Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases



Abstract
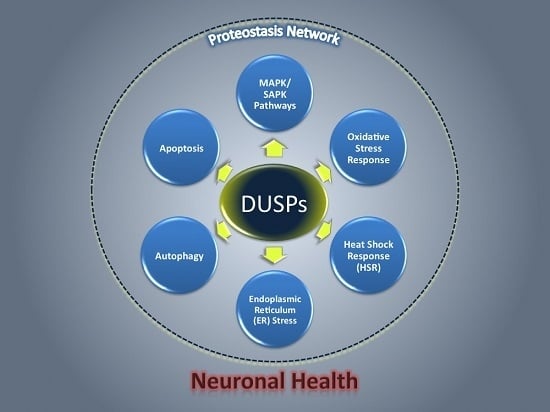
1. Introduction
2. Mechanisms by Which DUSPs May Affect Neuronal Proteostasis
2.1. DUSPs Act through Mitogen-Activated and Stress-Activated Protein Kinases
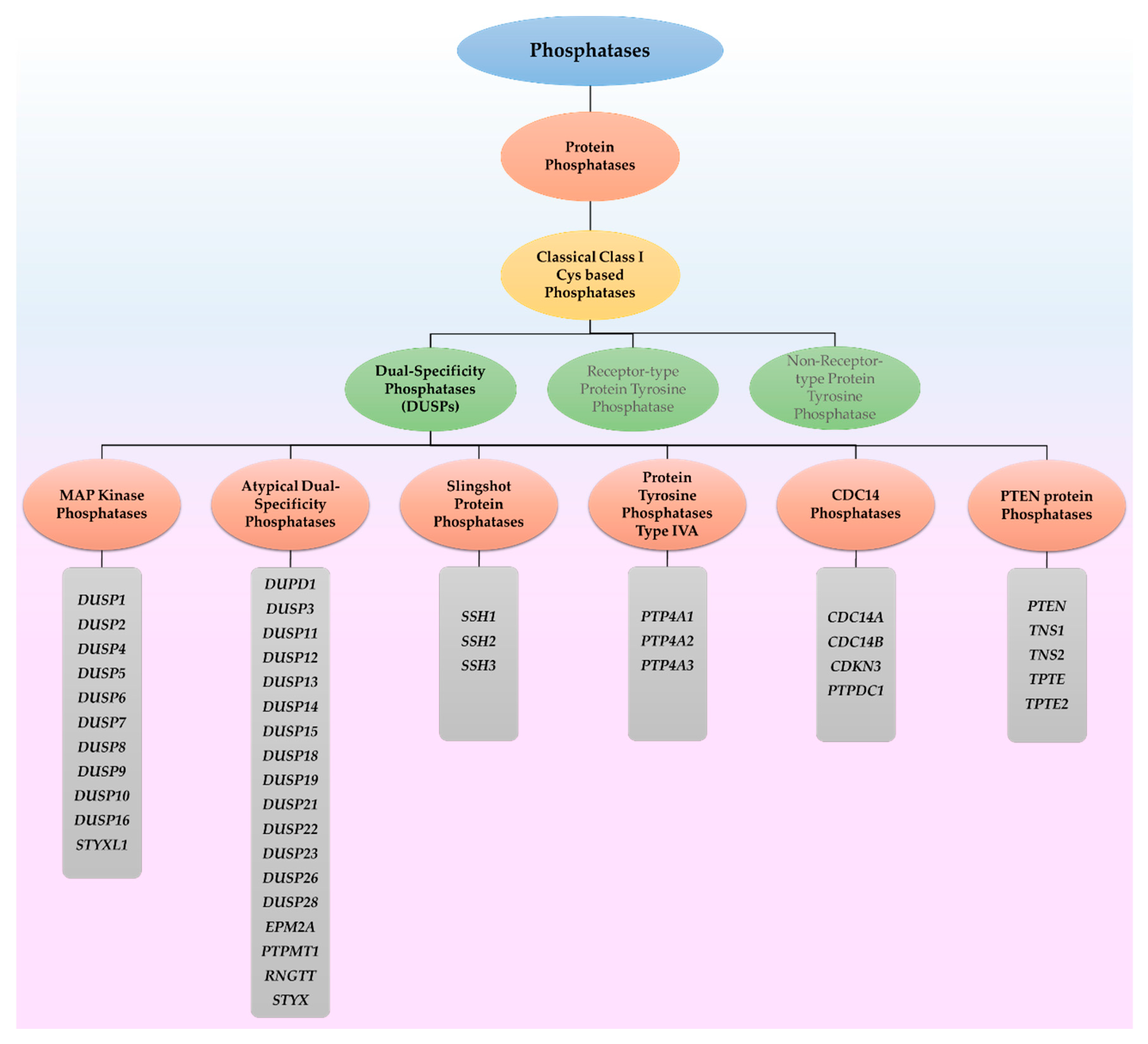
2.2. DUSPs Act through Other Mechanisms Based on Their Unique Functional Domains
3. DUSPs in Protein Aggregation Diseases
4. DUSPs in the Heat Shock Response Pathway
5. DUSPs in Oxidative Stress Response
6. DUSPs in Endoplasmic Reticulum Stress, Autophagy and Apoptosis
6.1. Endoplasmic Reticulum Stress
6.2. Autophagy
6.3. Apoptosis
7. Discussion
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-Hydroxydopamine |
| AD | Alzheimer’s disease |
| ADF | Actin depolymerizing factor |
| ALS | Amyotrophic lateral sclerosis |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATF | Activating transcription factor |
| ATG | Autophagy related |
| BCL | B-cell lymphoma |
| BIM | BCL-2 interacting mediator of cell death |
| CDC14 | Cell division cycle 14 |
| CDK | Cyclin-dependent kinase |
| CEBP/β | CCAAT/enhancer-binding protein β |
| CHOP | C/EBP homologous protein |
| DUSP | Dual-specificity phosphatase |
| eIF2α | Eukaryotic Initiation Factor 2 α |
| EGFR | Epidermal growth factor receptor |
| EPM2A | Epilepsy, Progressive Myoclonus type 2A (the gene encodes Laforin) |
| ERK | Extracellular signal–regulated kinase |
| HD | Huntington’s disease |
| HSF | Heat shock factor |
| Hsp | Heat shock protein |
| HSR | Heat shock response |
| IP3R | Inositol trisphosphate receptor |
| IRE1α | Inositol-requiring enzyme 1 |
| JIP | JNK-interacting protein-1 |
| JNK | c-Jun N-terminal kinase |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LIMK | Lin11, Isl-1 and Mec-3 domain kinase |
| MAPK | Mitogen-activated protein kinase |
| MK-2 | MAPK-activated protein kinase 2 |
| MKP | MAPK phosphatase |
| MOMP | Mitochondrial outer membrane permeabilization |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| OxR | Oxidative stress |
| PD | Parkinson’s disease |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PI(5)P | Phosphatidylinositol 5-phosphate |
| PI3K/AKT | Phosphoinositide 3-kinase/Protein kinase B |
| PIP3 | Phosphatidylinositol 3,4,5 trisphosphate |
| PN | Proteostasis network |
| PP2B | Protein phosphatase 2B |
| PTEN | Phosphatase and tensin homolog |
| PTP | Protein tyrosine phosphatase |
| PUMA | p53 Upregulated modulator of apoptosis |
| ROS | Reactive oxygen species |
| RYR | Ryanodine receptor |
| SSH | Slingshot protein phosphatase |
| STAT | Signal transducer and activator of transcription |
| STEP | Striatal-enriched protein tyrosine phosphatase |
| STYX(L1) | Serine/threonine/tyrosine-interacting-like protein |
| ULK | Unc-51 like autophagy activating kinase |
| VH1 | Vaccinia virus H1 phosphatase |
Appendix A

{kind=link}
{kind=link}
{kind=link}
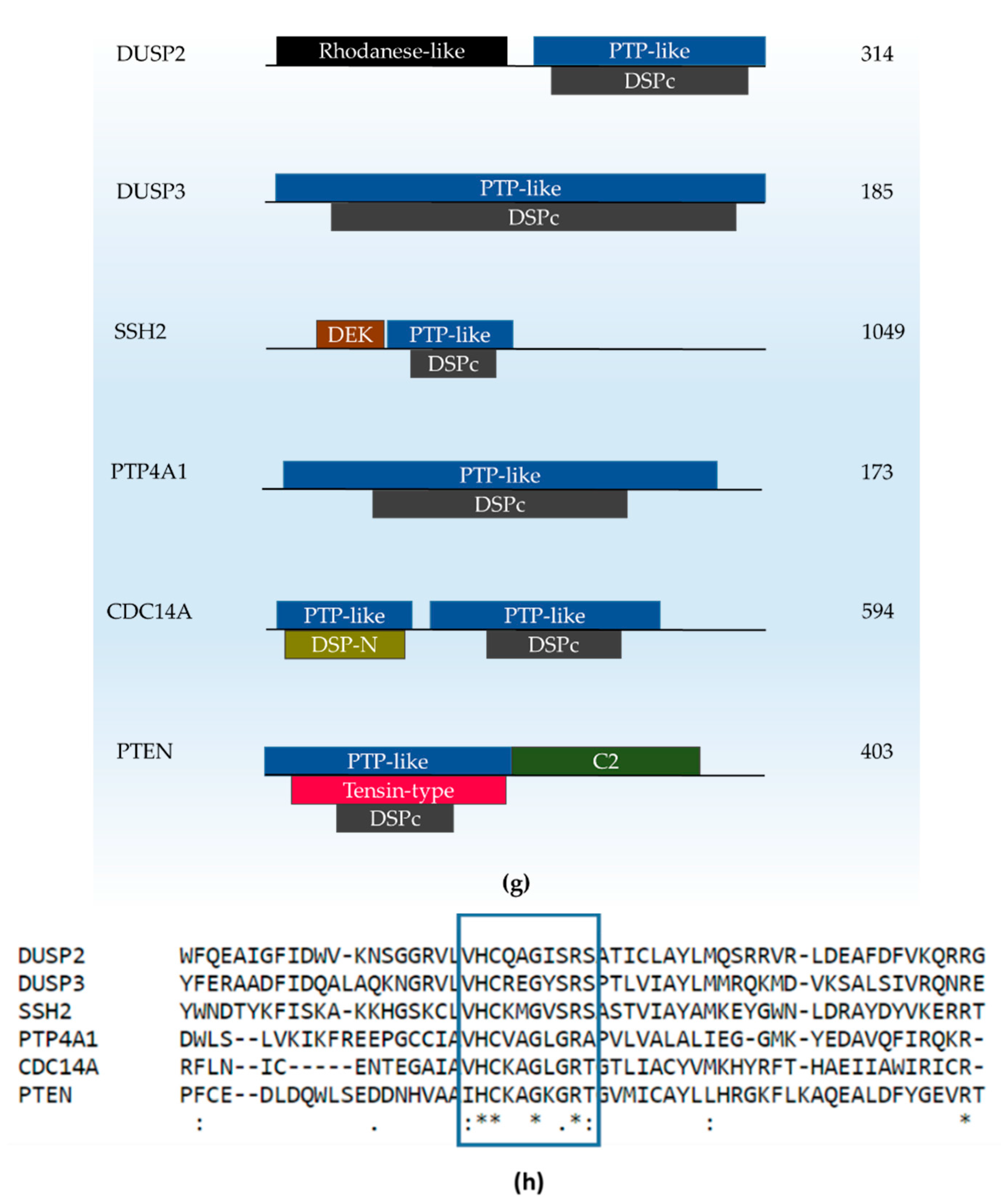{kind=link}
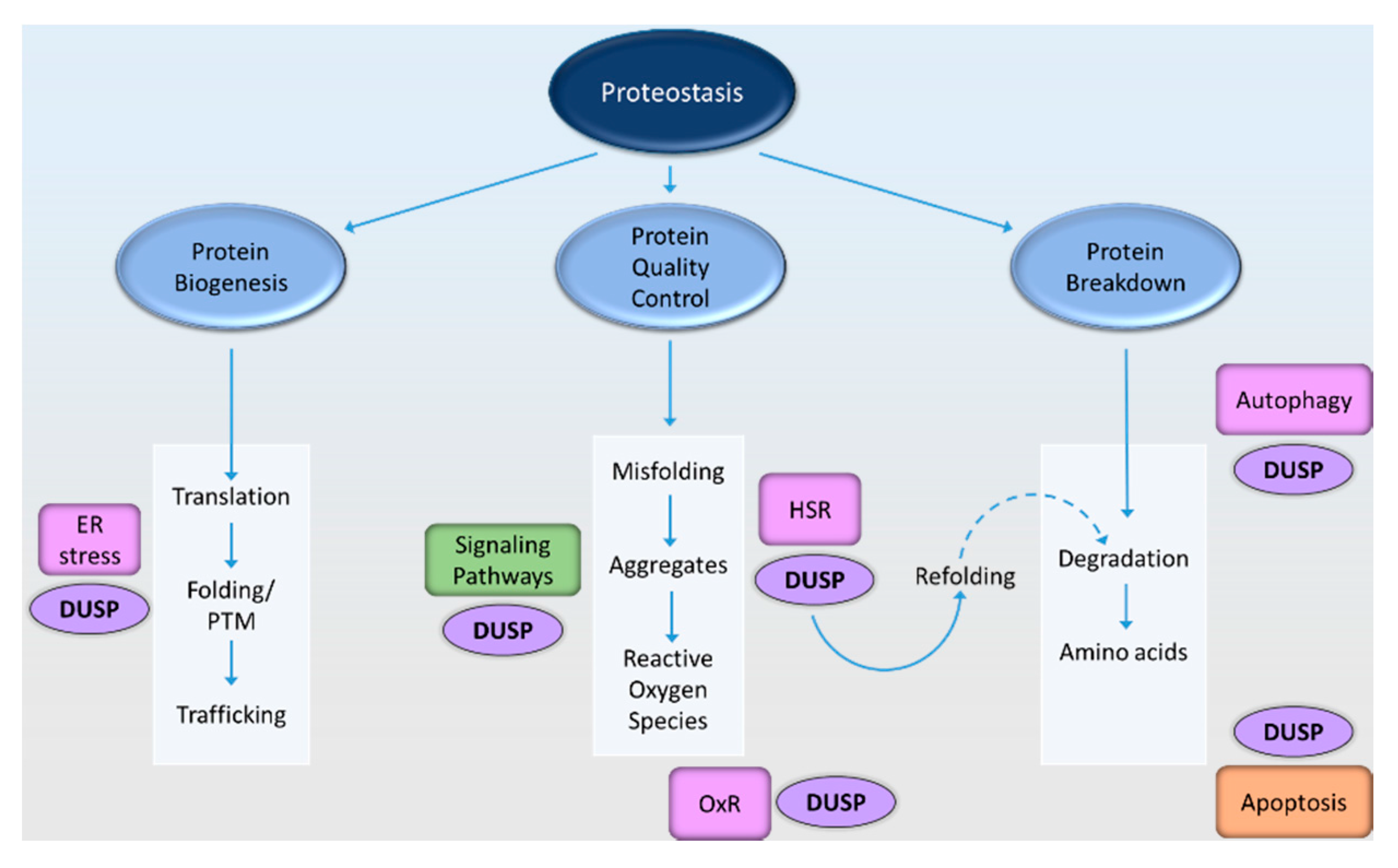{kind=link}
| No. | Gene Name | Entrez Gene ID * | UniProtKB § | Alternative Name (s) * |
|---|---|---|---|---|
| 1 | DUSP1 | 1843 | P28562 | HVH1; MKP1; CL100; MKP-1; PTPN10 |
| 2 | DUSP2 | 1844 | Q05923 | PAC-1 |
| 3 | DUSP4 | 1846 | Q13115 | TYP; HVH2; MKP2; MKP-2 |
| 4 | DUSP5 | 1847 | Q16690 | DUSP; HVH3 |
| 5 | DUSP6 | 1848 | Q16828 | HH19; MKP3; PYST1 |
| 6 | DUSP7 | 1849 | Q16829 | MKPX; PYST2 |
| 7 | DUSP8 | 1850 | Q13202 | HB5; HVH8; HVH-5; C11orf81 |
| 8 | DUSP9 | 1852 | Q99956 | MKP4; MKP-4 |
| 9 | DUSP10 | 11221 | Q9Y6W6 | MKP5; MKP-5 |
| 10 | DUSP16 | 80824 | Q9BY84 | MKP7; MKP-7 |
| 11 | STYXL1 | 51657 | Q9Y6J8 | DUSP24; MKSTYX; MK-STYX |
| 12 | DUPD1 | 338599 | Q68J44 | FMDSP; DUSP27 |
| 13 | DUSP3 | 1845 | P51452 | VHR |
| 14 | DUSP11 | 8446 | O75319 | PIR1 |
| 15 | DUSP12 | 11266 | Q9UNI6 | YVH1; DUSP1 |
| 16 | DUSP13 | 51207 | Q9UII6 | BEDP; MDSP; TMDP; SKRP4; DUSP13A; DUSP13B |
| 17 | DUSP14 | 11072 | O95147 | MKP6; MKP-L |
| 18 | DUSP15 | 128853 | Q9H1R2 | VHY; C20orf57 |
| 19 | DUSP18 | 150290 | Q8NEJ0 | DSP18; DUSP20; LMWDSP20 |
| 20 | DUSP19 | 142679 | Q8WTR2 | SKRP1; DUSP17; LMWDSP3; TS-DSP1 |
| 21 | DUSP21 | 63904 | Q9H596 | LMWDSP21 |
| 22 | DUSP22 | 56940 | Q9NRW4 | VHX; JKAP; JSP1; MKPX; JSP-1; MKP-x; LMWDSP2; LMW-DSP2 |
| 23 | DUSP23 | 54935 | Q9BVJ7 | VHZ; MOSP; LDP-3; DUSP25 |
| 24 | DUSP26 | 78986 | Q9BV47 | MKP8; NEAP; DSP-4; LDP-4; MKP-8; NATA1; SKRP3; DUSP24 |
| 25 | DUSP28 | 285193 | Q4G0W2 | VHP; DUSP26 |
| 26 | EPM2A | 7957 | O95278 | EPM2; MELF |
| 27 | PTPMT1 | 114971 | Q8WUK0 | PLIP; 1110001D10Rik; 2810004N20Rik |
| 28 | RNGTT | 8732 | O60942 | HCE; HCE1; hCAP; CAP1A |
| 29 | STYX | 6815 | Q8WUJ0 | STYX |
| 30 | SSH1 | 54434 | Q8WYL5 | SSH1L |
| 31 | SSH2 | 85464 | Q76I76 | SSH-2; SSH-2L |
| 32 | SSH3 | 54961 | Q8TE77 | SSH3L |
| 33 | PTP4A1 | 7803 | Q93096 | HH72; PRL1; PRL-1; PTPCAAX1; PTP(CAAX1) |
| 34 | PTP4A2 | 8073 | Q12974 | HH13; OV-1; PRL2; HH7-2; PRL-2; PTP4A; HU-PP-1; PTPCAAX2; ptp-IV1a; ptp-IV1b |
| 35 | PTP4A3 | 11156 | O75365 | PRL3; PRL-3; PRL-R |
| 36 | CDC14A | 8556 | Q9UNH5 | cdc14; hCDC14; DFNB105 |
| 37 | CDC14B | 8555 | O60729 | CDC14B3; Cdc14B1; Cdc14B2; hCDC14B |
| 38 | CDKN3 | 1033 | Q16667 | KAP; CDI1; CIP2; KAP1 |
| 39 | PTPDC1 | 138639 | A2A3K4 | Naa-1; Ptpcd1; AI843923; AW456874 |
| 40 | PTEN | 5728 | P60484 | BZS; DEC; CWS1; GLM2; MHAM; TEP1; MMAC1; PTEN1; 10q23del; PTEN β |
| 41 | TNS1 | 7145 | Q9HBL0 | TNS; MXRA6; MST091; MST122; MST127; MSTP091; MSTP122; MSTP127; PPP1R155 |
| 42 | TNS2 | 23371 | Q63HR2 | C1TEN; TENC1; C1-TEN |
| 43 | TPTE | 7179 | P56180 | CT44; PTEN2 |
| 44 | TPTE2 | 93492 | Q6XPS3 | TPIP |
References
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017, 216, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Weissman, J.S.; Dillin, A. Differential scales of protein quality control. Cell 2014, 157, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Balch, W.E. Diversity in the origins of proteostasis networks—A driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 2013, 14, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Nah, J.; Han, J.; Choi, S.G.; Kim, H.; Park, J.; Pyo, H.K.; Jung, Y.K. Dual-specificity phosphatase 26 (dusp26) stimulates abeta42 generation by promoting amyloid precursor protein axonal transport during hypoxia. J. Neurochem. 2016, 137, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Xie, Y.; Wang, X.; Khaleque, M.A.; Chou, S.D.; Murshid, A.; Prince, T.; Zhang, Y. Signal transduction pathways leading to heat shock transcription. Signal Transduct. Insights 2010, 2, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The role of mapk signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Balch, W.E. Expanding proteostasis by membrane trafficking networks. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Eckermann, K.; Outeiro, T.F. Protein phosphorylation in neurodegeneration: Friend or foe? Front. Mol. Neurosci. 2014, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Monteith, W.B.; Cohen, R.D.; Smith, A.E.; Guzman-Cisneros, E.; Pielak, G.J. Quinary structure modulates protein stability in cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Rojas, A.; Godzik, A.; Mustelin, T. The dual-specific protein tyrosine phosphatase family. In Protein Phosphatases; Ariño, J., Alexander, D.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 333–358. [Google Scholar]
- Alonso, A.; Bayón, Y. Atypical Dusps: 19 Phosphatases in Search of a Role; Transworld Research Network: Trivandrum, India, 2010. [Google Scholar]
