Statin and Bisphosphonate Induce Starvation in Fast-Growing Cancer Cell Lines

 ,
,
Abstract
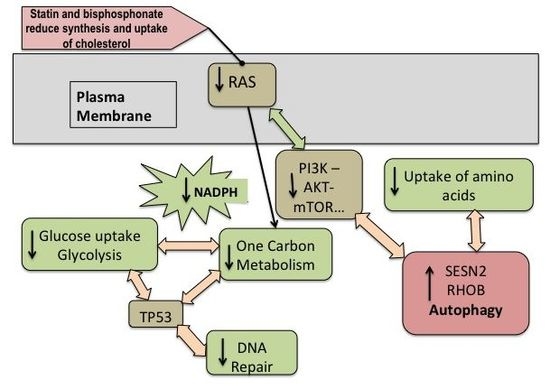
1. Introduction
2. Results
2.1. Cell Cycle
2.2. Influence of Statins and Bisphosphonates on NADP(H) Production
2.3. Autophagy
2.4. Histone Demethylation as a Sign of Starvation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and NADP+/NADPH Analyses
4.2. Analysis of Gene Expression and Transcriptomics
4.3. Flow Cytometry Analysis
4.4.Protein Isolation and Immunoblotting
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qi, X.F.; Zheng, L.; Lee, K.J.; Kim, D.H.; Kim, C.S.; Cai, D.Q.; Wu, Z.; Qin, J.W.; Yu, Y.H.; Kim, S.K. Hmg-coa reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. 2013, 4, e518. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Kim, D.H.; Yoon, Y.S.; Li, J.H.; Jin, D.; Teng, Y.C.; Kim, S.K.; Lee, K.J. Fluvastatin inhibits expression of the chemokine MDC/CCL22 induced by interferon-γ in HaCaT cells, a human keratinocyte cell line. Br. J. Pharmacol. 2009, 157, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Kotamraju, S.; Williams, C.L.; Kalyanaraman, B. Statin-induced breast cancer cell death: Role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007, 67, 7386–7394. [Google Scholar] [CrossRef] [PubMed]
- Koyuturk, M.; Ersoz, M.; Altiok, N. Simvastatin induces apoptosis in human breast cancer cells: P53 and estrogen receptor independent pathway requiring signalling through JNK. Cancer Lett. 2007, 250, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Thaler, R.; Gerner, C.; Grunt, T.; Proestling, K.; Haider, F.; Varga, F. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet. 2015, 208, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Seo, T.; Park, T.; Yagyu, H.; Hu, Y.; Son, N.H.; Augustus, A.S.; Vikramadithyan, R.K.; Ramakrishnan, R.; Pulawa, L.K.; et al. Effects of lipoprotein lipase and statins on cholesterol uptake into heart and skeletal muscle. J. Lipid Res. 2007, 48, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Trupp, M.; Zhu, H.; Wikoff, W.R.; Baillie, R.A.; Zeng, Z.B.; Karp, P.D.; Fiehn, O.; Krauss, R.M.; Kaddurah-Daouk, R. Metabolomics reveals amino acids contribute to variation in response to simvastatin treatment. PLoS ONE 2012, 7, e38386. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, H. Bisphosphonates: Mechanisms of action. Endocr. Rev. 1998, 19, 80–100. [Google Scholar] [CrossRef] [PubMed]
- Thaler, R.; Spitzer, S.; Karlic, H.; Berger, C.; Klaushofer, K.; Varga, F. Ibandronate increases the expression of the pro-apoptotic gene FAS by epigenetic mechanisms in tumor cells. Biochem. Pharmacol. 2013, 85, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ahern, T.P.; Lash, T.L.; Damkier, P.; Christiansen, P.M.; Cronin-Fenton, D.P. Statins and breast cancer prognosis: Evidence and opportunities. Lancet Oncol. 2014, 15, e461–468. [Google Scholar] [CrossRef]
