |
| GPCRs | G protein-coupled receptors |
| GTP | Guanosine triphosphate |
| H2S | Hydrogen sulfide |
| IKca | Intermediate conductance calcium-activated potassium channel |
| IP | Prostacyclin receptor |
| IP3 | Inositol triphosphate |
| iNOS | Inducible nitric oxide synthase |
| JNC7 | Seventh report of Joint National Committee |
| KATP | ATP-sensitive potassium channel |
| Kca | Calcium-activated potassium channel |
| Kir | Inwardly-rectifying potassium channel |
| Kv | Voltage-activated potassium channel |
| L-NAME | L-NG-Nitroarginine methyl ester |
| M3 | Muscarinic receptor M3 |
| MB | Methylene blue |
| MLCK | Myosin light-chain kinase |
| MLCP | Myosin light-chain phosphatase |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| nNOS | Neuronal nitric oxide synthase |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| ODQ | 1H-[1,2,4] oxadiazolo [4,3,-a] quinoxalin-1-one |
| PDE3 | Phosphodiesterase 3 |
| PDE5 | Phosphodiesterase 5 |
| PGH2 | Prostaglandin H2 |
| PGI2 | Prostacyclin |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PKG | Protein kinase G |
| PLA2 | Phospholipase A2 |
| PLC | Phospholipase C |
| ROCC | Receptor-operated calcium channel |
| RyRs | Ryanodine receptors |
| SERCA | Sacro/endoplasmic reticulum Ca2+-ATPase |
| sGC | Soluble guanylyl cyclase |
| SKca | Small conductance calcium-activated potassium channel |
| SOCC | Store-operated calcium channel |
| SR | Sacroplasmic reticulum |
| TRPV4 | Transient receptor potential vanilloid 4 |
| TxA2 | Thromboxane |
| VOCC | Voltage-operated calcium channel |
| VSMCs | Vascular smooth muscle cells |
| WHO | World Health Organization |
References
- Rahman, A.R.A. Clinical Practice Guidelines on Management of Hypertension, 4th ed.; Malaysian Society of Hypertension, Ministry of Health Malaysia, Academy of Medicine Malaysia: Putrajaya, Malaysia, 2013; p. 75. [Google Scholar]
- Loh, Y.C. New approaches for hypertension treatment. Int. J. Complement. Altern. Med. 2017. [Google Scholar] [CrossRef]
- Loh, Y.C.; Tan, C.S.; Ch’ng, Y.S.; Ahmad, M.; Asmawi, M.Z.; Yam, M.F. Overview of antagonists used for determining the mechanisms of action employed by potential vasodilators with their suggested signaling pathways. Molecules 2016, 21, 495. [Google Scholar] [CrossRef] [PubMed]
- Rameshrad, M.; Babaei, H.; Azarmi, Y.; Fouladia, D.F. Rat aorta as a pharmacological tool for in vitro and in vivo studies. Life Sci. 2016, 145, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.C.; Ch’ng, Y.S.; Tan, C.S.; Ahmad, M.; Asmawi, M.Z.; Yam, M.F. Mechanisms of action of Uncariarhyn chophylla ethanolic extract for its vasodilatory effects. J. Med. Food 2017, 20, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.C.; Tan, C.S.; Ch’ng, Y.S.; Ahmad, M.; Asmawi, M.Z.; Yam, M.F. Vasodilatory effects of combined traditional Chinese medicinal herbs in optimized ratio. J. Med. Food 2017, 20, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Jakala, P.; Pere, E.; Lehtinen, R.; Turpeinen, A.; Korpela, R.; Vapaatalo, H. Cardiovascular activity of milk casein-derived tripeptides and plant sterols in spontaneously hypertensive rats. J. Physiol. Pharmacol. 2009, 60, 11–20. [Google Scholar] [PubMed]
- Yildiz, O.; Gul, H.; Seyrek, M. Pharmacology of Arterial Grafts for Coronary Artery Bypass Surgery; INTECH Open Access Publisher: Rijeka, Croatia, 2013. [Google Scholar]