- Mocciaro, A.; Schiebel, E. Cdc14: A highly conserved family of phosphatases with non-conserved functions? J. Cell Sci. 2010, 123, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Rios, P.; Li, X.; Kohn, M. Molecular mechanisms of the prl phosphatases. Fed. Eur. Biochem. Soc. J. 2013, 280, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Haynie, D.T. Molecular physiology of the tensin brotherhood of integrin adaptor proteins. Proteins 2014, 82, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.M.; Gavin, A.M.; Walsh, S.; Sullivan, A.M.; Wyatt, S.L.; O’Keeffe, G.W.; Nolan, Y.M.; Toulouse, A. Expression of endogenous mkp1 in 6-ohda rat models of parkinson’s disease. Springerplus 2014, 3, 205. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; Moser, R.; Regulier, E.; Breuillaud, L.; Dixon, M.; Beesen, A.A.; Elliston, L.; Silva Santos Mde, F.; Kim, J.; Jones, L.; et al. Map kinase phosphatase 1 (mkp-1/dusp1) is neuroprotective in huntington’s disease via additive effects of jnk and p38 inhibition. J. Neurosci. 2013, 33, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.; Plotnikova, O.; Chaturvedi, G.; Yan, S.; Zeng, L.; Zhang, Q.; Zhou, M.M. Solution structure of the mapk phosphatase pac-1 catalytic domain. Insights into substrate-induced enzymatic activation of mkp. Structure 2003, 11, 155–164. [Google Scholar] [CrossRef]
- Wu, S.; Vossius, S.; Rahmouni, S.; Miletic, A.V.; Vang, T.; Vazquez-Rodriguez, J.; Cerignoli, F.; Arimura, Y.; Williams, S.; Hayes, T.; et al. Multidentate small-molecule inhibitors of vaccinia h1-related (vhr) phosphatase decrease proliferation of cervix cancer cells. J. Med. Chem. 2009, 52, 6716–6723. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.K.; Jeong, D.G.; Yoon, T.S.; Kim, J.H.; Ryu, S.E.; Kim, S.J. Crystal structure of human slingshot phosphatase 2. Proteins 2007, 68, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.G.; Kim, S.J.; Kim, J.H.; Son, J.H.; Park, M.R.; Lim, S.M.; Yoon, T.S.; Ryu, S.E. Trimeric structure of prl-1 phosphatase reveals an active enzyme conformation and regulation mechanisms. J. Mol. Biol. 2005, 345, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.H.; Good, V.M.; Tonks, N.K.; Barford, D. The structure of the cell cycle protein cdc14 reveals a proline-directed protein phosphatase. EMBO J. 2003, 22, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the pten tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. Interpro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. Fed. Eur. Biochem. Soc. J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [PubMed]
- Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169.
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]
- Boschert, U.; Muda, M.; Camps, M.; Dickinson, R.; Arkinstall, S. Induction of the dual specificity phosphatase pac1 in rat brain following seizure activity. NeuroReport 1997, 8, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rahman, N.Z.; Greenwood, S.M.; Brett, R.R.; Tossell, K.; Ungless, M.A.; Plevin, R.; Bushell, T.J. Mitogen-activated protein kinase phosphatase-2 deletion impairs synaptic plasticity and hippocampal-dependent memory. J. Neurosci. 2016, 36, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Mengozzi, M.; Cervellini, I.; Villa, P.; Erbayraktar, Z.; Gokmen, N.; Yilmaz, O.; Erbayraktar, S.; Manohasandra, M.; Van Hummelen, P.; Vandenabeele, P.; et al. Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc. Natl. Acad. Sci. USA 2012, 109, 9617–9622. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liao, W.; Huang, Y.; Jiang, M.; Chen, J.; Wang, M.; Lin, H.; Guan, S.; Liu, J. Neuroprotective effect of dual specificity phosphatase 6 against glutamate-induced cytotoxicity in mouse hippocampal neurons. Biomed. Pharmacother. 2017, 91, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kudo, L.C.; Parfenova, L.; Vi, N.; Lau, K.; Pomakian, J.; Valdmanis, P.; Rouleau, G.A.; Vinters, H.V.; Wiedau-Pazos, M.; Karsten, S.L. Integrative gene-tissue microarray-based approach for identification of human disease biomarkers: Application to amyotrophic lateral sclerosis. Hum. Mol. Genet. 2010, 19, 3233–3253. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, Y.; Zhu, J.; Wu, H.; Guo, J. Involvement of the dual-specificity phosphatase m3/6 in c-jun n-terminal kinase inactivation following cerebral ischemia in the rat hippocampus. Int. J. Neurosci. 2013, 123, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fei, T.; Zhang, J.; Zhu, G.; Wang, L.; Lu, D.; Chi, X.; Teng, Y.; Hou, N.; Yang, X.; et al. Bmp4 signaling acts via dual-specificity phosphatase 9 to control erk activity in mouse embryonic stem cells. Cell Stem Cell 2012, 10, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gobert, R.P.; Joubert, L.; Curchod, M.L.; Salvat, C.; Foucault, I.; Jorand-Lebrun, C.; Lamarine, M.; Peixoto, H.; Vignaud, C.; Fremaux, C.; et al. Convergent functional genomics of oligodendrocyte differentiation identifies multiple autoinhibitory signaling circuits. Mol. Cell Biol. 2009, 29, 1538–1553. [Google Scholar] [CrossRef] [PubMed]
- Maor-Nof, M.; Romi, E.; Sar Shalom, H.; Ulisse, V.; Raanan, C.; Nof, A.; Leshkowitz, D.; Lang, R.; Yaron, A. Axonal degeneration is regulated by a transcriptional program that coordinates expression of pro- and anti-degenerative factors. Neuron 2016, 92, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Flowers, B.M.; Rusnak, L.E.; Wong, K.E.; Banks, D.A.; Munyikwa, M.R.; McFarland, A.G.; Hinton, S.D. The pseudophosphatase mk-styx induces neurite-like outgrowths in pc12 cells. PLoS ONE 2014, 9, e114535. [Google Scholar] [CrossRef] [PubMed]
- West, R.; Waddell, D. Dual specificity phosphatase and pro isomerase domain containing 1 (dupd1) is upregulated during neurogenic skeletal muscle atrophy and is differentially expressed in murf1-null mice. Fed. Am. Soc. Exp. Biol. J. 2017, 31 (Suppl. 1021.10), 1. [Google Scholar]
- Kim, S.H.; Markham, J.A.; Weiler, I.J.; Greenough, W.T. Aberrant early-phase erk inactivation impedes neuronal function in fragile x syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, M.; Orr-Urtreger, A. Expression changes in mouse brains following nicotine-induced seizures: The modulation of transcription factor networks. Physiol. Genom. 2007, 30, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Coggins, G.E.; Maris, J.M. Genetic susceptibility to neuroblastoma. Curr. Opin. Genet. Dev. 2017, 42, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Park, B.C.; Kim, H.A.; Song, M.; Park, S.G.; Lee, D.H.; Kim, H.J.; Choi, H.K.; Kim, J.T.; Cho, S. Positive regulation of apoptosis signal-regulating kinase 1 by dual-specificity phosphatase 13a. Cell Mol. Life Sci. 2010, 67, 2619–2629. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; van den Eijnden, M.; Pescini Gobert, R.; Saborio, G.P.; Carboni, S.; Alliod, C.; Pouly, S.; Staugaitis, S.M.; Dutta, R.; Trapp, B.; et al. Identification of vhy/dusp15 as a regulator of oligodendrocyte differentiation through a systematic genomics approach. PLoS ONE 2012, 7, e40457. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Hou, J.; Wang, F.; Zhang, Y.; Zhang, T.; Sun, T. Comparative analysis of molecular mechanism of spinal cord injury with time based on bioinformatics data. Spinal Cord. 2016, 54, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Duric, V.; Banasr, M.; Licznerski, P.; Schmidt, H.D.; Stockmeier, C.A.; Simen, A.A.; Newton, S.S.; Duman, R.S. A negative regulator of map kinase causes depressive behavior. Nat. Med. 2010, 16, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Heyn, H.; Matsuda, T.; Bock, C.; Ferrer, I.; Esteller, M. Promoter hypermethylation of the phosphatase dusp22 mediates pka-dependent tau phosphorylation and creb activation in alzheimer’s disease. Hippocampus 2014, 24, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Oh, M.; Lee, K.S.; Kim, W.K.; Oh, K.J.; Lee, S.C.; Bae, K.H.; Han, B.S. Profiling analysis of protein tyrosine phosphatases during neuronal differentiation. Neurosci. Lett. 2016, 612, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.S.; Wood, N.W.; Houlden, H. Late-onset lafora disease with prominent parkinsonism due to a rare mutation in epm2a. Neurol. Genet. 2016, 2, e101. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Kember, R.L.; Brown, C.D.; Bucan, M. Increased burden of deleterious variants in essential genes in autism spectrum disorder. Proc. Natl. Acad. Sci. USA 2016, 113, 15054–15059. [Google Scholar] [CrossRef] [PubMed]
