Tumor Inhibitory Effect of IRCR201, a Novel Cross-Reactive c-Met Antibody Targeting the PSI Domain

Abstract
1. Introduction
2. Results
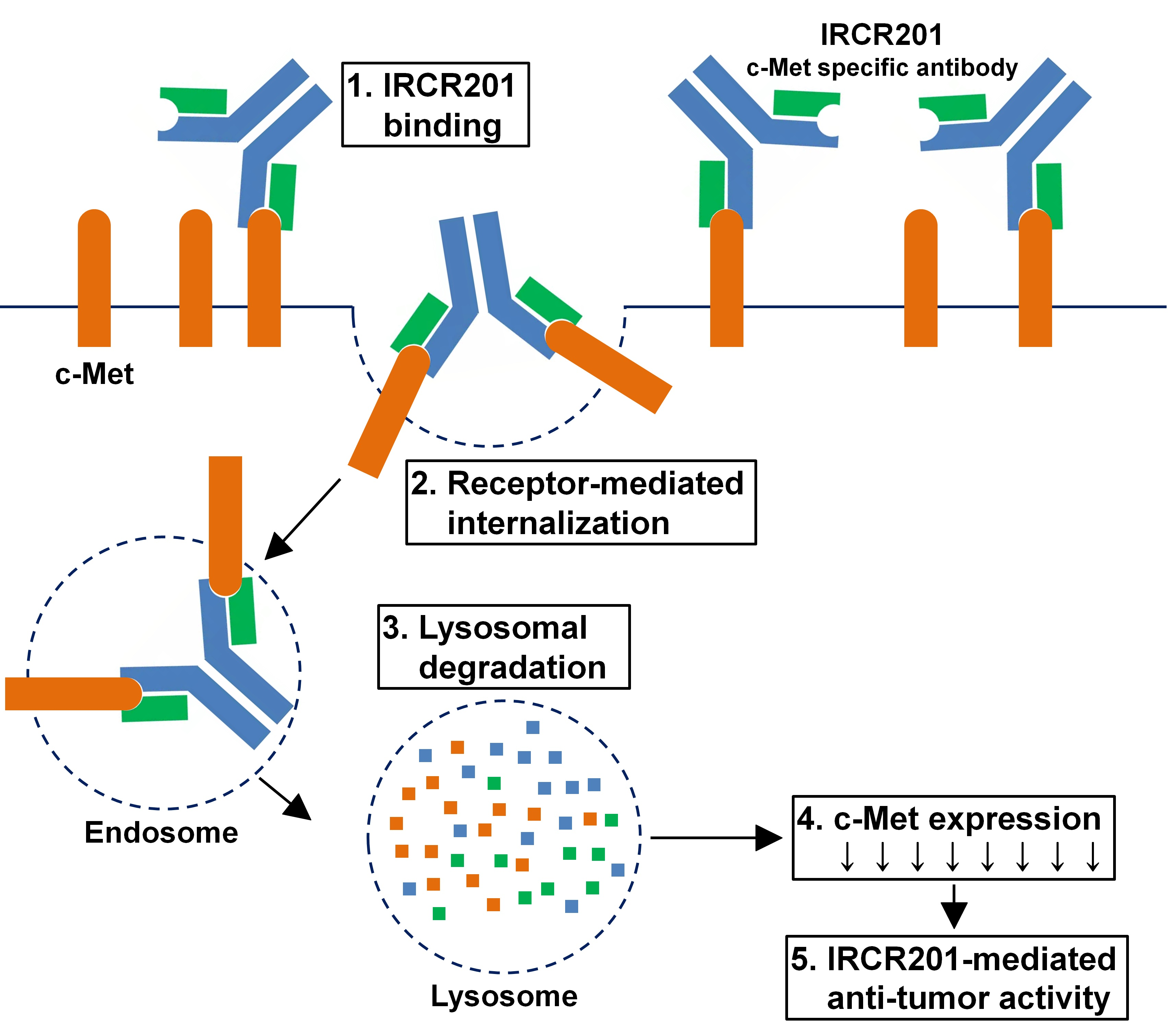
2.1. IRCR201 Exhibits High Affinity to Both Human and Mouse c-Met
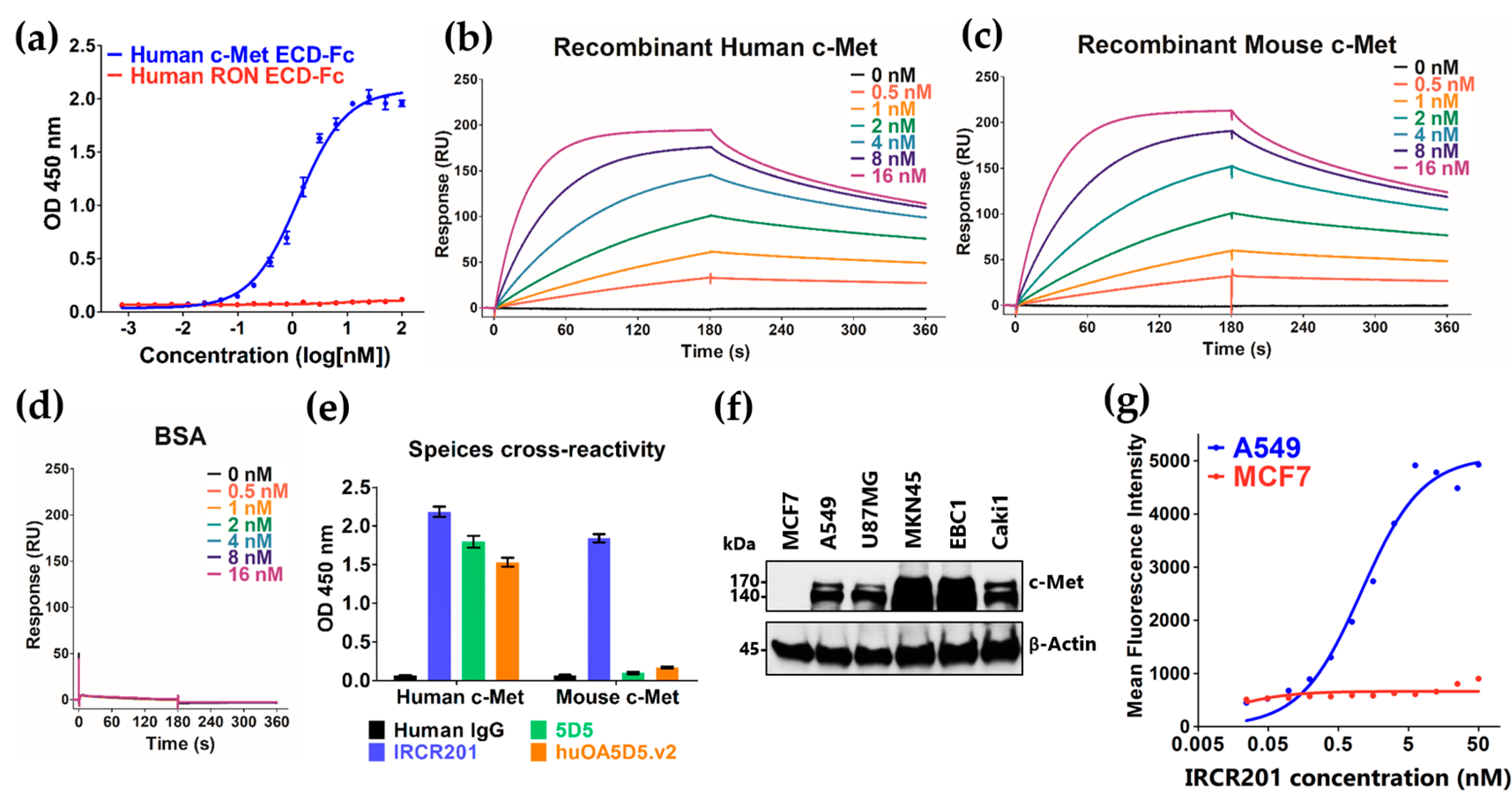
2.2. IRCR201 Specifically Binds to the PSI Domain of c-Met
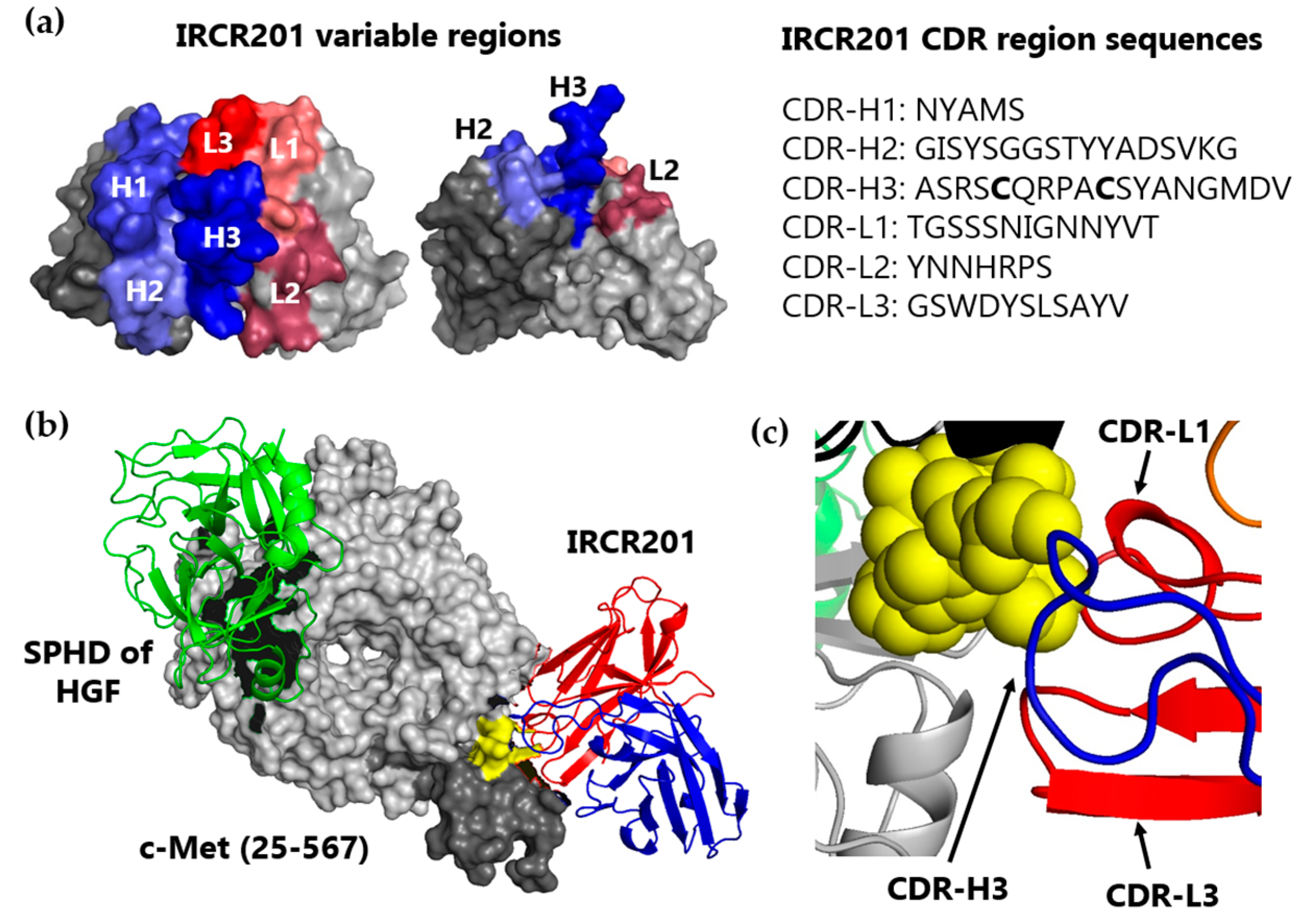
2.3. IRCR201 Docks onto the PSI Domain of c-Met in Computational Modeling Analysis
2.4. IRCR201 Exhibits Low Agonistic Activity
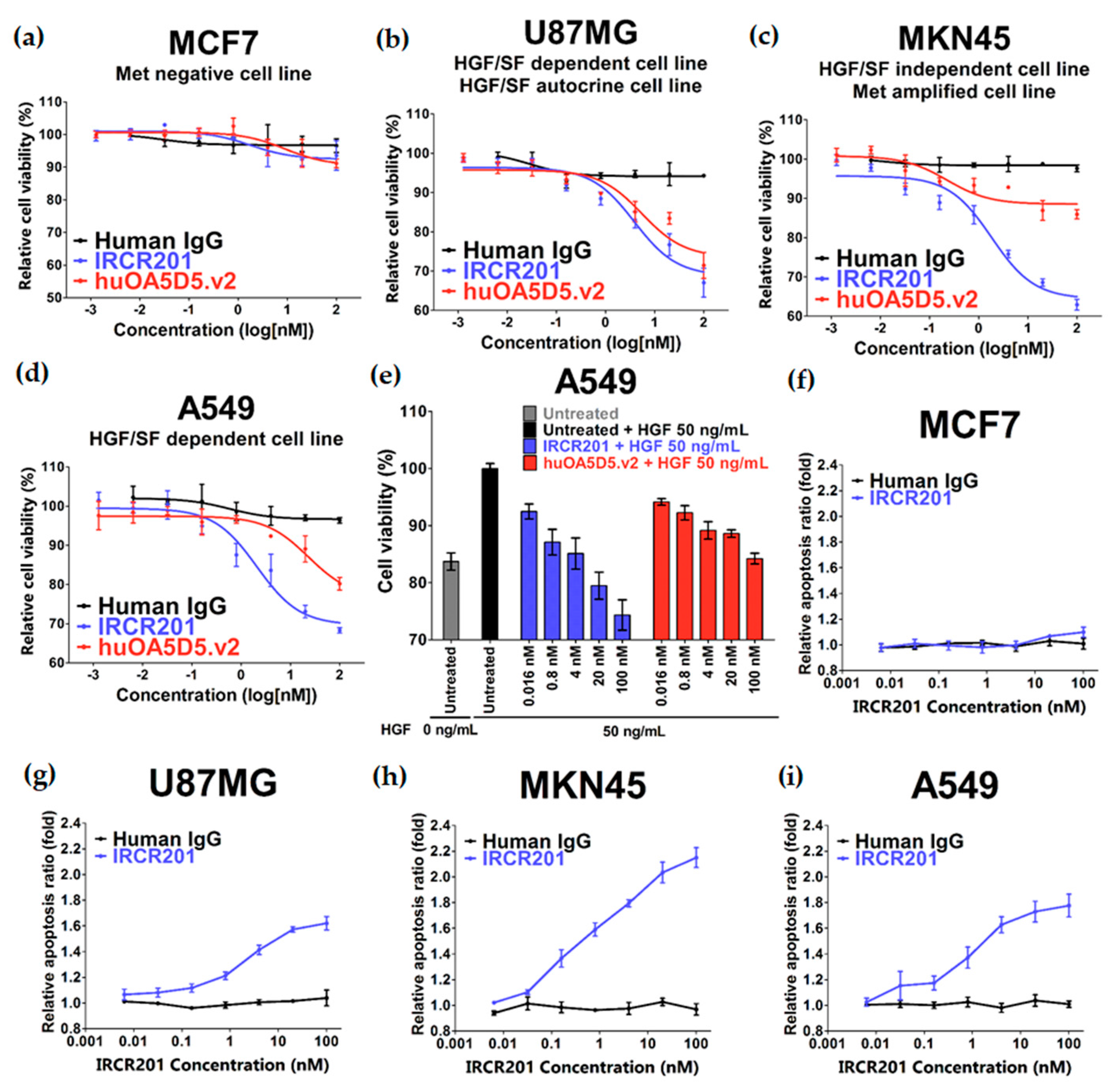
2.5. IRCR201 Impedes Tumor Growth and Induces Cellular Apoptosis
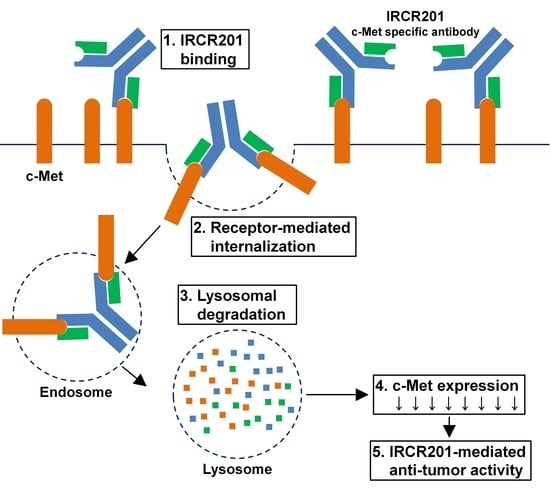
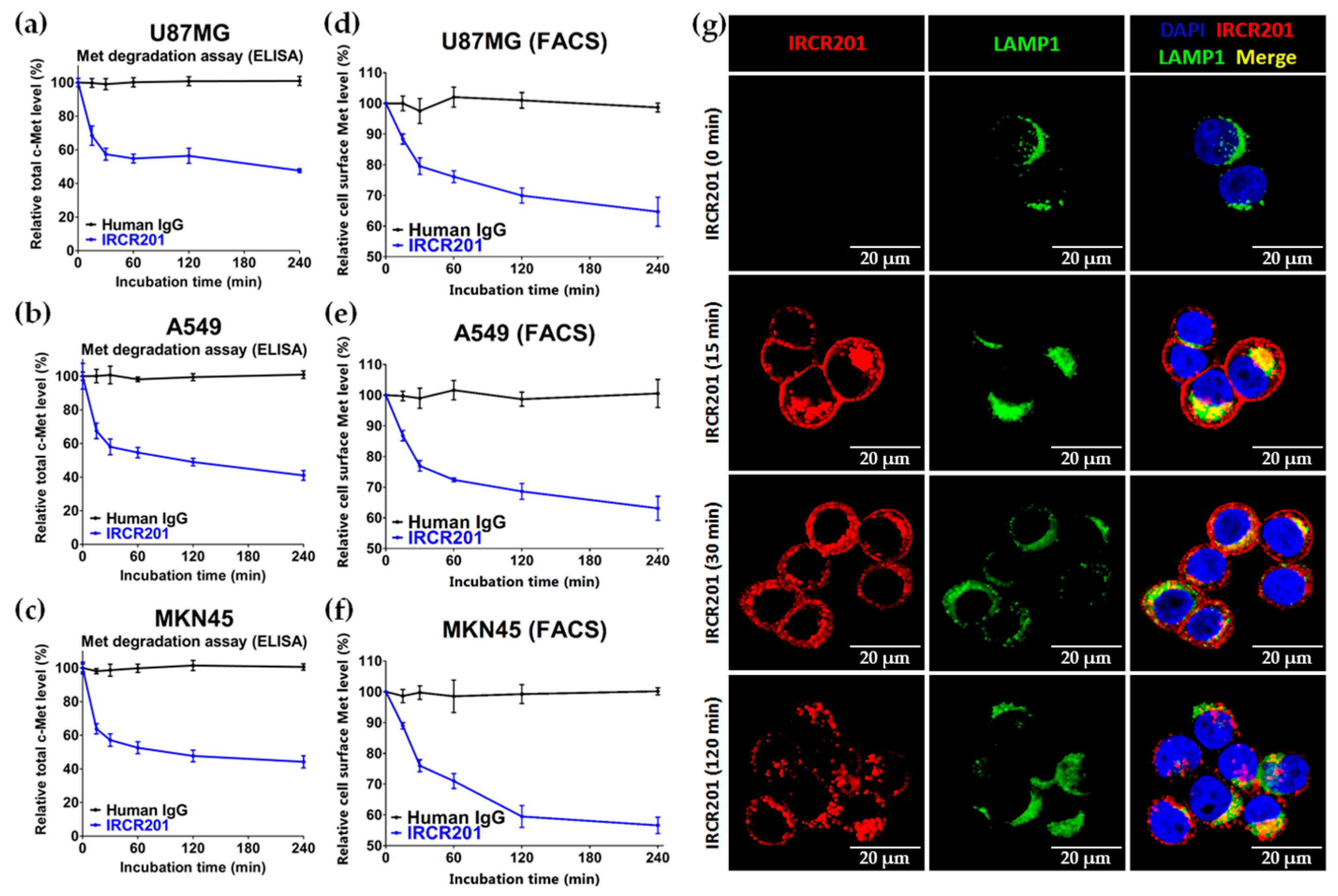
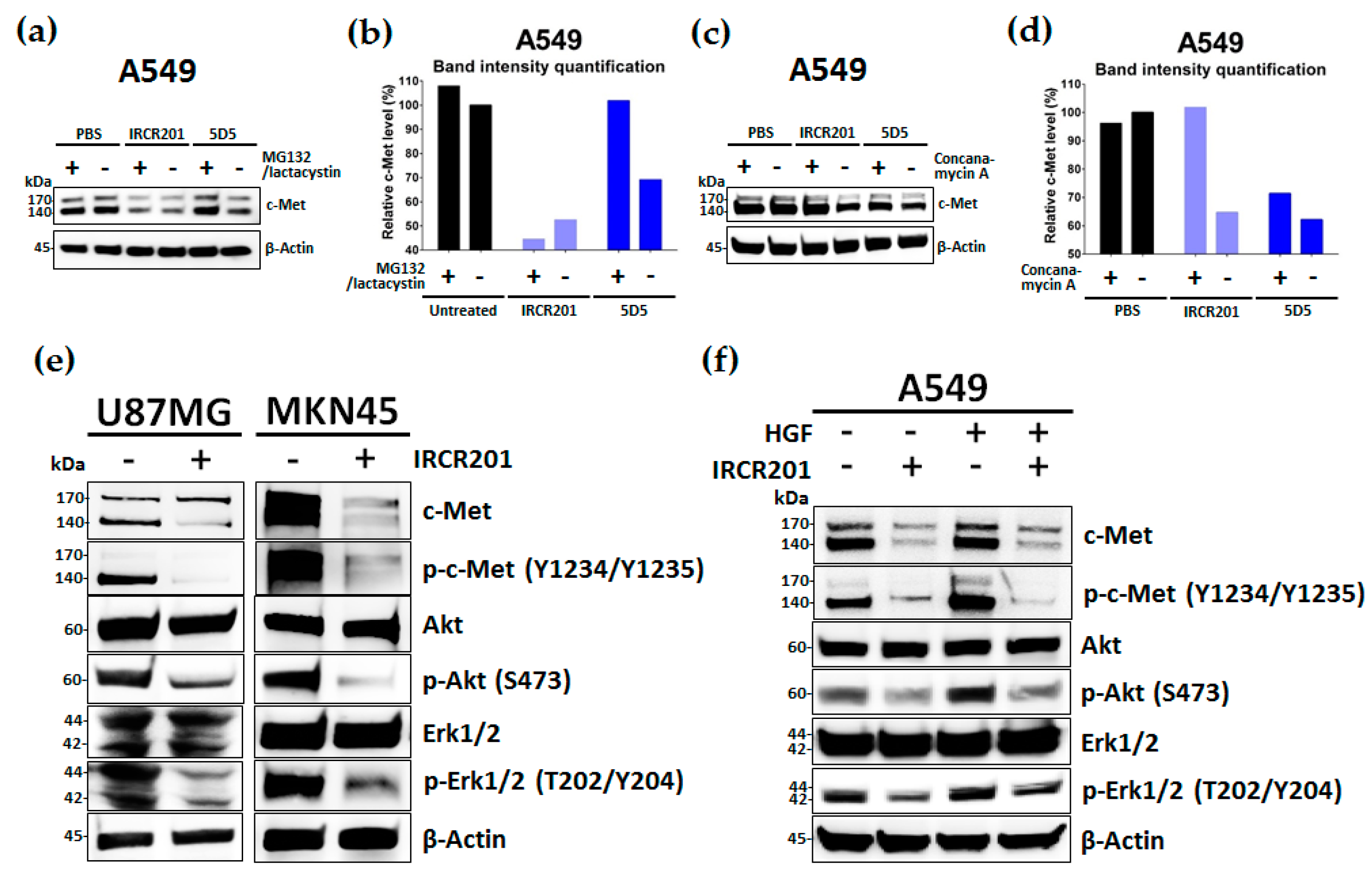
2.6. IRCR201 Induces Rapid c-Met Internalization and Lysosomal Degradation
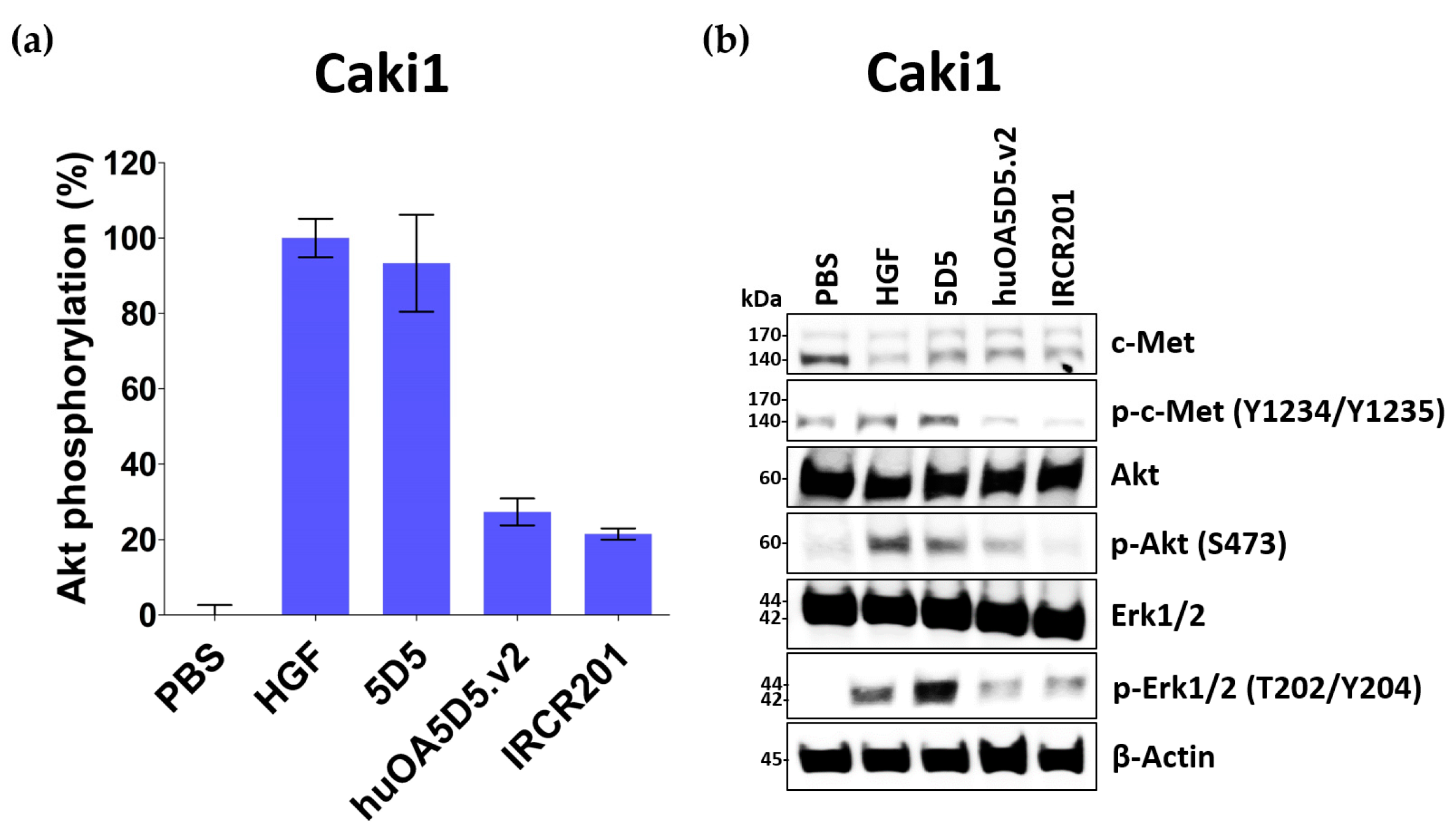
2.7. IRCR201 Suppresses the c-Met Signaling Pathway via the Degradation of c-Met
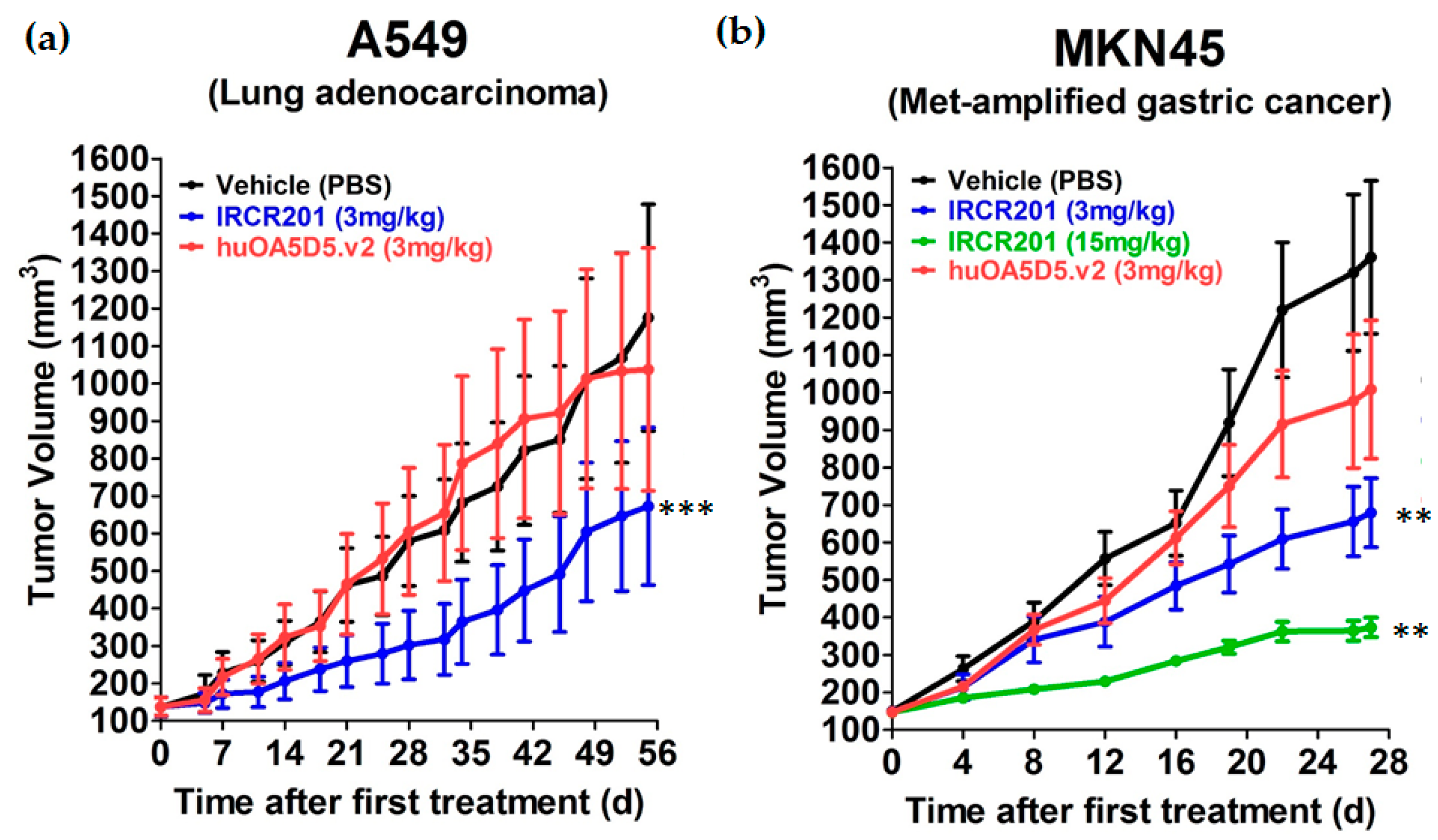
2.8. IRCR201 Impedes Tumor Growth In Vivo
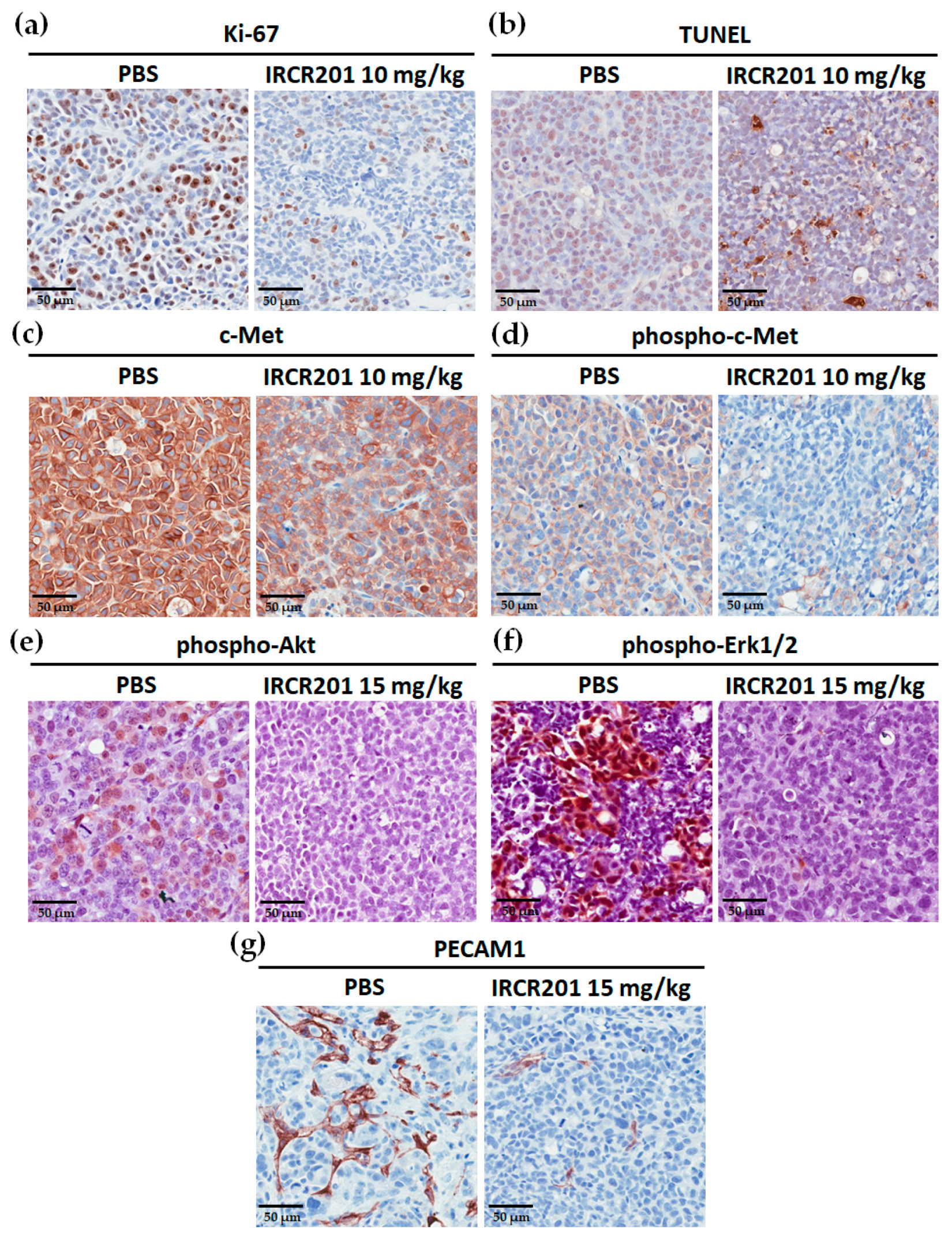
2.9. IRCR201 Inhibits Tumor Cell Proliferation and Angiogenesis through the Downregulation of c-Met
3. Discussion
4. Materials and Methods
4.1. Generation of Antibodies
4.2. Cells and Cell Cultures
4.3. Animals
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. HGF Ligand-Blocking ELISA
4.6. Surface Plasmon Resonance Assay
4.7. Epitope Mapping
4.8. Protein Modeling and Docking
4.9. Agonism Analysis
4.10. c-Met Degradation Assay
4.11. Proliferation Assay
4.12. Caspase 3/7 Activity Assay
4.13. Western Blot Analysis
4.14. Laser Scanning Microscopy
4.15. In Vivo Therapeutic Efficacy
4.16. Immunohistochemistry
4.17. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADC | Antibody-drug conjugates |