- Quillon, A.; Fromy, B.; Debret, R. Endothelium microenvironment sensing leading to nitric oxide mediated vasodilation: A review of nervous and biomechanical signals. Nitric Oxide 2015, 45, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S. A2. Nitric oxide and bioenergetics: Physiology and pathophysiology. Nitric Oxide 2007, 17, 9. [Google Scholar] [CrossRef]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zou, A.P.; Campbell, W.B. Regulation of potassium channels in coronary arterial smooth muscle by endothelium-derived vasodilators. Hypertension 1997, 29, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-J.; Tod, M.L.; Rubin, L.J.; Blaustein, M.P. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 10489–10494. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L.; Kelly, R.A.; Marsden, P.A.; Smith, T.W.; Michel, T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc. Natl. Acad. Sci. USA 1993, 90, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, S.; Leopold, E.; Schmidt, K.; Brunner, F.; Mayer, B. Inhibition of nitric oxide synthesis by NG-nitro-l-arginine methyl ester (L-name): Requirement for bioactivation to the free acid, NG-nitro-l-arginine. Br. J. Pharmacol. 1996, 118, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Berumen, L.C.; Rodriguez, A.; Miledi, R.; Garcia-Alcocer, G. Serotonin receptors in hippocampus. Sci. World J. 2012, 2012, 823493. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.E.; Nichols, C.D. Serotonin receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Dannhardt, G.; Kiefer, W. Cyclooxygenase inhibitors--current status and future prospects. Eur. J. Med. Chem. 2001, 36, 109–126. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Scotland, R.S.; Madhani, M.; Chauhan, S.; Moncada, S.; Andresen, J.; Nilsson, H.; Hobbs, A.J.; Ahluwalia, A. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: Key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation 2005, 111, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’sa crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Dams Ramos, C.M.; Matin, N.; Dorrance, A.M. The effects of hypertension on the cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1598–H1614. [Google Scholar] [CrossRef] [PubMed]
- Quyyumi, A.A.; Ozkor, M. Vasodilation by hyperpolarization: Beyond no. Hypertension 2006, 48, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Hydrogen sulfide: A new EDRF. Kidney Int. 2009, 76, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Menice, C.B.; Laporte, R.; Morgan, K.G. Mechanisms of smooth muscle contraction. Physiol. Rev. 1996, 76, 967–1003. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; Russel, R.J.; Cellek, S.; Lizasoain, I.; Su, Y.; Darley-Usmar, V.M.; Radomski, M.W.; Moncada, S. cGMP mediates the vascular and platelet actions of nitric oxide: Confirmation using an inhibitor of the soluble guanylyl cyclase. Proc. Natl. Acad. Sci. USA 1996, 93, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.J.; Knych, E.T., Jr.; Herzig, T.C.; Drewett, J.G. Selective guanylyl cyclase inhibitor reverses nitric oxide-induced vasorelaxation. Hypertension 1997, 29, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Schrammel, A.; Behrends, S.; Schmidt, K.; Koesling, D.; Mayer, B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol. Pharmacol. 1996, 50, 1–5. [Google Scholar] [PubMed]
- Kontos, H.A.; Wei, E.P. Hydroxyl radical-dependent inactivation of guanylate cyclase in cerebral arterioles by methylene blue and by LY83583. Stroke 1993, 24, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Brunner, F.; Schmidt, K. Inhibition of nitric oxide synthesis by methylene blue. Biochem. Pharmacol. 1993, 45, 367–374. [Google Scholar] [CrossRef]
- Bowen, R.; Haslam, R.J. Effects of nitrovasodilators on platelet cyclic nucleotide levels in rabbit blood; role for cyclic AMP in synergistic inhibition of platelet function by sin-1 and prostaglandin E1. J. Cardiovasc. Pharmacol. 1991, 17, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Bouschet, T.; Perez, V.; Fernandez, C.; Bockaert, J.; Eychene, A.; Journot, L. Stimulation of the ERK pathway by GTP-loaded rap1 requires the concomitant activation of RAS, protein kinase c, and protein kinase a in neuronal cells. J. Biol. Chem. 2003, 278, 4778–4785. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.P. The mechanism of protein kinase c activation. Trends Neurosci. 1989, 12, 425–432. [Google Scholar] [CrossRef]