- Wang, A.; Aragaki, A.K.; Tang, J.Y.; Kurian, A.W.; Manson, J.E.; Chlebowski, R.T.; Simon, M.; Desai, P.; Wassertheil-Smoller, S.; Liu, S.; et al. Statin use and all-cancer survival: Prospective results from the women’s health initiative. Br. J. Cancer 2016, 115, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Manthravadi, S.; Shrestha, A.; Madhusudhana, S. Impact of statin use on cancer recurrence and mortality in breast cancer: A systematic review and meta-analysis. Int. J. Cancer 2016, 139, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Mansourian, M.; Haghjooy-Javanmard, S.; Eshraghi, A.; Vaseghi, G.; Hayatshahi, A.; Thomas, J. Statins use and risk of breast cancer recurrence and death: A systematic review and meta-analysis of observational studies. J. Pharm. Pharm. Sci. 2016, 19, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, N.; Schmidt, R.; Stockler, M. Bisphosphonates for breast cancer. Cochrane Database Syst. Rev. 2005, CD003474. [Google Scholar]
- Han, Y.; Lian, S.; Cui, X.; Meng, K.; Gyorffy, B.; Jin, T.; Huang, D. Potential options for managing LOX+ ER- breast cancer patients. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Liao, Y.B.; Xu, P.; Wei, W.R.; Wang, J. Statin use and mortality of patients with prostate cancer: A meta-analysis. Onco. Targets Ther. 2016, 9, 1689–1696. [Google Scholar] [PubMed]
- Sun, Q.; Arnold, R.S.; Sun, C.Q.; Petros, J.A. A mitochondrial DNA mutation influences the apoptotic effect of statins on prostate cancer. Prostate 2015, 75, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Morote, J.; Celma, A.; Planas, J.; Placer, J.; de Torres, I.; Olivan, M.; Carles, J.; Reventos, J.; Doll, A. Role of serum cholesterol and statin use in the risk of prostate cancer detection and tumor aggressiveness. Int. J. Mol. Sci. 2014, 15, 13615–13623. [Google Scholar] [CrossRef] [PubMed]
- Kuoppala, J.; Lamminpaa, A.; Pukkala, E. Statins and cancer: A systematic review and meta-analysis. Eur. J. Cancer 2008, 44, 2122–2132. [Google Scholar] [CrossRef] [PubMed]
- Reyes, C.; Hitz, M.; Prieto-Alhambra, D.; Abrahamsen, B. Risks and benefits of bisphosphonate therapies. J. Cell Biochem. 2016, 117, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.R.; Galick, H.; Wu, J.M.; Siperstein, M.D. The effect of mevalonic acid deprivation on enzymes of DNA replication in cells emerging from quiescence. Biochem. J. 1992, 288, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lu, X.C.; Yang, H.Y.; Liu, X.F.; Cao, J.; Fan, L. The molecular mechanism of the anticancer effect of atorvastatin: DNA microarray and bioinformatic analyses. Int. J. Mol. Med. 2012, 30, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Li, W.; Sheng, Y.; Li, L.; Huang, Y.; Zhang, Z.; Zhu, T.; Peace, D.; Quigley, J.G.; Wu, W.; et al. The transcription factor FOXM1 is essential for the quiescence and maintenance of hematopoietic stem cells. Nat. Immunol. 2015, 16, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Wang, P.; Cui, J.; Gao, Y.; Xie, K. The roles of foxm1 in pancreatic stem cells and carcinogenesis. Mol. Cancer 2013, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Abraham, B.A. Subtractive cloning of a hybrid human endogenous retrovirus and calbindin gene in the prostate cell line PC3. Cancer Res. 1991, 51, 4107–4110. [Google Scholar] [PubMed]
- Wang, N.P.; To, H.; Lee, W.H.; Lee, E.Y. Tumor suppressor activity of RB and p53 genes in human breast carcinoma cells. Oncogene 1993, 8, 279–288. [Google Scholar] [PubMed]