| ADCC | Antibody-dependent cell cytotoxicity |
| ATCC | American type culture collection |
| BSA | Bovine serum albumin |
| CDR | Complementarity determining regions |
| DAPI | 4′,6-diamidino-2-phenylindole |
| ECD | Extracellular domain |
| EGFR | Epidermal growth factor receptor |
| ELISA | Enzyme-linked immunosorbent assay |
| EMEM | Eagle’s Minimum Essential Medium |
| FBS | Fetal bovine serum |
| HBSS | Hank’s balanced salt solution |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| HGF | Hepatocyte growth factor |
| HGFR | Hepatocyte growth factor receptor |
| HRP | Horseradish Peroxidase |
| IgG1 | Immunoglobulin G1 |
| IHC | Immunohistochemistry |
| IMAC | Immobilized metal affinity chromatography |
| IPT | Immunoglobulin–plexin–transcription |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| MET | Mesenchymal-epithelial transition |
| MFI | Mean fluorescence intensity |
| Ni-NTA | Nickel–nitrilotriacetic acid |
| NSCLC | Non-small cell lung cancer |
| PBS | Phosphate-buffered saline |
| PBST | Phosphate-buffered saline with Tween 20 |
| PECAM1 | Platelet endothelial cell adhesion molecule 1 |
| PSI | Plexin–semaphorin–integrin |
| PVDF | Polyvinylidene difluoride |
| RPMI | Roswell Park Memorial Institute |
| RON | Recepteur d’origine nantais |
| RTK | Receptor tyrosine kinase |
| scFv | Single-chain variable fragment |
| SEM | Standard error of mean |
| SPR | Surface plasmon resonance |
| TBST | Tris-buffered saline with Tween 20 |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VEGF | Vascular endothelial growth factor |
References
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Goetsch, L.; Caussanel, V.; Corvaia, N. Biological significance and targeting of c-Met tyrosine kinase receptor in cancer. Front. Biosci. 2013, 18, 454–473. [Google Scholar] [CrossRef]