- Wall, M.E.; Francis, S.H.; Corbin, J.D.; Grimes, K.; Richie-Jannetta, R.; Kotera, J.; Macdonald, B.A.; Gibson, R.R.; Trewhella, J. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 2380–2385. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Valtcheva, N.; Nestorov, P.; Beck, A.; Russwurm, M.; Hillenbrand, M.; Weinmeister, P.; Feil, R. The commonly used cGMP-dependent protein kinase type i (cGKI) inhibitor rp-8-br-pet-cgmps can activate cGKI in vitro and in intact cells. J. Biol. Chem. 2009, 284, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Walaas, O.; Jahnsen, T.; Walaas, S.I.; Hansson, V. The 1992 Nobel Prize in physiology and medicine. Tidsskrift Den Norske Laegeforening 1992, 112, 3775. [Google Scholar] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Yuen, J.W.; Poon, L.S.; Chan, A.S.; Yu, F.W.; Lo, R.K.; Wong, Y.H. Activation of stat3 by specific G-alpha subunits and multiple G-betagamma dimers. Int. J. Biochem. Cell Biol. 2010, 42, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Claeysen, S.; Bécamel, C.; Dumuis, A.; Marin, P. Neuronal 5-HT metabotropic receptors: Fine-tuning of their structure, signaling, and roles in synaptic modulation. Cell Tissue Res. 2006, 326, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yildiz, O.; Purdy, R.E. Phenylephrine precontraction increases the sensitivity of rabbit femoral artery to serotonin by enabling 5-HT1-like receptors. J. Cardiovasc. Pharmacol. 2000, 35, 863–870. [Google Scholar] [CrossRef] [PubMed]
- De Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Goodman, L.S.; Hardman, J.G.; Limbird, L.E.; Gilman, A.G. Goodman and Gilman’s the Pharmacological Basis of Therapeutics; McGraw-Hill Medical Pub: New York, NY, USA, 2001. [Google Scholar]
- Ishii, M.; Kurachi, Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 2006, 12, 3573–3581. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, R. Cardiovascular Physiology Concepts; Wolters Kluwer Health: Philadelphia, PA, USA, 2011. [Google Scholar]
- Qin, K.; Sethi, P.R.; Lambert, N.A. Abundance and stability of complexes containing inactive G protein-coupled receptors and g proteins. FASEB J. 2008, 22, 2920–2927. [Google Scholar] [CrossRef] [PubMed]
- Littleton, J.T.; Ganetzky, B. Ion channels and synaptic organization: Analysis of the drosophila genome. Neuron 2000, 26, 35–43. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Hollenberg, M.; Triggle, C.; Ding, H. The contribution of D-tubocurarine-sensitive and apamin-sensitive K-channels to EDHF-mediated relaxation of mesenteric arteries from eNOS−/− mice. J. Cardiovasc. Pharmacol. 2012, 59, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Eichler, I.; Wibawa, J.; Grgic, I.; Knorr, A.; Brakemeier, S.; Pries, A.R.; Hoyer, J.; Köhler, R. Selective blockade of endothelial Ca2+-activated small-and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 2003, 138, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Marchenko, S.M.; Sage, S.O. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 1996, 492, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Vanhoutte, P. EDHF: The Complete Story; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Gautam, M.; Gojova, A.; Barakat, A.I. Flow-activated ion channels in vascular endothelium. Cell Biochem. Biophys. 2006, 46, 277–284. [Google Scholar] [CrossRef]
- Robertson, B.E.; Schubert, R.; Hescheler, J.; Nelson, M.T. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am. J. Physiol. 1993, 265, C299–C303. [Google Scholar] [CrossRef] [PubMed]
- Scornik, F.S.; Codina, J.; Birnbaumer, L.; Toro, L. Modulation of coronary smooth muscle Kca channels by Gs alpha independent of phosphorylation by protein kinase A. Am. J. Physiol. 1993, 265, H1460–H1465. [Google Scholar] [CrossRef] [PubMed]