- Dahal, A.; Hinton, S.D. Antagonistic roles for styx pseudophosphatases in neurite outgrowth. Biochem. Soc. Trans. 2017, 45, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Yuen, E.Y.; Liu, W.; Kafri, T.; van Praag, H.; Yan, Z. Regulation of ampa receptor channels and synaptic plasticity by cofilin phosphatase slingshot in cortical neurons. J. Physiol. 2010, 588, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Ohashi, K.; Mizuno, K. Lim kinase and slingshot are critical for neurite extension. J. Biol. Chem. 2007, 282, 13692–13702. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Kousaka, K.; Nagata-Ohashi, K.; Ohashi, K.; Muramoto, A.; Shima, Y.; Niwa, R.; Uemura, T.; Mizuno, K. Differential activities, subcellular distribution and tissue expression patterns of three members of slingshot family phosphatases that dephosphorylate cofilin. Genes Cells 2003, 8, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Fukuyama, H.; Fukumoto, M.; Kimura, J.; Xue, J.H.; Ohashi, H.; Fujita, J. Prl-1, a protein tyrosine phosphatase, is expressed in neurons and oligodendrocytes in the brain and induced in the cerebral cortex following transient forebrain ischemia. Brain Res. Mol. Brain Res. 1996, 40, 105–115. [Google Scholar] [CrossRef]
- Von Schantz, C.; Saharinen, J.; Kopra, O.; Cooper, J.D.; Gentile, M.; Hovatta, I.; Peltonen, L.; Jalanko, A. Brain gene expression profiles of cln1 and cln5 deficient mice unravels common molecular pathways underlying neuronal degeneration in ncl diseases. BMC Genom. 2008, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Pajer, K.; Andrus, B.M.; Gardner, W.; Lourie, A.; Strange, B.; Campo, J.; Bridge, J.; Blizinsky, K.; Dennis, K.; Vedell, P.; et al. Discovery of blood transcriptomic markers for depression in animal models and pilot validation in subjects with early-onset major depression. Transl. Psychiatry 2012, 2, e101. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zhou, H.; Tao, Y.; Guo, Z.; Zhang, S.; Zhang, Y.; Huang, Y.; Tang, Y.; Hu, R.; Dong, Q. Hcdc14a is involved in cell cycle regulation of human brain vascular endothelial cells following injury induced by high glucose, free fatty acids and hypoxia. Cell Signal. 2015, 27, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, W.; Wei, J.; Sun, D.; Gutala, R.; Gu, J.; Li, M.D. Genome-wide expression analysis reveals diverse effects of acute nicotine exposure on neuronal function-related genes and pathways. Front. Psychiatry 2011, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Partridge, V.; Du, L. The role of cdkn3 in neuroblastoma differentiation. Fed. Am. Soc. Exp. Biol. J. 2017, 31 (Suppl. 933.7), 1. [Google Scholar]
- Hu, Y.; Deng, L.; Zhang, J.; Fang, X.; Mei, P.; Cao, X.; Lin, J.; Wei, Y.; Zhang, X.; Xu, R. A pooling genome-wide association study combining a pathway analysis for typical sporadic Parkinson’s disease in the Han population of Chinese mainland. Mol. Neurobiol. 2016, 53, 4302–4318. [Google Scholar] [CrossRef] [PubMed]
- Ihle, N.T.; Abraham, R.T. The pten-parkin axis: At the nexus of cancer and neurodegeneration. Mol. Cell 2017, 65, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, S.; Smith, L.J.; Schutz, S.; Hafizi, S. Interaction of disc1 with the ptb domain of tensin2. Cell Mol. Life Sci. 2013, 70, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ma, S.; Wang, Y.; Huang, T.; Zhu, Z.; Zhao, G. Mus musculus-microrna-449a ameliorates neuropathic pain by decreasing the level of kcnma1 and trpa1, and increasing the level of tpte. Mol. Med. Rep. 2017, 16, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Füllgrabe, A.; Fuentes, A.M.-P.; Jupp, S.; Koskinen, S.; et al. Expression atlas update—An integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef] [PubMed]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Wilkie, N. Mapks: New targets for neurodegeneration. Expert Opin. Ther. Targets 2003, 7, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (map) kinase/map kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’Amato, L.; Perrone-Capano, C.; di Porzio, U. Chronic activation of erk and neurodegenerative diseases. Bioessays 2003, 25, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.D.; Cruz, F. The erk 1 and 2 pathway in the nervous system: From basic aspects to possible clinical applications in pain and visceral dysfunction. Curr. Neuropharmacol. 2007, 5, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Haeusgen, W.; Boehm, R.; Zhao, Y.; Herdegen, T.; Waetzig, V. Specific activities of individual c-jun n-terminal kinases in the brain. Neuroscience 2009, 161, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ichijo, H. Neuronal p38 mapk signalling: An emerging regulator of cell fate and function in the nervous system. Genes Cells 2002, 7, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, K.L.; Camps, M.; Rommel, C.; Mackay, C.R. Targeting dual-specificity phosphatases: Manipulating map kinase signalling and immune responses. Nat. Rev. Drug Discov. 2007, 6, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wirths, O.; Stuber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brustle, O.; et al. Phosphorylation of the amyloid beta-peptide at ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, F.; Pennuto, M. Post-translational modifications and protein quality control in motor neuron and polyglutamine diseases. Front. Mol. Neurosci. 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, S.P.; Stock, J.B.; Lombroso, P.J.; Nairn, A.C. Protein phosphatases and Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 106, 343–379. [Google Scholar] [PubMed]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Deinhardt, K.; Miyoshi, G.; Bennett, A.M.; Chao, M.V. The map kinase phosphatase mkp-1 regulates bdnf-induced axon branching. Nat. Neurosci. 2010, 13, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, F.A.; Sousa, M.M.; Cardoso, I.; do Amaral, J.B.; Guimaraes, A.; Saraiva, M.J. Activation of erk1/2 map kinases in familial amyloidotic polyneuropathy. J. Neurochem. 2006, 97, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. Microrna-125b induces tau hyperphosphorylation and cognitive deficits in alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, E.A.; Collins, M.K. Dynamic interaction between the dual specificity phosphatase mkp7 and the jnk3 scaffold protein beta-arrestin 2. J. Biol. Chem. 2005, 280, 25651–25658. [Google Scholar] [CrossRef] [PubMed]
- Thathiah, A.; Horre, K.; Snellinx, A.; Vandewyer, E.; Huang, Y.; Ciesielska, M.; De Kloe, G.; Munck, S.; De Strooper, B. Beta-arrestin 2 regulates abeta generation and gamma-secretase activity in alzheimer’s disease. Nat. Med. 2013, 19, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Dunys, J.; Pardossi-Piquard, R.; Alves da Costa, C. P53 is regulated by and regulates members of the gamma-secretase complex. Neurodegener. Dis. 2010, 7, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Lokareddy, R.K.; Bhardwaj, A.; Cingolani, G. Atomic structure of dual-specificity phosphatase 26, a novel p53 phosphatase. Biochemistry 2013, 52, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Tsuji, S.; Ikeda, O.; Sato, N.; Aoki, N.; Aoyama, K.; Sugiyama, K.; Matsuda, T. Regulation of stat3-mediated signaling by lmw-dsp2. Oncogene 2006, 25, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K. Roles of cofilin in development and its mechanisms of regulation. Dev. Growth Differ. 2015, 57, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bernstein, B.W.; Davis, R.C.; Flynn, K.C.; Goldsbury, C.; Jensen, J.R.; Maloney, M.T.; Marsden, I.T.; Minamide, L.S.; Pak, C.W.; et al. Adf/cofilin-actin rods in neurodegenerative diseases. Curr. Alzheimer Res. 2010, 7, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Nagata-Ohashi, K.; Takeichi, M.; Mizuno, K.; Uemura, T. Control of actin reorganization by slingshot, a family of phosphatases that dephosphorylate adf/cofilin. Cell 2002, 108, 233–246. [Google Scholar] [CrossRef]