- Kentsis, A.; Reed, C.; Rice, K.L.; Sanda, T.; Rodig, S.J.; Tholouli, E.; Christie, A.; Valk, P.J.; Delwel, R.; Ngo, V.; et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Grugan, K.D.; Miller, C.G.; Yao, Y.; Michaylira, C.Z.; Ohashi, S.; Klein-Szanto, A.J.; Diehl, J.A.; Herlyn, M.; Han, M.; Nakagawa, H.; et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc. Natl. Acad. Sci. USA 2010, 107, 11026–11031. [Google Scholar] [CrossRef] [PubMed]
- Ferracini, R.; Di Renzo, M.F.; Scotlandi, K.; Baldini, N.; Olivero, M.; Lollini, P.; Cremona, O.; Campanacci, M.; Comoglio, P.M. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995, 10, 739–749. [Google Scholar] [PubMed]
- Gao, C.F.; Vande Woude, G.F. HGF/SF-Met signaling in tumor progression. Cell Res. 2005, 15, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, E.; Maeshima, A.; Nakajima, T.; Nakamura, T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn. J. Cancer Res. 1996, 87, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Han, S.U.; Lee, J.H.; Kim, W.H.; Cho, Y.K.; Kim, M.W. Significant correlation between serum level of hepatocyte growth factor and progression of gastric carcinoma. World J. Surg. 1999, 23, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Burrows, J.; Salgia, R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Vigna, E.; Comoglio, P.M. Targeting the oncogenic Met receptor by antibodies and gene therapy. Oncogene 2015, 34, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 2017, 49, e307. [Google Scholar] [CrossRef] [PubMed]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res 2006, 66, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Burgess, T.L.; Sun, J.; Meyer, S.; Tsuruda, T.S.; Sun, J.; Elliott, G.; Chen, Q.; Haniu, M.; Barron, W.F.; Juan, T.; et