- Barfod, E.T.; Moore, A.L.; Lidofsky, S.D. Cloning and functional expression of a liver isoform of the small conductance Ca2+-activated K+ channel SK3. Am. J. Physiol. Cell Physiol. 2001, 280, C836–C842. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, B.; Maylie, J.; Adelman, J.P.; Marrion, N.V. Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 1998, 111, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.M.; Fakler, B.; Rivard, A.; Wayman, G.; Johnson-Pais, T.; Keen, J.E.; Ishii, T.; Hirschberg, B.; Bond, C.T.; Lutsenko, S.; et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 1998, 395, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Garcia Pascual, A.; Labadia, A.; Jimenez, E.; Costa, G. Endothelium-dependent relaxation to acetylcholine in bovine oviductal arteries: Mediation by nitric oxide and changes in apamin-sensitive K+ conductance. Br. J. Pharmacol. 1995, 115, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Rivard, A.F.; Bachinger, H.P.; Adelman, J.P. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 2001, 410, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995, 268, C799–C822. [Google Scholar] [CrossRef] [PubMed]
- Robertson, B.E.; Nelson, M.T. Aminopyridine inhibition and voltage dependence of K+ currents in smooth muscle cells from cerebral arteries. Am. J. Physiol. 1994, 267, C1589–C1597. [Google Scholar] [CrossRef] [PubMed]
- Aiello, E.A.; Walsh, M.P.; Cole, W.C. Phosphorylation by protein kinase A enhances delayed rectifier K+ current in rabbit vascular smooth muscle cells. Am. J. Physiol. 1995, 268, H926–H934. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.C.; Clement-Chomienne, O.; Aiello, E.A. Regulation of 4-aminopyridine-sensitive, delayed rectifier K+ channels in vascular smooth muscle by phosphorylation. Biochem. Cell Biol. 1996, 74, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ganong, W.F. The general and cellular basis of medical physiology. Rev. Med. Physiol. 1993, 16, 36–39. [Google Scholar]
- Tucker, S.J.; Baukrowitz, T. How highly charged anionic lipids bind and regulate ion channels. J. Gen. Physiol. 2008, 131, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Dora, K.; Gardener, M.; Garland, C.; Weston, A. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998, 396, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Weston, A.H. Pharmacology of the potassium channel openers. Cardiovasc. Drugs Ther. 1995, 9, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, S.J.; Ashcroft, F.M. Properties and functions of ATP-sensitive K-channels. Cell. Signal. 1990, 2, 197–214. [Google Scholar] [CrossRef]
- Boyd, A.E., 3rd; Aguilar-Bryan, L.; Nelson, D.A. Molecular mechanisms of action of glyburide on the beta cell. Am. J. Med. 1990, 89, 3S–10S. [Google Scholar] [CrossRef]
- Standen, N.B.; Quayle, J.M.; Davies, N.W.; Brayden, J.E.; Huang, Y.; Nelson, M.T. Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science 1989, 245, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kawano, K.; Yoshiyama, S.; Kawamichi, H.; Wang, X.; Nakamura, A.; Kohama, K. Myosin light chain kinase stimulates smooth muscle myosin ATPase activity by binding to the myosin heads without phosphorylating the myosin light chain. Biochem. Biophys. Res. Commun. 2003, 305, 16–21. [Google Scholar] [CrossRef]
- Webb, R.C. Smooth muscle contraction and relaxation. Adv. Physiol. Educ. 2003, 27, 201–206. [Google Scholar] [CrossRef] [PubMed]