- Zafar, S.; Younas, N.; Sheikh, N.; Tahir, W.; Shafiq, M.; Schmitz, M.; Ferrer, I.; Andréoletti, O.; Zerr, I. Cytoskeleton-associated risk modifiers involved in early and rapid progression of sporadic creutzfeldt-jakob disease. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kreis, P.; Leondaritis, G.; Lieberam, I.; Eickholt, B.J. Subcellular targeting and dynamic regulation of pten: Implications for neuronal cells and neurological disorders. Front. Mol. Neurosci. 2014, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, S.; Wang, Z.; Wang, Z.; Sun, C.; Zhang, Y. Inhibition of pten attenuates endoplasmic reticulum stress and apoptosis via activation of pi3k/akt pathway in alzheimer’s disease. Neurochem. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, F.; Bulloj, A.; Zhang, Y.W.; Tong, G.; Zhang, Z.; Liao, F.F.; Xu, H. Tumor-suppressor pten affects tau phosphorylation, aggregation, and binding to microtubules. Fed. Am. Soc. Exp. Biol. J. 2006, 20, 1272–1274. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Heat shock proteins: Multiple neuroprotective functions and implications for neurologic disease. Neurology 2011, 76, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Gan, Y.; Zhang, W.; Liou, A.K.; Gao, Y.; Cao, G.; Chen, J. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol. 2010, 92, 184–211. [Google Scholar] [CrossRef] [PubMed]
- Bakthisaran, R.; Tangirala, R.; Rao Ch, M. Small heat shock proteins: Role in cellular functions and pathology. Biochim. Biophys. Acta 2015, 1854, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Li, W.; Cheetham, M.E. Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 2015, 40, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, J.; O’Callaghan-Sunol, C.; Gabai, V.; Sherman, M.Y. Inactivation of dual-specificity phosphatases is involved in the regulation of extracellular signal-regulated kinases by heat shock and hsp72. Mol. Cell Biol. 2003, 23, 3813–3824. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Tabira, T.; Yamamura, T.; Kim, S.U. Hsp72 induction by heat stress is not universal in mammalian neural cell lines. J. Neurosci. Res. 1994, 37, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Palacios, C.; Collins, M.K.; Perkins, G.R. The jnk phosphatase m3/6 is inhibited by protein-damaging stress. Curr. Biol. 2001, 11, 1439–1443. [Google Scholar] [CrossRef]
- Merienne, K.; Helmlinger, D.; Perkin, G.R.; Devys, D.; Trottier, Y. Polyglutamine expansion induces a protein-damaging stress connecting heat shock protein 70 to the jnk pathway. J. Biol. Chem. 2003, 278, 16957–16967. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kim, W.; Kim, S.H.; Kim, K.T. Vrk3-mediated nuclear localization of hsp70 prevents glutamate excitotoxicity-induced apoptosis and abeta accumulation via enhancement of erk phosphatase vhr activity. Sci. Rep. 2016, 6, 38452. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mivechi, N.F. Association and regulation of heat shock transcription factor 4b with both extracellular signal-regulated kinase mitogen-activated protein kinase and dual-specificity tyrosine phosphatase dusp26. Mol. Cell Biol. 2006, 26, 3282–3294. [Google Scholar] [CrossRef] [PubMed]
- Sharda, P.R.; Bonham, C.A.; Mucaki, E.J.; Butt, Z.; Vacratsis, P.O. The dual-specificity phosphatase hyvh1 interacts with hsp70 and prevents heat-shock-induced cell death. Biochem. J. 2009, 418, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Woodford, M.R.; Truman, A.W.; Dunn, D.M.; Jensen, S.M.; Cotran, R.; Bullard, R.; Abouelleil, M.; Beebe, K.; Wolfgeher, D.; Wierzbicki, S.; et al. Mps1 mediated phosphorylation of hsp90 confers renal cell carcinoma sensitivity and selectivity to hsp90 inhibitors. Cell Rep. 2016, 14, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of map kinases by map kinase phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.P.; Reynolds, D.N.; Kraguljac, A.P.; Smith, G.S.; Mosser, D.D. Overexpression of hsp70 inhibits cofilin phosphorylation and promotes lymphocyte migration in heat-stressed cells. J. Cell Sci. 2011, 124, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The role of reactive oxygen species in the pathogenesis of alzheimer’s disease, parkinson’s disease, and huntington’s disease: A mini review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.G.; Wei, C.H.; Ku, B.; Jeon, T.J.; Chien, P.N.; Kim, J.K.; Park, S.Y.; Hwang, H.S.; Ryu, S.Y.; Park, H.; et al. The family-wide structure and function of human dual-specificity protein phosphatases. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Levinthal, D.J.; Kulich, S.M.; Chalovich, E.M.; DeFranco, D.B. Oxidative neuronal injury. The dark side of erk1/2. Eur. J. Biochem. 2004, 271, 2060–2066. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic ras/erk signalling by dual-specificity mitogen activated protein kinase phosphatases (mkps). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Kojima, S.; Kishimoto, T.; Kuwabara, S.; Yamaguchi, A. Over-expression of map kinase phosphatase-1 (mkp-1) suppresses neuronal death through regulating jnk signaling in hypoxia/re-oxygenation. Brain Res. 2012, 1436, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Mosca, L.; d’Erme, M. Parp-1 involvement in neurodegeneration: A focus on alzheimer’s and parkinson’s diseases. Mech. Ageing Dev. 2015, 146, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Hocsak, E.; Szabo, V.; Kalman, N.; Antus, C.; Cseh, A.; Sumegi, K.; Eros, K.; Hegedus, Z.; Gallyas, F., Jr.; Sumegi, B.; et al. Parp inhibition protects mitochondria and reduces ros production via parp-1-atf4-mkp-1-mapk retrograde pathway. Free Radic. Biol. Med. 2017, 108, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Oehrl, W.; Cotsiki, M.; Panayotou, G. Differential regulation of m3/6 (dusp8) signaling complexes in response to arsenite-induced oxidative stress. Cell Signal. 2013, 25, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.Q.; Xiao, F.J.; Sun, H.Y.; Shi, X.F.; Wang, H.; Yang, Y.F.; Li, Y.X.; Wang, L.S.; Ge, R.L. Ptpmt1 induced by hif-2alpha regulates the proliferation and glucose metabolism in erythroleukemia cells. Biochem. Biophys. Res. Commun. 2016, 471, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Ezerskiy, L.A.; Bertelsen, S.; Goate, A.M. Alzheimer’s disease risk polymorphisms regulate gene expression in the zcwpw1 and the celf1 loci. PLoS ONE 2016, 11, e0148717. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Zhang, L.J.; Chen, Z.; Liu, L.B. The pten inhibitor bpv(pic) promotes neuroprotection against amyloid beta-peptide (25-35)-induced oxidative stress and neurotoxicity. Neurol. Res. 2017, 39, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Kelly, U.; Ebright, J.N.; Malek, G.; Saloupis, P.; Rickman, D.W.; McKay, B.S.; Arshavsky, V.Y.; Bowes Rickman, C. Oxidative stress-induced expression and modulation of phosphatase of regenerating liver-1 (prl-1) in mammalian retina. Biochim. Biophys. Acta 2007, 1773, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Karkali, K.; Panayotou, G. The Drosophila dusp puckered is phosphorylated by jnk and p38 in response to arsenite-induced oxidative stress. Biochem. Biophys. Res. Commun. 2012, 418, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Huang, T.Y.; Bokoch, G.M. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell 2009, 20, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Rininger, A.; Dejesus, C.; Totten, A.; Wayland, A.; Halterman, M.W. Mkp-1 antagonizes c/ebpbeta activity and lowers the apoptotic threshold after ischemic injury. Cell Death Differ. 2012, 19, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Neo, S.P.; Gunaratne, J.; Poulsen, A.; Boping, L.; Ong, E.H.; Sangthongpitag, K.; Pendharkar, V.; Hill, J.; Cohen, S.M. Feedback regulation on pten/akt pathway by the er stress kinase perk mediated by interaction with the vault complex. Cell Signal. 2015, 27, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Esclatine, A.; Chaumorcel, M.; Codogno, P. Macroautophagy signaling and regulation. Curr. Top. Microbiol. Immunol. 2009, 335, 33–70. [Google Scholar] [PubMed]