- Izumi, H.; Yasuniwa, Y.; Akiyama, M.; Yamaguchi, T.; Kuma, A.; Kitamura, N.; Kohno, K. Forced expression of ZNF143 restrains cancer cell growth. Cancers 2011, 3, 3909–3920. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.; Medina, V.; Cricco, G.; Croci, M.; Cocca, C.; Rivera, E.; Bergoc, R.; Martin, G. Glibenclamide inhibits cell growth by inducing G0/G1 arrest in the human breast cancer cell line MDA-MB-231. BMC Pharmacol. Toxicol. 2013, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Holzel, M.; Kohlhuber, F.; Schlosser, I.; Holzel, D.; Luscher, B.; Eick, D. Myc/Max/Mad regulate the frequency but not the duration of productive cell cycles. EMBO Rep. 2001, 2, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luther, G.; Zhang, W.; Nan, G.; Wagner, E.R.; Liao, Z.; Wu, N.; Zhang, H.; Wang, N.; Wen, S.; et al. The EF hand calcium-binding protein S100A4 regulates the proliferation, survival and differentiation potential of human osteosarcoma cells. Cell Physiol. Biochem. 2013, 32, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Lin, C.W.; Yang, J.S.; Hsieh, M.J.; Yang, S.F.; Lu, K.H. Zoledronate blocks geranylgeranylation not farnesylation to suppress human osteosarcoma U2OS cells metastasis by EMT via Rho A activation and FAK-inhibited JNK and p38 pathways. Oncotarget 2016, 7, 9742–9758. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Christofk, H.R. Cell cycle: Division enzyme regulates metabolism. Nature 2017, 546, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M.; et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017, 546, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.A.; Lee, D.Y.; Roman, L.J.; Khazim, K.; Gorin, Y. Sestrin 2 and AMPK connect hyperglycemia to NOX4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol. Cell Biol. 2013, 33, 3439–3460. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhu, J.; Huang, H.; Xiang, D.; Li, Y.; Zhang, D.; Li, J.; Wang, Y.; Jin, H.; Jiang, G.; et al. SESN2/Sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy 2016, 12, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sala, D.; Boya, P.; Ramos, I.; Herrera, M.; Stamatakis, K. The C-terminal sequence of RhoB directs protein degradation through an endo-lysosomal pathway. PLoS ONE 2009, 4, e8117. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an APG1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.Y.; Lo, H.L.; Yang, P.M. EZH2 inhibitors transcriptionally upregulate cytotoxic autophagy and cytoprotective unfolded protein response in human colorectal cancer cells. Am. J. Cancer Res. 2016, 6, 1661–1680. [Google Scholar] [PubMed]
- Croonquist, P.A.; Van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant Ras phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K. Ras regulation of DNA-methylation and cancer. Exp. Cell Res. 2008, 314, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Callegari, S.; Oeljeklaus, S.; Warscheid, B.; Dennerlein, S.; Thumm, M.; Rehling, P.; Dudek, J. Phospho-ubiquitin-PARK2 complex as a marker for mitophagy defects. Autophagy 2017, 13, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Pinweha, P.; Rattanapornsompong, K.; Charoensawan, V.; Jitrapakdee, S. Micrornas and oncogenic transcriptional regulatory networks controlling metabolic reprogramming in cancers. Comput. Struct. Biotechnol. J. 2016, 14, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, F.; de Vita, G.; Salvatore, M.; Laukkanen, M.O. Ras oncogene-mediated progressive silencing of extracellular superoxide dismutase in tumorigenesis. BioMed Res. Int. 2015, 2015, 780409. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Hayashi, H.; Kinoshita, K.; Abe, M.; Kuroki, H.; Tokunaga, R.; Tomiyasu, S.; Tanaka, H.; Sugita, H.; Arita, T.; et al. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal cancer. Int. J. Cancer 2014, 135, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Karantanos, T.; Bardhan, K.; Boussiotis, V.A. Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget 2016, 7, 85624–85640. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Cai, J.; Hou, Y.; Huang, Z.; Wang, Z. Role of EZH2 in cancer stem cells: From biological insight to a therapeutic target. Oncotarget 2017, 8, 37974. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Muramatsu, M.; Wang, F.; Tsuchida, R.; Kodama, T.; Minami, T.; Shibuya, M. Increased expression of histone demethylase JHDM1D under nutrient starvation suppresses tumor growth via down-regulating angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 20725–20729. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Hypoxia-inducible histone lysine demethylases: Impact on the aging process and age-related diseases. Aging Dis. 2016, 7, 180–200. [Google Scholar] [PubMed]
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, J.W.; Chun, Y.S. Jumonji histone demethylases as emerging therapeutic targets. Pharmacol. Res. 2016, 105, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Kretzmer, H.; Holdt, L.M.; Juhling, F.; Ammerpohl, O.; Bergmann, A.K.; Northoff, B.H.; Doose, G.; Siebert, R.; Stadler, P.F.; et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016, 6, 37393. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Wu, Y.; Zheng, J.; Linden, J.; Holoshitz, J. Genoprotective pathways: II. Attenuation of oxidative DNA damage by isopentenyl diphosphate. Mutat. Res. 2004, 554, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wong, C.W. Statins enhance peroxisome proliferator-activated receptor γ coactivator-1α activity to regulate energy metabolism. J. Mol. Med. 2010, 88, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Kotsikorou, E.; Sahota, G.; Oldfield, E. Bisphosphonate inhibition of phosphoglycerate kinase: Quantitative structure-activity relationship and pharmacophore modeling investigation. J. Med. Chem. 2006, 49, 6692–6703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.H.; Xu, Z.R.; Zhang, Q.; Yang, Y.M. Simvastatin protects human osteosarcoma cells from oxidative stress-induced apoptosis through mitochondrial-mediated signaling. Mol. Med. Rep. 2012, 5, 483–488. [Google Scholar] [PubMed]