al. Biochemical characterization of AMG 102: A neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol. Cancer Ther. 2010, 9, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Mittra, E.S.; Fan-Minogue, H.; Lin, F.I.; Karamchandani, J.; Sriram, V.; Han, M.; Gambhir, S.S. Preclinical efficacy of the anti-hepatocyte growth factor antibody ficlatuzumab in a mouse brain orthotopic glioma model evaluated by bioluminescence, PET, and MRI. Clin. Cancer Res. 2013, 19, 5711–5721. [Google Scholar] [CrossRef] [PubMed]
- Schelter, F.; Kobuch, J.; Moss, M.L.; Becherer, J.D.; Comoglio, P.M.; Boccaccio, C.; Kruger, A. A disintegrin and metalloproteinase-10 (ADAM-10) mediates DN30 antibody-induced shedding of the met surface receptor. J. Biol. Chem. 2010, 285, 26335–26340. [Google Scholar] [CrossRef] [PubMed]
- Pacchiana, G.; Chiriaco, C.; Stella, M.C.; Petronzelli, F.; de Santis, R.; Galluzzo, M.; Carminati, P.; Comoglio, P.M.; Michieli, P.; Vigna, E. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J. Biol. Chem. 2010, 285, 36149–36157. [Google Scholar] [CrossRef] [PubMed]
- Prat, M.; Crepaldi, T.; Pennacchietti, S.; Bussolino, F.; Comoglio, P.M. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J. Cell Sci. 1998, 111, 237–247. [Google Scholar] [PubMed]
- Martens, T.; Schmidt, N.O.; Eckerich, C.; Fillbrandt, R.; Merchant, M.; Schwall, R.; Westphal, M.; Lamszus, K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin. Cancer Res. 2006, 12, 6144–6152. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Ma, X.; Maun, H.R.; Zheng, Z.; Peng, J.; Romero, M.; Huang, A.; Yang, N.Y.; Nishimura, M.; Greve, J.; et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. USA 2013, 110, E2987–E2996. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manro, J.R.; et al. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin. Cancer Res. 2014, 20, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, B.; Lee, S.B.; Jeong, Y.; Oh, Y.M.; Song, Y.J.; Jung, S.; Choi, J.; Lee, S.; Cheong, K.H.; et al. Cbl-independent degradation of Met: Ways to avoid agonism of bivalent Met-targeting antibody. Oncogene 2014, 33, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Genentech to salvage anti-MET antibody with subgroup analysis. Nat. Biotechnol. 2014, 32, 399–400. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Singh, R.K.; Fidler, I.J.; Raz, A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am. J. Pathol. 2007, 170, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Hoey, T.; Yen, W.C.; Axelrod, F.; Basi, J.; Donigian, L.; Dylla, S.; Fitch-Bruhns, M.; Lazetic, S.; Park, I.K.; Sato, A.; et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 2009, 5, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tan, Y.; Tang, Q.; Liu, X.; Guan, X.; Feng, Z.; Zhu, J. A high-affinity human/mouse cross-reactive monoclonal antibody, specific for VEGFR-2 linear and conformational epitopes. Cytotechnology 2010, 62, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.C.; Wu, X.; Peale, F.V.; Lee, C.V.; Meng, Y.G.; Gutierrez, J.; Fu, L.; Malik, A.K.; Gerber, H.P.; Ferrara, N.; et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J. Biol. Chem. 2006, 281, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Wagh, P.K.; Peace, B.E.; Waltz, S.E. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv. Cancer Res. 2008, 100, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.P.; Zhou, Y.Q.; Zhang, R.; Wang, M.H. MSP-RON signalling in cancer: Pathogenesis and therapeutic potential. Nat. Rev. Cancer 2013, 13, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Lee, W.; Luo, Y.L.; Weis, M.T.; Yao, H.P. Altered expression of the RON receptor tyrosine kinase in various epithelial cancers and its contribution to tumourigenic phenotypes in thyroid cancer cells. J. Pathol. 2007, 213, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF β-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Weitzner, B.D.; Jeliazkov, J.R.; Lyskov, S.; Marze, N.; Kuroda, D.; Frick, R.; Adolf-Bryfogle, J.; Biswas, N.; Dunbrack, R.L., Jr.; Gray, J.J. Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 2017, 12, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Naka, D.; Shimomura, T.; Yoshiyama, Y.; Sato, M.; Sato, M.; Ishii, T.; Hara, H. Internalization and degradation of hepatocyte growth factor in hepatocytes with down-regulation of the receptor/c-Met. FEBS Lett. 1993, 329, 147–152. [Google Scholar] [CrossRef]
- Jeffers, M.; Taylor, G.A.; Weidner, K.M.; Omura, S.; Vande Woude, G.F. Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol. Cell Biol. 1997, 17, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Hammond, D.E.; Urbe, S.; Vande Woude, G.F.; Clague, M.J. Down-regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001, 20, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- McCarty, J.H. Glioblastoma resistance to anti-VEGF therapy: Has the challenge been MET? Clin. Cancer Res. 2013, 19, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Chao, K.L.; Tsai, I.W.; Chen, C.; Herzberg, O. Crystal structure of the Sema-PSI extracellular domain of human RON receptor tyrosine kinase. PLoS ONE 2012, 7, e41912. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Perreault, A.; Schrag, J.D.; Park, M.; Cygler, M.; Gehring, K.; Ekiel, I. Insights into function of PSI domains from structure of the Met receptor PSI domain. Biochem. Biophys. Res. Commun. 2004, 321, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Basilico, C.; Hultberg, A.; Blanchetot, C.; de Jonge, N.; Festjens, E.; Hanssens, V.; Osepa, S.I.; de Boeck, G.; Mira, A.; Cazzanti, M.; et al. Four individually druggable MET hotspots mediate HGF-driven tumor progression. J. Clin. Investig. 2014, 124, 3172–3186. [Google Scholar] [CrossRef] [PubMed]
- Greenall, S.A.; Gherardi, E.; Liu, Z.; Donoghue, J.F.; Vitali, A.A.; Li, Q.; Murphy, R.; Iamele, L.; Scott, A.M.; Johns, T.G. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PLoS ONE 2012, 7, e34658. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, J.W.; Jun, H.J.; Ki, C.S.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Choi, M.G.; Bae, J.M.; Sohn, T.S.; et al. Impact of MET amplification on gastric cancer: Possible roles as a novel prognostic marker and a potential therapeutic target. Oncol. Rep. 2011, 25, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Surati, M.; Patel, P.; Peterson, A.; Salgia, R. Role of MetMAb (OA-5D5) in c-MET active lung malignancies. Expert Opin. Biol. Ther. 2011, 11, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Goetsch, L.; Tucker, L.; Zhang, Q.; Gonzalez, A.; Vaidya, K.S.; Oleksijew, A.; Boghaert, E.; Song, M.; Sokolova, I.; et al. Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification. BMC Cancer 2016, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Kang, K.J.; Chung, J.E.; Shim, H. Construction of a large synthetic human scFv library with six diversified CDRs and high functional diversity. Mol. Cells 2009, 27, 225–235. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Ka (1/Ms) | Kd (1/s) | KD (nM) |