- Subramaniam, S.; Unsicker, K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience 2006, 138, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. Mapk/jnk signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [PubMed]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. P38 and jnk mapk pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.Y.; Kho, D.; Reiners, J.J., Jr.; Wu, G.S. Role for dusp1 (dual-specificity protein phosphatase 1) in the regulation of autophagy. Autophagy 2016, 12, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.-M.; Nirschl, J.J.; Holzbaur, E.L.F. Lc3 binding to the scaffolding protein jip1 regulates processive dynein-driven transport of autophagosomes. Dev. Cell 2014, 29, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Yeasmin, A.M.; Waliullah, T.M.; Kondo, A.; Ushimaru, T. Yvh1 protein phosphatase is required for pre-autophagosomal structure formation after torc1 inactivation. Biosci. Biotechnol. Biochem. 2015, 79, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Zhang, P.; Chen, W.D.; Li, D.D.; Wu, X.Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.K.; et al. Atm-mediated pten phosphorylation promotes pten nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Aguado, C.; Sarkar, S.; Korolchuk, V.I.; Criado, O.; Vernia, S.; Boya, P.; Sanz, P.; de Cordoba, S.R.; Knecht, E.; Rubinsztein, D.C. Laforin, the most common protein mutated in lafora disease, regulates autophagy. Hum. Mol. Genet. 2010, 19, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Okouchi, M.; Ekshyyan, O.; Maracine, M.; Aw, T.Y. Neuronal apoptosis in neurodegeneration. Antioxid. Redox Signal. 2007, 9, 1059–1096. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Hughes, R.; Patel, P.; Jacques, T.S.; Clark, A.R.; Ham, J. Mkp1 is a c-jun target gene that antagonizes jnk-dependent apoptosis in sympathetic neurons. J. Neurosci. 2010, 30, 10820–10832. [Google Scholar] [CrossRef] [PubMed]
- Niemi, N.M.; Lanning, N.J.; Klomp, J.A.; Tait, S.W.; Xu, Y.; Dykema, K.J.; Murphy, L.O.; Gaither, L.A.; Xu, H.E.; Furge, K.A.; et al. Mk-styx, a catalytically inactive phosphatase regulating mitochondrially dependent apoptosis. Mol. Cell Biol. 2011, 31, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Huyer, G.; Liu, S.; Kelly, J.; Moffat, J.; Payette, P.; Kennedy, B.; Tsaprailis, G.; Gresser, M.J.; Ramachandran, C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997, 272, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Rios, P.; Nunes-Xavier, C.E.; Tabernero, L.; Kohn, M.; Pulido, R. Dual-specificity phosphatases as molecular targets for inhibition in human disease. Antioxid. Redox Signal. 2014, 20, 2251–2273. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Cho, S. Dual-specificity phosphatase 8 promotes the degradation of the polyglutamine protein ataxin-1. Bull. Korean Chem. Soc. 2014, 35, 297–300. [Google Scholar] [CrossRef]
- Zhang, Z.; Pinto, A.M.; Wan, L.; Wang, W.; Berg, M.G.; Oliva, I.; Singh, L.N.; Dengler, C.; Wei, Z.; Dreyfuss, G. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 19348–19353. [Google Scholar] [CrossRef] [PubMed]
- Isrie, M.; Zamani Esteki, M.; Peeters, H.; Voet, T.; Van Houdt, J.; Van Paesschen, W.; Van Esch, H. Homozygous missense mutation in styxl1 associated with moderate intellectual disability, epilepsy and behavioural complexities. Eur. J. Med. Genet. 2015, 58, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, A.; Di Francesco, L.; Principe, S.; Mignogna, G.; Sennels, L.; Mancone, C.; Alonzi, T.; Sbriccoli, M.; De Pascalis, A.; Rappsilber, J.; et al. Proteomic profiling of prp27-30-enriched preparations extracted from the brain of hamsters with experimental scrapie. Proteomics 2009, 9, 3802–3814. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. Drugbank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of pten function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, Z.; Murphy, A.N.; Wiley, S.E.; Perkins, G.A.; Worby, C.A.; Engel, J.L.; Heacock, P.; Nguyen, O.K.; Wang, J.H.; et al. Mitochondrial phosphatase ptpmt1 is essential for cardiolipin biosynthesis. Cell Metab. 2011, 13, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Molina, G.; Vogt, A.; Bakan, A.; Dai, W.; Queiroz de Oliveira, P.; Znosko, W.; Smithgall, T.E.; Bahar, I.; Lazo, J.S.; Day, B.W.; et al. Zebrafish chemical screening reveals an inhibitor of dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 2009, 5, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; McDonald, P.R.; Tamewitz, A.; Sikorski, R.P.; Wipf, P.; Skoko, J.J., III; Lazo, J.S. A cell-active inhibitor of mitogen-activated protein kinase phosphatases restores paclitaxel-induced apoptosis in dexamethasone-protected cancer cells. Mol. Cancer Ther. 2008, 7, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Lazo, J.S.; Nunes, R.; Skoko, J.J.; de Oliveira, P.E.Q.; Vogt, A.; Wipf, P. Novel benzofuran inhibitors of human mitogen-activated protein kinase phosphatase-1. Bioorg. Med. Chem. 2006, 14, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Tamewitz, A.; Skoko, J.; Sikorski, R.P.; Giuliano, K.A.; Lazo, J.S. The benzo[c]phenanthridine alkaloid, sanguinarine, is a selective, cell-active inhibitor of mitogen-activated protein kinase phosphatase-1. J. Biol. Chem. 2005, 280, 19078–19086. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, H.; Rumzhum, N.N.; Rahman, M.M.; FitzPatrick, M.; de Pedro, M.; Oliver, B.G.; Bourke, J.E.; Ammit, A.J. Tlr2 activation causes tachyphylaxis to beta2-agonists in vitro and ex vivo: Modelling bacterial exacerbation. Allergy 2014, 69, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Manetsch, M.; Ramsay, E.E.; King, E.M.; Seidel, P.; Che, W.; Ge, Q.; Hibbs, D.E.; Newton, R.; Ammit, A.J. Corticosteroids and beta(2)-agonists upregulate mitogen-activated protein kinase phosphatase 1: In vitro mechanisms. Br. J. Pharmacol. 2012, 166, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Landry, R.P.; Martinez, E.; DeLeo, J.A.; Romero-Sandoval, E.A. Spinal cannabinoid receptor type 2 agonist reduces mechanical allodynia and induces mitogen-activated protein kinase phosphatases in a rat model of neuropathic pain. J. Pain 2012, 13, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Hamamura, K.; Nishimura, A.; Chen, A.; Takigawa, S.; Sudo, A.; Yokota, H. Salubrinal acts as a dusp2 inhibitor and suppresses inflammation in anti-collagen antibody-induced arthritis. Cell Signal. 2015, 27, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, K.L.; Brummer, T.; Rolph, M.S.; Liu, S.M.; Callejas, N.A.; Grumont, R.J.; Gillieron, C.; Mackay, F.; Grey, S.; Camps, M.; et al. Positive regulation of immune cell function and inflammatory responses by phosphatase pac-1. Nat. Immunol. 2006, 7, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jeon, T.J.; Chien, P.N.; Park, S.Y.; Oh, S.M.; Kim, S.J.; Ryu, S.E. Discovery of novel dusp4 inhibitors through the virtual screening with docking simulations. Bull. Korean Chem. Soc. 2014, 35, 2655–2659. [Google Scholar] [CrossRef]
- Neumann, T.S.; Span, E.A.; Kalous, K.S.; Bongard, R.; Gastonguay, A.; Lepley, M.A.; Kutty, R.G.; Nayak, J.; Bohl, C.; Lange, R.G.; et al. Identification of inhibitors that target dual-specificity phosphatase 5 provide new insights into the binding requirements for the two phosphate pockets. BMC Biochem. 2015, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.E.; Bernard, J.; Al-Shami, A.; Liu, C.; Bollenbacher-Reilley, J.; Young, L.; Pise-Masison, C.; Spolski, R.; Leonard, W.J. T-cell development and function are modulated by dual specificity phosphatase dusp5. J. Biol. Chem. 2008, 283, 17362–17369. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Cooley, K.A.; Brisson, M.; Tarpley, M.G.; Wipf, P.; Lazo, J.S. Cell-active dual specificity phosphatase inhibitors identified by high-content screening. Chem. Biol. 2003, 10, 733–742. [Google Scholar] [CrossRef]
- Saha, M.; Skopelja, S.; Martinez, E.; Alvarez, D.L.; Liponis, B.S.; Romero-Sandoval, E.A. Spinal mitogen-activated protein kinase phosphatase-3 (mkp-3) is necessary for the normal resolution of mechanical allodynia in a mouse model of acute postoperative pain. J. Neurosci. 2013, 33, 17182–17187. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Scott, D.A.; Hatch, E.; Tian, X.; Mansour, S.L. Dusp6(mkp3) is a negative feedback regulator of fgf stimulated erk signaling during mouse development. Development 2007, 134, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jeon, J.Y.; Ryu, S.E.; Kim, S.J. Discovery of novel inhibitors of dual-specificity phosphatase pyst2 with structure-based virtual screening. Bull. Korean Chem. Soc. 2011, 32, 2167–2168. [Google Scholar] [CrossRef]