- Ho, W.P.; Chan, W.P.; Hsieh, M.S.; Chen, R.M. Runx2-mediated bcl-2 gene expression contributes to nitric oxide protection against hydrogen peroxide-induced osteoblast apoptosis. J. Cell Biochem. 2009, 108, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Zabala-Letona, A.; Arruabarrena-Aristorena, A.; Martin-Martin, N.; Fernandez-Ruiz, S.; Sutherland, J.D.; Clasquin, M.; Tomas-Cortazar, J.; Jimenez, J.; Torres, I.; Quang, P.; et al. mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 2017, 547, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Laxman, S.; Estill, S.J.; Wang, C.; Wang, Y.; Wang, Y.; Bulut, G.B.; Gao, J.; Huang, L.J.; Tu, B.P. Regulation of hematopoiesis and methionine homeostasis by mTORC1 inhibitor NPRL2. Cell Rep. 2015, 12, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Statins impair glucose uptake in tumor cells. Cancer Biol. Ther. 2013, 14, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Parmigiani, A.; Divakaruni, A.S.; Archer, K.; Murphy, A.N.; Budanov, A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016, 6, 22538. [Google Scholar] [CrossRef] [PubMed]
- Toulis, K.A.; Nirantharakumar, K.; Ryan, R.; Marshall, T.; Hemming, K. Bisphosphonates and glucose homeostasis: A population-based, retrospective cohort study. J. Clin. Endocrinol. Metab. 2015, 100, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Dirat, B.; Laurent, K.; Puissant, A.; Auberger, P.; Budanov, A.; Tanti, J.F.; Bost, F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 2013, 20, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Teets, N.M.; Denlinger, D.L. Autophagy in antarctica: Combating dehydration stress in the world’s southernmost insect. Autophagy 2013, 9, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Wu, C.H.; Huang, C.N.; Lan, K.P.; Chang, W.C.; Wang, C.J. Simvastatin inhibits glucose-stimulated vascular smooth muscle cell migration involving increased expression of rhob and a block of Ras/Akt signal. Cardiovasc. Ther. 2012, 30, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Sun, J.; Pradines, A.; Favre, G.; Adnane, J.; Sebti, S.M. Both farnesylated and geranylgeranylated RhoB inhibit malignant transformation and suppress human tumor growth in nude mice. J. Biol. Chem. 2000, 275, 17974–17978. [Google Scholar] [CrossRef] [PubMed]
- Lebowitz, P.F.; Prendergast, G.C. Non-Ras targets of farnesyltransferase inhibitors: Focus on Rho. Oncogene 1998, 17, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.; Mordec, K.; Waszczuk, J.; Wang, Z.; Lai, J.; Fridlib, M.; Buckley, D.; Kemble, G.; Heuer, T.S. Inhibition of de novo palmitate synthesis by fatty acid synthase induces apoptosis in tumor cells by remodeling cell membranes, inhibiting signaling pathways, and reprogramming gene expression. EBioMedicine 2015, 2, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Jeremiah, S.; Wagstaff, J. Ibandronate reduces endogenous reactive oxygen species levels in cultured prostate cancer and endothelial cells. Microvasc. Res. 2009, 78, 141. [Google Scholar] [CrossRef] [PubMed]
- Itzstein, C.; Coxon, F.P.; Rogers, M.J. The regulation of osteoclast function and bone resorption by small GTPases. Small GTPases 2011, 2, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of nadph oxidases: The role of RAC proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kim, J.H.; Seo, J.H.; Lee, J.; Choi, W.S.; Kim, Y.S. Matrix metalloproteinase-3 causes dopaminergic neuronal death through NOX1-regenerated oxidative stress. PLoS ONE 2014, 9, e115954. [Google Scholar] [CrossRef] [PubMed]