|---|---|---|---|
| Human c-Met ECD-Fc | 4.226 × 106 | 3.045 × 10−3 | 0.7207 |
| Mouse c-Met ECD-Fc | 3.826 × 106 | 3.232 × 10−3 | 0.8448 |
| Spot | Peptide Sequence | Amino Acid Positions | Binding Activity |
|---|---|---|---|
| C-7 | LNGLGCRHFQSCSQC | 515–529 | − |
| D-7 | GCRHFQSCSQCLSAP | 519–533 | − |
| E-7 | FQSCSQCLSAPPFVQ | 523–537 | +++ |
| F-7 | SQCLSAPPFVQCGWC | 527–541 | ++ |
| G-7 | SAPPFVQCGWCHDKC | 531–545 | + |
| H-7 | FVQCGWCHDKCVRSE | 535–549 | − |
| I-7 | GWCHDKCVRSEECLS | 539–553 | − |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Kim, D.; Kim, E.; Sa, J.K.; Lee, H.W.; Yu, S.; Oh, J.; Kim, S.-H.; Yoon, Y.; Nam, D.-H. Tumor Inhibitory Effect of IRCR201, a Novel Cross-Reactive c-Met Antibody Targeting the PSI Domain. Int. J. Mol. Sci. 2017, 18, 1968. https://doi.org/10.3390/ijms18091968
Park H, Kim D, Kim E, Sa JK, Lee HW, Yu S, Oh J, Kim S-H, Yoon Y, Nam D-H. Tumor Inhibitory Effect of IRCR201, a Novel Cross-Reactive c-Met Antibody Targeting the PSI Domain. International Journal of Molecular Sciences. 2017; 18(9):1968. https://doi.org/10.3390/ijms18091968
Chicago/Turabian StylePark, Hyunkyu, Donggeon Kim, Eunmi Kim, Jason K. Sa, Hee Won Lee, Suji Yu, Jiwon Oh, Seok-Hyung Kim, Yeup Yoon, and Do-Hyun Nam. 2017. "Tumor Inhibitory Effect of IRCR201, a Novel Cross-Reactive c-Met Antibody Targeting the PSI Domain" International Journal of Molecular Sciences 18, no. 9: 1968. https://doi.org/10.3390/ijms18091968
APA StylePark, H., Kim, D., Kim, E., Sa, J. K., Lee, H. W., Yu, S., Oh, J., Kim, S.-H., Yoon, Y., & Nam, D.-H. (2017). Tumor Inhibitory Effect of IRCR201, a Novel Cross-Reactive c-Met Antibody Targeting the PSI Domain. International Journal of Molecular Sciences, 18(9), 1968. https://doi.org/10.3390/ijms18091968