- Theodosiou, A.; Ashworth, A. Differential effects of stress stimuli on a jnk-inactivating phosphatase. Oncogene 2002, 21, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; van Berlo, J.H.; York, A.J.; Maillet, M.; Vagnozzi, R.J.; Molkentin, J.D. Dusp8 regulates cardiac ventricular remodeling by altering erk1/2 signaling. Circ. Res. 2016, 119, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jeon, J.Y.; Ryu, S.E. Virtual screening and biochemical evaluation of mitogen-activated protein kinase phosphatase 4 inhibitors. Bull. Korean Chem. Soc. 2012, 33, 3772–3776. [Google Scholar] [CrossRef]
- Ryu, S.E.; Kim, S.J. Targeting allosteric sites for protein tyrosine phosphatase inhibition. Bio Des. 2014, 2, 81–90. [Google Scholar]
- Christie, G.R.; Williams, D.J.; MacIsaac, F.; Dickinson, R.J.; Rosewell, I.; Keyse, S.M. The dual-specificity protein phosphatase dusp9/mkp-4 is essential for placental function but is not required for normal embryonic development. Mol. Cell Biol. 2005, 25, 8323–8333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Blattman, J.N.; Kennedy, N.J.; Duong, J.; Nguyen, T.; Wang, Y.; Davis, R.J.; Greenberg, P.D.; Flavell, R.A.; Dong, C. Regulation of innate and adaptive immune responses by map kinase phosphatase 5. Nature 2004, 430, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, S.Y.; Nam, S.-W.; Ryu, S.E. Discovery of novel dusp16 phosphatase inhibitors through virtual screening with homology modeled protein structure. J. Biomol. Screen. 2014, 19, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Devi, Y.S.; Seibold, A.M.; Shehu, A.; Maizels, E.; Halperin, J.; Le, J.; Binart, N.; Bao, L.; Gibori, G. Inhibition of mapk by prolactin signaling through the short form of its receptor in the ovary and decidua: Involvement of a novel phosphatase. J. Biol. Chem. 2011, 286, 7609–7618. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Sudo, T.; Osada, H. Rk-682, a potent inhibitor of tyrosine phosphatase, arrested the mammalian cell cycle progression at g1phase. Fed. Eur. Biochem. Soc. Lett. 1995, 372, 54–58. [Google Scholar] [CrossRef]
- Musumeci, L.; Kuijpers, M.J.; Gilio, K.; Hego, A.; Theatre, E.; Maurissen, L.; Vandereyken, M.; Diogo, C.V.; Lecut, C.; Guilmain, W.; et al. Dual-specificity phosphatase 3 deficiency or inhibition limits platelet activation and arterial thrombosis. Circulation 2015, 131, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Usui, T.; Nakayama, H.; Ueki, M.; Takio, K.; Ubukata, M.; Osada, H. 4-isoavenaciolide covalently binds and inhibits vhr, a dual-specificity phosphatase. Fed. Eur. Biochem. Soc. Lett. 2002, 525, 48–52. [Google Scholar] [CrossRef]
- Amand, M.; Erpicum, C.; Bajou, K.; Cerignoli, F.; Blacher, S.; Martin, M.; Dequiedt, F.; Drion, P.; Singh, P.; Zurashvili, T.; et al. Dusp3/vhr is a pro-angiogenic atypical dual-specificity phosphatase. Mol. Cancer 2014, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, T.; Takagi, T.; Hao, L.; Buratowski, S.; Charbonneau, H. Human pir1 of the protein-tyrosine phosphatase superfamily has RNA 5′-triphosphatase and diphosphatase activities. J. Biol. Chem. 1999, 274, 16590–16594. [Google Scholar] [CrossRef] [PubMed]
- Nallaparaju, K.C.; Zhang, Y.; Liu, X.; Reynolds, J.M.; Nurieva, R.I.; Dong, C. Dusp11 is a critical regulator of innate immune responses mediated by dendritic cells. Cytokine 2013, 63, 286. [Google Scholar] [CrossRef]
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Tilley, D.G.; Sabri, A. Dusps as critical regulators of cardiac hypertrophy. Clin. Sci. 2017, 131, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Youn, D.; Cho, S. Inhibition of dusp13a activity by ptp inhibitor v. Bull. Korean Chem. Soc. 2013, 34, 3912–3914. [Google Scholar] [CrossRef]
- Park, J.E.; Park, B.C.; Song, M.; Park, S.G.; Lee, D.H.; Park, S.Y.; Kim, J.H.; Cho, S. Ptp inhibitor iv protects jnk kinase activity by inhibiting dual-specificity phosphatase 14 (dusp14). Biochem. Biophys. Res. Commun. 2009, 387, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Cho, S. Specific inhibition of dusp14 by NSC-95397 in vitro. Bull. Korean Chem. Soc. 2011, 32, 4435–4437. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Li, J.-P.; Chiu, L.-L.; Lan, J.-L.; Chen, D.-Y.; Chuang, H.-C.; Huang, C.-Y.; Tan, T.-H. Dual-specificity phosphatase 14 (dusp14/mkp6) negatively regulates tcr signaling by inhibiting tab1 activation. J. Immunol. 2014, 192, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, H.S.; Kim, S.J. Virtual screening with docking simulations and biochemical evaluation of vhy phosphatase inhibitors. Chem. Pharm. Bull. 2015, 63, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Muth, K.N.; Piefke, S.; Weider, M.; Sock, E.; Hermans-Borgmeyer, I.; Wegner, M.; Küspert, M. The dual-specificity phosphatase dusp15 is regulated by sox10 and myrf in myelinating oligodendrocytes. Glia 2016, 64, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.L.; Tobin, J.F.; Yoon, C. Identification and characterization of two novel low-molecular-weight dual specificity phosphatases. Biochem. Biophys. Res. Commun. 2002, 298, 545–551. [Google Scholar] [CrossRef]
- Wu, Q.; Gu, S.; Dai, J.; Dai, J.; Wang, L.; Li, Y.; Zeng, L.; Xu, J.; Ye, X.; Zhao, W.; et al. Molecular cloning and characterization of a novel dual-specificity phosphatase18 gene from human fetal brain. Biochim. Biophys. Acta Gene Struct. Expr. 2003, 1625, 296–304. [Google Scholar] [CrossRef]
- Zama, T.; Aoki, R.; Kamimoto, T.; Inoue, K.; Ikeda, Y.; Hagiwara, M. A novel dual specificity phosphatase skrp1 interacts with the mapk kinase mkk7 and inactivates the jnk mapk pathway: Implication for the precise regulation of the particular mapk pathway. J. Biol. Chem. 2002, 277, 23909–23918. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1b. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Ju, A.N.; Cho, S.Y. Inhibition of dual-specificity phosphatase 22 (dusp22) by prl-3 inhibitor i. Bull. Korean Chem. Soc. 2012, 33, 3142–3144. [Google Scholar] [CrossRef]
- Li, J.-P.; Yang, C.-Y.; Chuang, H.-C.; Lan, J.-L.; Chen, D.-Y.; Chen, Y.-M.; Wang, X.; Chen, A.J.; Belmont, J.W.; Tan, T.-H. The phosphatase jkap/dusp22 inhibits T-cell receptor signalling and autoimmunity by inactivating lck. Nat. Commun. 2014, 5, 3618. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, Y.; Gu, S.; Li, N.; Zheng, D.; Li, D.; Zheng, Z.; Ji, C.; Xie, Y.; Mao, Y. Molecular cloning and characterization of a novel dual-specificity phosphatase 23 gene from human fetal brain. Int. J. Biochem. Cell Biol. 2004, 36, 1542–1553. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, S.Y.; Oh, J.J.; Ryu, S.E. Identification of potent vhz phosphatase inhibitors with structure-based virtual screening. J. Biomol. Screen. 2013, 18, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ma, I.T.; Patel, R.H.; Shang, X.; Chen, Z.; Zhao, Y.; Cheng, J.; Fan, Y.; Rojas, Y.; Barbieri, E.; et al. Nsc-87877 inhibits dusp26 function in neuroblastoma resulting in p53-mediated apoptosis. Cell Death Dis. 2015, 6, e1841. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Cho, S. Inhibition of dual-specificity phosphatase 26 by ethyl-3,4-dephostatin: Ethyl-3,4-dephostatin as a multiphosphatase inhibitor. Die Pharm. Int. J. Pharm. Sci. 2016, 71, 196–200. [Google Scholar]
- Park, H.; Kyung, A.; Lee, H.J.; Kang, S.; Yoon, T.S.; Ryu, S.E.; Jeong, D.G. Virtual screening and biochemical evaluation of the inhibitors of dual-specificity phosphatase 26. Med. Chem. Res. 2013, 22, 3905–3910. [Google Scholar] [CrossRef]
- Lee, J.; Hun Yun, J.; Lee, J.; Choi, C.; Hoon Kim, J. Blockade of dual-specificity phosphatase 28 decreases chemo-resistance and migration in human pancreatic cancer cells. Sci. Rep. 2015, 5, 12296. [Google Scholar] [CrossRef] [PubMed]
- Toyota, R.; Honjo, Y.; Imajo, R.; Satoh, A. S-nitrosylation of laforin inhibits its phosphatase activity and is implicated in lafora disease. Matters 2016, 2, e201606000014. [Google Scholar]