- Ory, B.; Blanchard, F.; Battaglia, S.; Gouin, F.; Redini, F.; Heymann, D. Zoledronic acid activates the DNA S-phase checkpoint and induces osteosarcoma cell death characterized by apoptosis-inducing factor and endonuclease-G translocation independently of p53 and retinoblastoma status. Mol. Pharmacol. 2007, 71, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Gavin, M.; Dahiya, A.; Postigo, A.A.; Ma, D.; Luo, R.X.; Harbour, J.W.; Dean, D.C. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 2000, 101, 79–89. [Google Scholar] [CrossRef]
- Murayama, T.; Kawasoe, Y.; Yamashita, Y.; Ueno, Y.; Minami, S.; Yokouchi, M.; Komiya, S. Efficacy of the third-generation bisphosphonate risedronate alone and in combination with anticancer drugs against osteosarcoma cell lines. Anticancer Res. 2008, 28, 2147–2154. [Google Scholar] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, T.P.; Bjorklund, M. The mevalonate pathway as a metabolic requirement for autophagy-implications for growth control, proteostasis, and disease. Mol. Cell Oncol. 2016, 3, e1143546. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Linher-Melville, K.; Tsakiridis, T.; Singh, G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS ONE 2012, 7, e32035. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Hu, J.; Jiang, S.; Liu, Y.; Sahin, E.; Zhuang, L.; Fletcher-Sananikone, E.; Colla, S.; Wang, Y.A.; Chin, L.; et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010, 468, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 promotes Lkb1-mediated AMPK activation in the ischemic heart. FASEB J. 2015, 29, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D.; Saunders, T.L.; Morrison, S.J. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 2010, 468, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The Lkb1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Verlinden, L.; Meyer, M.B.; Gijsbers, R.; Pike, J.W.; Bouillon, R.; Verstuyf, A. 1,25-Dihydroxyvitamin D3 and the aging-related Forkhead Box O and Sestrin proteins in osteoblasts. J. Steroid Biochem. Mol. Biol. 2013, 136, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. P53 target genes Sestrin1 and Sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Ro, S.H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective role of Sestrin2 against chronic er stress. Nat. Commun. 2014, 5, 4233. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated Sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Saveljeva, S.; Cleary, P.; Mnich, K.; Ayo, A.; Pakos-Zebrucka, K.; Patterson, J.B.; Logue, S.E.; Samali, A. Endoplasmic reticulum stress-mediated induction of Sestrin 2 potentiates cell survival. Oncotarget 2016, 7, 12254–12266. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.X.; Papadia, S.; Bell, K.F.; Hardingham, G.E. Role of histone acetylation in the activity-dependent regulation of sulfiredoxin and Sestrin 2. Epigenetics 2009, 4, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Weber, J.D. Deacetylation of the retinoblastoma tumour suppressor protein by SIRT1. Biochem. J. 2007, 407, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Cam, M.; Bid, H.K.; Xiao, L.; Zambetti, G.P.; Houghton, P.J.; Cam, H. p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J. Biol. Chem. 2014, 289, 4083–4094. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Zhang, P.; Chen, W.D.; Li, D.D.; Wu, X.Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.K.; et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Bharati, I.S.; Fogal, V.; Pastorino, S.; Cong, X.; Nomura, N.; Gallagher, M.; Abbasi, T.; et al. Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med. 2014, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Pandi, N.S.; Manimuthu, M.; Harunipriya, P.; Murugesan, M.; Asha, G.V.; Rajendran, S. In silico analysis of expression pattern of a Wnt/β-catenin responsive gene anln in gastric cancer. Gene 2014, 545, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yue, W.; Zhu, M.J.; Sreejayan, N.; Du, M. Amp-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of β-catenin at Ser 552. Biochem. Biophys. Res. Commun. 2010, 395, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, K.; Gremel, G.; Ryden, L.; Ponten, V.; Uhlen, M.; Dimberg, A.; Jirstrom, K.; Ponten, F. ANLN is a prognostic biomarker independent of Ki-67 and essential for cell cycle progression in primary breast cancer. BMC Cancer 2016, 16, 904. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Parmigiani, A.; Yang, C.; Budanov, A.V. Sestrin2 facilitates death receptor-induced apoptosis in lung adenocarcinoma cells through regulation of XIAP degradation. Cell Cycle 2015, 14, 3231–3241. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mo, Y.; Midorikawa, K.; Zhang, Z.; Huang, G.; Ma, N.; Zhao, W.; Hiraku, Y.; Oikawa, S.; Murata, M. The potent tumor suppressor miR-497 inhibits cancer phenotypes in nasopharyngeal carcinoma by targeting ANLN and HSPA4L. Oncotarget 2015, 6, 35893–35907. [Google Scholar] [PubMed]
- Baxter, J.; Sauer, S.; Peters, A.; John, R.; Williams, R.; Caparros, M.L.; Arney, K.; Otte, A.; Jenuwein, T.; Merkenschlager, M.; et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 2004, 23, 4462–4472. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Van Iersel, M.P.; Kelder, T.; Pico, A.R.; Hanspers, K.; Coort, S.; Conklin, B.R.; Evelo, C. Presenting and exploring biological pathways with pathvisio. BMC Bioinform. 2008, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Varga, F.; Luegmayr, E.; Fratzl-Zelman, N.; Glantschnig, H.; Ellinger, A.; Prinz, D.; Rumpler, M.; Klaushofer, K. Tri-iodothyronine inhibits multilayer formation of the osteoblastic cell line, MC3T3-E1, by promoting apoptosis. J. Endocrinol. 1999, 160, 57–65. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
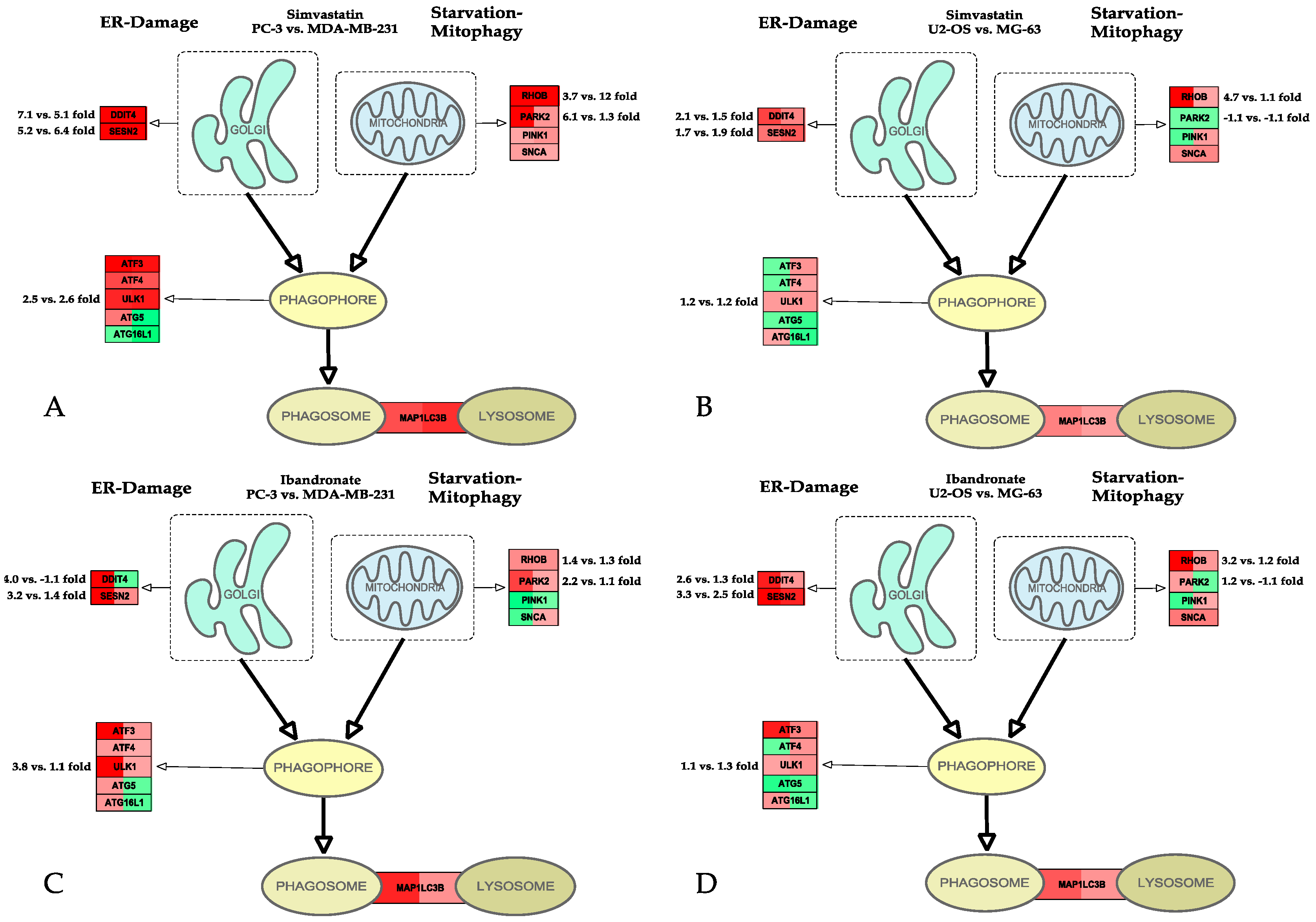{kind=link}
{kind=link}
{kind=link}
| Cell-line | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −2.88 | −3.15 |
| MDA-MB-231 | −2.28 | −1.33 |
| U-2 OS | −1.24 | −1.70 |
| MG-63 | −1.13 | −1.52 |
| CCNA2 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −12.71 | −2.47 |
| MDA-MB-231 | −15.84 | −1.05 |
| U-2 OS | −1.16 | −1.13 |