- McFadzean, I.; Gibson, A. The developing relationship between receptor-operated and store-operated calcium channels in smooth muscle. Br. J. Pharmacol. 2002, 135, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Furberg, C.D.; Psaty, B.M.; Meyer, J.V. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation 1995, 92, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Elementary and global aspects of calcium signalling. J. Physiol. 1997, 499, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.; McFadzean, I.; Wallace, P.; Wayman, C.P. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends Pharmacol. Sci. 1998, 19, 266–269. [Google Scholar] [PubMed]
- Landsberg, J.W.; Yuan, J.X. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol. Sci. 2004, 19, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr.; Broad, L.M.; Braun, F.J.; Lievremont, J.P.; Bird, G.S. Mechanisms of capacitative calcium entry. J. Cell Sci. 2001, 114, 2223–2229. [Google Scholar] [PubMed]
- Chemaly, E.R.; Bobe, R.; Adnot, S.; Hajjar, R.J.; Lipskaia, L. Sarco (endo) plasmic reticulum calcium ATPases (SERCA) isoforms in the normal and diseased cardiac, vascular and skeletal muscle. J. Cardiovasc. Dis. Diagn. 2013, 1, 113. [Google Scholar]
- Swietach, P.; Spitzer, K.W.; Vaughan-Jones, R.D. Ca2+-mobility in the sarcoplasmic reticulum of ventricular myocytes is low. Biophys. J. 2008, 95, 1412–1427. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.W. Physiology, Biophysics, and Biomedical Engineering; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Rogers, T.B.; Inesi, G.; Wade, R.; Lederer, W. Use of thapsigargin to study Ca2+ homeostasis in cardiac cells. Biosci. Rep. 1995, 15, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Loh, Y.C.; Ng, C.H.; Ch’ng, Y.S.; Asmawi, M.Z.; Ahmad, M.; Yam, M.F. Anti-hypertensive and vasodilatory effects of amended banxia baizhu tianma tang. Biomed. Pharmacother. 2017, 97, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Ch’ng, Y.S.; Loh, Y.C.; ZainiAsmawi, M.; Ahmad, M.; Yam, M.F. Vasorelaxation effect of Glycyrrhiza euralensis through the endothelium-dependent pathway. J. Ethnopharmacol. 2017, 199, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.C.; Tan, C.S.; Ch’ng, Y.S.; Ahmad, M.; Ng, C.H.; Yam, M.F. Overview of signaling mechanism pathways employed by BPaid in vasodilatory activity. J. Med. Food 2017, 20, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Ch’ng, Y.S.; Loh, Y.C.; Tan, C.S.; Ahmad, M.; Asmawi, M.Z.; Wan Omar, W.M.; Yam, M.F. Vasorelaxant properties of Vernonia amygdalina ethanol extract and its possible mechanism. Pharm. Biol. 2017, 55, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.F.; Tan, C.S.; Ahmad, M.; Shibao, R. Mechanism of vasorelaxation induced by eupatorin in the rats aortic ring. Eur. J. Pharmacol. 2016, 789, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.F.; Tan, C.S.; Ahmad, M.; Ruan, S. Vasorelaxant action of the chloroform fraction of Orthosiphon stamineus via NO/cGMP pathway, potassium and calcium channels. Am. J. Chin. Med. 2016, 44, 1413–1439. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, Y.C.; Tan, C.S.; Ch’ng, Y.S.; Yeap, Z.Q.; Ng, C.H.; Yam, M.F. Overview of the Microenvironment of Vasculature in Vascular Tone Regulation. Int. J. Mol. Sci. 2018, 19, 120. https://doi.org/10.3390/ijms19010120
Loh YC, Tan CS, Ch’ng YS, Yeap ZQ, Ng CH, Yam MF. Overview of the Microenvironment of Vasculature in Vascular Tone Regulation. International Journal of Molecular Sciences. 2018; 19(1):120. https://doi.org/10.3390/ijms19010120
Chicago/Turabian StyleLoh, Yean Chun, Chu Shan Tan, Yung Sing Ch’ng, Zhao Qin Yeap, Chiew Hoong Ng, and Mun Fei Yam. 2018. "Overview of the Microenvironment of Vasculature in Vascular Tone Regulation" International Journal of Molecular Sciences 19, no. 1: 120. https://doi.org/10.3390/ijms19010120
APA StyleLoh, Y. C., Tan, C. S., Ch’ng, Y. S., Yeap, Z. Q., Ng, C. H., & Yam, M. F. (2018). Overview of the Microenvironment of Vasculature in Vascular Tone Regulation. International Journal of Molecular Sciences, 19(1), 120. https://doi.org/10.3390/ijms19010120