- Wang, W.; Roach, P.J. Glycogen and related polysaccharides inhibit the laforin dual-specificity protein phosphatase. Biochem. Biophys. Res. Commun. 2004, 325, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Delgado-Escueta, A.V.; Sakamoto, T.; Avila, M.R.; Machado-Salas, J.; Hoshii, Y.; Akagi, T.; Gomi, H.; Suzuki, T.; Amano, K.; et al. Targeted disruption of the epm2a gene causes formation of lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet. 2002, 11, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Doughty-Shenton, D.; Joseph, J.D.; Zhang, J.; Pagliarini, D.J.; Kim, Y.; Lu, D.; Dixon, J.E.; Casey, P.J. Pharmacological targeting of the mitochondrial phosphatase ptpmt1. J. Pharmacol. Exp. Ther. 2010, 333, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, S.Y.; Kyung, A.; Yoon, T.S.; Ryu, S.E.; Jeong, D.G. Structure-based virtual screening approach to the discovery of novel ptpmt1 phosphatase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Picard-Jean, F.; Bougie, I.; Shuto, S.; Bisaillon, M. The immunosuppressive agent mizoribine monophosphate is an inhibitor of the human RNA capping enzyme. PLoS ONE 2013, 8, e54621. [Google Scholar] [CrossRef] [PubMed]
- Wishart, M.J.; Denu, J.M.; Williams, J.A.; Dixon, J.E. A single mutation converts a novel phosphotyrosine binding domain into a dual-specificity phosphatase. J. Biol. Chem. 1995, 270, 26782–26785. [Google Scholar] [CrossRef] [PubMed]
- Li, K.-S.; Xiao, P.; Zhang, D.-L.; Hou, X.-B.; Ge, L.; Yang, D.-X.; Liu, H.-D.; He, D.-F.; Chen, X.; Han, K.-R.; et al. Identification of para-substituted benzoic acid derivatives as potent inhibitors of the protein phosphatase slingshot. Chem. Med. Chem. 2015, 10, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, W.; Lee, Y.G.; Kang, H.J.; Lee, S.H.; Park, S.Y.; Min, J.K.; Lee, S.R.; Chung, S.J. Identification of sennoside a as a novel inhibitor of the slingshot (ssh) family proteins related to cancer metastasis. Pharmacol. Res. 2017, 119, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Mui, M.K.-H. Identification of Specific Inhibitors for a Dual-Specificity Phosphatase SSH-2; University of California: San Diego, CA, USA, 2011. [Google Scholar]
- Kousaka, K.; Kiyonari, H.; Oshima, N.; Nagafuchi, A.; Shima, Y.; Chisaka, O.; Uemura, T. Slingshot-3 dephosphorylates adf/cofilin but is dispensable for mouse development. Genesis 2008, 46, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Daouti, S.; Li, W.-H.; Qian, H.; Huang, K.-S.; Holmgren, J.; Levin, W.; Reik, L.; McGady, D.L.; Gillespie, P.; Perrotta, A.; et al. A selective phosphatase of regenerating liver phosphatase inhibitor suppresses tumor cell anchorage-independent growth by a novel mechanism involving p130cas cleavage. Cancer Res. 2008, 68, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Hoeger, B.; Diether, M.; Ballester, P.J.; Kohn, M. Biochemical evaluation of virtual screening methods reveals a cell-active inhibitor of the cancer-promoting phosphatases of regenerating liver. Eur. J. Med. Chem. 2014, 88, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.K.; Dhawan, D.; Lindner, D.J.; Borden, E.C.; Farver, C.; Yi, T. Pentamidine is an inhibitor of prl phosphatases with anticancer activity 1 supported in part by nih grants r01ca79891 and r01mg58893 (to T.Y.) and ca90914 (to E.C.B.). Mol. Cancer Ther. 2002, 1, 1255–1264. [Google Scholar] [PubMed]
- Jiao, Y.; Ye, D.Z.; Li, Z.; Teta-Bissett, M.; Peng, Y.; Taub, R.; Greenbaum, L.E.; Kaestner, K.H. Protein tyrosine phosphatase of liver regeneration-1 is required for normal timing of cell cycle progression during liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G85–G91. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbe, D.P.; Begin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase prl-2 interacts with the magnesium transporter cnnm3 to promote oncogenesis. Oncogene 2015, 34, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; McQueeney, K.E.; Isenberg, J.S.; Pitt, B.R.; Wasserloos, K.A.; Homanics, G.E.; Lazo, J.S. Protein-tyrosine phosphatase 4a3 (ptp4a3) promotes vascular endothelial growth factor signaling and enables endothelial cell motility. J. Biol. Chem. 2014, 289, 5904–5913. [Google Scholar] [CrossRef] [PubMed]
- Salamoun, J.M.; McQueeney, K.E.; Patil, K.; Geib, S.J.; Sharlow, E.R.; Lazo, J.S.; Wipf, P. Photooxygenation of an amino-thienopyridone yields a more potent ptp4a3 inhibitor. Org. Biomol. Chem. 2016, 14, 6398–6402. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Kim, S.J.; Park, W.S.; Cho, S.Y.; Ha, J.D.; Kim, S.S.; Kang, S.K.; Jeong, D.G.; Jung, S.K.; Lee, S.H.; et al. Synthesis and biological evaluation of rhodanine derivatives as prl-3 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 2996–2999. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; Homanics, G.E.; Lazo, J.S. Targeted deletion of the metastasis-associated phosphatase ptp4a3 (prl-3) suppresses murine colon cancer. PLoS ONE 2013, 8, e58300. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ha, K.; Lu, G.; Fang, X.; Cheng, R.; Zuo, Q.; Zhang, P. Cdc14a and cdc14b redundantly regulate DNA double-strand break repair. Mol. Cell. Biol. 2015, 35, 3657–3668. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Peddibhotla, S.; Lin, H.; Fang, X.; Li, M.; Rosen, J.M.; Zhang, P. Early-onset aging and defective DNA damage response in cdc14b-deficient mice. Mol. Cell. Biol. 2011, 31, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J.; Casso, D.; Beach, D. Kap: A dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Cerabona, D.; He, Y.; Nalepa, G. Cdkn3 knockout mice develop hematopoietic malignancies. Blood 2016, 128, 1537. [Google Scholar]
- Takasuga, A.; Sato, K.; Nakamura, R.; Saito, Y.; Sasaki, S.; Tsuji, T.; Suzuki, A.; Kobayashi, H.; Matsuhashi, T.; Setoguchi, K.; et al. Non-synonymous fgd3 variant as positional candidate for disproportional tall stature accounting for a carcass weight qtl (cw-3) and skeletal dysplasia in japanese black cattle. PLoS Genet. 2015, 11, e1005433. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Lindsay, Y.E.; Leslie, N.R. Pten inhibitors: An evaluation of current compounds. Adv. Biol. Regul. 2015, 57, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.C.; Byrne, R.D.; Vilar, R.; Woscholski, R. Bisperoxovanadium compounds are potent pten inhibitors. Fed. Eur. Biochem. Soc. Lett. 2004, 566, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, M.C.; Sunnen, C.N.; Lugo, J.N.; Anderson, A.E.; D’Arcangelo, G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis. Models Mech. 2009, 2, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-P.; Sun, P.; Wang, A.; Lo, S.H. Tensin1 positively regulates rhoa activity through its interaction with dlc1. Biochim. Biophys. Acta 2015, 1853, 3258–3265. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.H.; Lee, J.; Jeong, H.; Koh, A. Pharmaceutical Compositions for Preventing or Treating Diabetic Nephropathy Comprising the Activity Inhibitor of Tenc1. U.S. Patent 15/176,064, 31 May 2016. [Google Scholar]
- Sasaki, H.; Marusugi, K.; Kimura, J.; Kitamura, H.; Nagasaki, K.-I.; Torigoe, D.; Agui, T.; Sasaki, N. Genetic background-dependent diversity in renal failure caused by the tensin2 gene deficiency in the mouse. Biomed. Res. 2015, 36, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar]




| No. | Gene Name | Family & Domains | Possible Association with Neurological Deficits or Affected Neuronal Functions | Gene Expression in Indicative Brain Regions |
|---|---|---|---|---|
| 1 | DUSP1 | a, b, c, d, e, Δ | HD [19] | CCx x, CbCx x, H x, A y, Sn y |
| 2 | DUSP2 | a, b, c, d, Δ | Seizure [31] | CCx x, CbCx y, H y, A y, Sn y |
| 3 | DUSP4 | a, b, c, d, Δ | Hippocampal synaptic function [32] | CCx y, CbCx y, H y, A y |
| 4 | DUSP5 | a, b, c, d, Δ | Cerebral ischemia [33] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 5 | DUSP6 | a, b, c, d, Δ | Glutamate-induced cytotoxicity [34] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 6 | DUSP7 | a, b, c, d, Δ | ALS [35] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 7 | DUSP8 | a, b, c, d, Δ | Cerebral ischemia [36] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 8 | DUSP9 | a, b, c, d, Δ | Neural fate commitment [37] | H y, A y, NAc y |
| 9 | DUSP10 | a, b, c, d, e, Δ | Oligodendrocyte differentiation [38] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 10 | DUSP16 | a, b, c, d, Δ | Axonal degeneration [39] | CCx x, CbCx x, H y, A y, SN y, NAc y |