| MG-63 | −1.12 | −1.03 |
| CCNB1 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −6.68 | −1.87 |
| MDA-MB-231 | −9.2 | −1.1 |
| U-2 OS | −1.02 | −1.62 |
| MG-63 | −1.2 | −1.56 |
| FOXM1 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −5.25 | −2.43 |
| MDA-MB-231 | −4.00 | 1.07 |
| U-2 OS | −1.14 | −1.13 |
| MG-63 | −1.22 | −1.02 |
| Cell Line | Used Conc. (EC50) | Simvastatin NADP+/NADPH Vs. Co | Doubling Time Citation |
|---|---|---|---|
| PC-3 | 1 µM | 1.54 | 13.2 h [27] |
| MDA-MB-231 | 0.5 µM | 2.42 | 24 h [28] |
| U-2 OS | 3 µM | 2.43 | 28 h [29] |
| MG-63 | 10 µM | 4.56 | 38 h [30] |
| Ibandronate | |||
| PC-3 | 50 µM | 1.07 | 13.2 h [27] |
| MDA-MB-231 | 50 µM | 0.95 | 24 h [28] |
| U-2 OS | 50 µM | 2.71 | 28 h [29] |
| MG-63 | 50 µM | 3.57 | 38 h [30] |
| Cell Line—Regulation | Simvastatin | Ibandronate | Overlap |
|---|---|---|---|
| MDA—upregulation | 516 | 35 | 4 |
| MDA—downregulation | 1450 | 60 | 26 |
| MG63—upregulation | 81 | 278 | 37 |
| MG63—downregulation | 252 | 574 | 123 |
| PC3—upregulation | 572 | 290 | 216 |
| PC3—downregulation | 637 | 334 | 228 |
| U2OS—upregulation | 322 | 320 | 78 |
| U2OS—downregulation | 74 | 175 | 21 |
| NOX4 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | 1.53 | 1.22 |
| MDA-MB-231 | 1.52 | −1.07 |
| U-2 OS | −3.22 | 1.83 |
| MG-63 | −1.29 | 1.11 |
| NOS1 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −1.18 | 1.22 |
| MDA-MB-231 | 1.08 | −1.07 |
| U-2 OS | 3.56 | 1.83 |
| MG-63 | 1.19 | 1.11 |
| PDSS1 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −2.43 | −2.65 |
| MDA-MB-231 | −2.61 | −1.03 |
| U-2 OS | −1.31 | 1.00 |
| MG-63 | −1.07 | −1.15 |
| MIR21 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | 1.3 | −1.2 |
| MDA-MB-231 | 1.1 | 1.0 |
| U-2 OS | 1.0 | 3.2 |
| MG-63 | 1.1 | 1.0 |
| EZH2 | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | −1.9 | −1.3 |
| MDA-MB-231 | −2.2 | −1.6 |
| U-2 OS | −1.2 | −1.1 |
| MG-63 | −1.2 | −1.1 |
| KDM7A | Simvastatin | Ibandronate |
|---|---|---|
| PC-3 | 5.6 | 3.8 |
| MDA-MB-231 | 2.0 | 1.0 |
| U-2 OS | 3.2 | 4.0 |
| MG-63 | 1.5 | 1.7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karlic, H.; Haider, F.; Thaler, R.; Spitzer, S.; Klaushofer, K.; Varga, F. Statin and Bisphosphonate Induce Starvation in Fast-Growing Cancer Cell Lines. Int. J. Mol. Sci. 2017, 18, 1982. https://doi.org/10.3390/ijms18091982
Karlic H, Haider F, Thaler R, Spitzer S, Klaushofer K, Varga F. Statin and Bisphosphonate Induce Starvation in Fast-Growing Cancer Cell Lines. International Journal of Molecular Sciences. 2017; 18(9):1982. https://doi.org/10.3390/ijms18091982
Chicago/Turabian StyleKarlic, Heidrun, Florian Haider, Roman Thaler, Silvia Spitzer, Klaus Klaushofer, and Franz Varga. 2017. "Statin and Bisphosphonate Induce Starvation in Fast-Growing Cancer Cell Lines" International Journal of Molecular Sciences 18, no. 9: 1982. https://doi.org/10.3390/ijms18091982
APA StyleKarlic, H., Haider, F., Thaler, R., Spitzer, S., Klaushofer, K., & Varga, F. (2017). Statin and Bisphosphonate Induce Starvation in Fast-Growing Cancer Cell Lines. International Journal of Molecular Sciences, 18(9), 1982. https://doi.org/10.3390/ijms18091982