| 11 | STYXL1 | a, b, d, Δ | Neuronal differentiation [40] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 12 | DUPD1 | a, b, e, Δ | Skeletal muscle atrophy [41] | CCx x, CbCx x, H x |
| 13 | DUSP3 | a, b, e, Δ | Glutamate-induced cytotoxicity [42] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 14 | DUSP11 | a, b, Δ | Seizure [43] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 15 | DUSP12 | a, b, f, Δ | Neuroblastoma GWAS [44] | CCx x, CbCx y, H y, A y, SN y, NAc y |
| 16 | DUSP13 | a, b, e, Δ | Neuron development [45] | Some regions of CCx z |
| 17 | DUSP14 | a, b, e, Δ | HD [19] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 18 | DUSP15 | a, b, e, g, Δ | Oligodendrocyte differentiation [46] | Low expression |
| 19 | DUSP18 | a, b, e, Δ | SCI [47] | CCx x, CbCx x, H x |
| 20 | DUSP19 | a, b, e, Δ | Depression [48] | CCx x |
| 21 | DUSP21 | a, b, e, Δ | Not defined | Not defined |
| 22 | DUSP22 | a, b, e, Δ | AD [49] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 23 | DUSP23 | a, b, Δ | Neuronal differentiation [50] | CCx x, CbCx y, H x, A y, SN y, NAc y |
| 24 | DUSP26 | a, b, e, Δ | AD [6] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 25 | DUSP28 | a, b, Δ | Not defined | Low expression |
| 26 | EPM2A | a, b, h, i, j, Δ | Lafora disease [51] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 27 | PTPMT1 | a, b, Δ | AD GWAS [52] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 28 | RNGTT | a, b, k, l, Δ | ASD RNA-Seq [53] | CCx x, CbCx y, H x, A y, SN y, NAc y |
| 29 | STYX | a, b, Δ | Golgi fragmentation [54] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 30 | SSH1 | a, b, m, o, Δ | Synaptic plasticity [55] | CCx x, CbCx y, H x, A y, SN y, NAc y |
| 31 | SSH2 | a, b, n, o, Δ | Neurite extension [56] | CCx x, CbCx y, H x, A y, SN y, NAc y |
| 32 | SSH3 | a, b, n, o, Δ | Actin reorganization [57] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 33 | PTP4A1 | a, b, Δ | Cerebral ischemia [58] | CCx y, CbCx y, H x, A y, SN y, NAc y |
| 34 | PTP4A2 | a | NCL [59] | CCx y, CbCx x, H x, A y, SN y, NAc y |
| 35 | PTP4A3 | a, b, Δ | MDD, Stress [60] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 36 | CDC14A | a, b, Δ | Diabetic stroke [61] | CCx x, CbCx x, H x, A y |
| 37 | CDC14B | a, b, Δ | Addictive behavior [62] | CCx x, CbCx x, H x, A y, SN y, NA y |
| 38 | CDKN3 | a, p | Neuroblastoma [63] | Low expression |
| 39 | PTPDC1 | a, b, Δ | PD GWAS [64] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 40 | PTEN | a, q, r, s, Δ | PD [65] | CCx x, CbCx x, H x, A y, SN y, NAc y |
| 41 | TNS1 | a, r, s, t, u, v | Not defined | CCx x, CbCx x, H y, A y, SN y, NAc y |
| 42 | TNS2 | a, r, s, t, u, v, w | Schizophrenia [66] | CCx y, CbCx y, H y, A y, SN y, NAc y |
| 43 | TPTE | a, r, s, Δ | Neuropathic pain [67] | Not defined |
| 44 | TPTE2 | a, r, s, Δ | Not defined | Not defined |
| S.No. | Gene Name | Inhibitors Validated in Biomedical Literature | Activators Validated in Biomedical Literature | Mouse Model Employed in Biomedical Literature * |
|---|---|---|---|---|
| 1 | DUSP1 | BCI Φ [156], NSC 95397 Φ [157], NU-126 [158], Sanguinarine chloride Φ [159] | Salbutamol Φ [160], Formoretol Φ [160], Dexamethasone Φ [161], JWH015 Φ [162] | KO; Neuronal death [145] |
| 2 | DUSP2 | Salubrinal Φ [163] | Not defined | KO; Arthritis [164] |
| 3 | DUSP4 | Y [165] | Not defined | KO; Synaptic plasticity [32] |
| 4 | DUSP5 | CSDDD2320, RR505, RR506, SM1842 [166] | Not defined | Transgenic; Inflammation [167] |
| 5 | DUSP6 | BCI Φ [156], NSC 95397 Φ [157], NSC 45382 Φ [168], NSC 295642 Φ [168], NSC 357756 [168] | JWH015 Φ [162] | KO; Allodynia [169], Transgenic; FGFR signaling [170] |
| 6 | DUSP7 | Y [171] | Not defined | Not defined |
| 7 | DUSP8 | Arsenite Φ, Anisomycin Φ inhibit the mouse ortholog M3/6 [172] | Not defined | KO, Transgenic; Ventricular remodeling [173] |
| 8 | DUSP9 | Y [174,175] | Not defined | KO; Placental organogenesis [176] |
| 9 | DUSP10 | AS077234-4 Φ [38] | Not defined | KO; Immune response [177] |
| 10 | DUSP16 | Y [178] | Not defined | KO; Axon degeneration [39] |
| 11 | STYXL1 | Not defined | Not defined | Not defined |
| 12 | DUPD1 | NSC 95397 Φ [179], NSC 663284 Φ [179] | Not defined | Not defined |
| 13 | DUSP3 | RK-682 Φ [180], MLS-0437605 [181], NU-126 [158], Isovenaciolide [182] | Not defined | KO; Angiogenesis [183] |
| 14 | DUSP11 | Sodium (ortho)vanadate Φ [184], Magnesium Chloride Φ [184] | Not defined | KO; Immune response [185] |
| 15 | DUSP12 | Zinc chelators (Possibly) [186] | Not defined | KO; Cardiac hypertrophy [187] |
| 16 | DUSP13 | PTP inhibitor V Φ [188] | Not defined | Not defined |
| 17 | DUSP14 | PTP inhibitor IV Φ [189], NSC-95397 Φ [190] | Not defined | KO; Immune response [191] |
| 18 | DUSP15 | Y [192] | Not defined | Transgenic; Myelination [193] |
| 19 | DUSP18 | Sodium orthovanadate Φ [194], Iodoaretic acid Φ [195] | Not defined | Not defined |
| 20 | DUSP19 | Sodium (ortho)vanadate Φ [196] | Not defined | Not defined |
| 21 | DUSP21 | Sodium orthovanadate Φ [194] | Not defined | Not defined |
| 22 | DUSP22 | Sodium (ortho)vanadate Φ [196], BML-260 Φ [197], PRL-3 Inhibitor 1 Φ [198] | Not defined | KO; Immune response [199] |
| 23 | DUSP23 | Sodium orthovanadate Φ [200], EDTA Φ [200], N-ethylmaleimide Φ [200], Y [201] | Not defined | Not defined |
| 24 | DUSP26 | NSC-87877 Φ [202], Ethyl-3,4-dephostatin Φ [203], Y [204] | Not defined | Not defined |
| 25 | DUSP28 | U0216 Φ [205] | Not defined | Not defined |
| 26 | EPM2A | Nitric oxide Φ [206], Glycogen Φ [207], polysaccharides Φ [207] | Not defined | KO; Lafora disease [208] |
| 27 | PTPMT1 | Alexidine dihydrochloride Φ [209], Y [210] | Not defined | KO; Cardiolipin biosynthesis [155] |
| 28 | RNGTT | Mizoribine Monophosphate Φ [211] | Not defined | Not defined |
| 29 | STYX | Vandate (Sodium orthovanadate) Φ [212] | Not defined | Not defined |
| 30 | SSH1 | Slingshot Inhibitor D3 Φ [213], Sennoside A Φ [214] | Not defined | Not defined |
| 31 | SSH2 | Slingshot Inhibitor D3 Φ [213], Sennoside A Φ [214], ZINC04307500 [215] | Not defined | Not defined |
| 32 | SSH3 | Sennoside A Φ [214] | Not defined | KO; Unknown [216] |
| 33 | PTP4A1 | Thienopyridone Φ [217], Analog 3 Φ [218], Pentamidine Φ [219] | Not defined | CKO; Liver regeneration [220] |
| 34 | PTP4A2 | Thienopyridone Φ [217], Analog 3 Φ [218], Pentamidine Φ [219] | Not defined | KO; Oncogenesis [221] |
| 35 | PTP4A3 | BR-1 Φ [222], Analog 13 [223], PRL-3 inhibitor 1Φ [224], Thienopyridone Φ [217], Analog 3 Φ [218], Pentamidine Φ [219] | Not defined | KO; Colon cancer [225] |
| 36 | CDC14A | Not defined | Not defined | Double KO; DDR [226] |
| 37 | CDC14B | Not defined | Not defined | CKO; DDR [227] |
| 38 | CDKN3 | Sodium orthovanadate Φ [228] | Not defined | KO; Cancer [229] |
| 39 | PTPDC1 | Not defined | Not defined | KO; Unknown [230] |
| 40 | PTEN | bpV(phen) Φ [231], bpV(pic) Φ [231], VO-Ohpic Φ [231], SF1670 Φ [231], bpV(HOpic) Φ [232] | Not defined | KO; Cortical dysplasia [233] |
| 41 | TNS1 | Not defined | Not defined | KO; Angiogenesis [234] |
| 42 | TNS2 | DHTS Φ [235] | Not defined | KO; Renal failure [236] |
| 43 | TPTE | Not defined | Not defined | Not defined |
| 44 | TPTE2 | Not defined | Not defined | Not defined |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhore, N.; Wang, B.-J.; Chen, Y.-W.; Liao, Y.-F. Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases. Int. J. Mol. Sci. 2017, 18, 1963. https://doi.org/10.3390/ijms18091963
Bhore N, Wang B-J, Chen Y-W, Liao Y-F. Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases. International Journal of Molecular Sciences. 2017; 18(9):1963. https://doi.org/10.3390/ijms18091963
Chicago/Turabian StyleBhore, Noopur, Bo-Jeng Wang, Yun-Wen Chen, and Yung-Feng Liao. 2017. "Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases" International Journal of Molecular Sciences 18, no. 9: 1963. https://doi.org/10.3390/ijms18091963
APA StyleBhore, N., Wang, B.-J., Chen, Y.-W., & Liao, Y.-F. (2017). Critical Roles of Dual-Specificity Phosphatases in Neuronal Proteostasis and Neurological Diseases. International Journal of Molecular Sciences, 18(9), 1963. https://doi.org/10.3390/ijms18091963