“Autoimmune(-Like)” Drug and Herb Induced Liver Injury: New Insights into Molecular Pathogenesis

Abstract
1. Introduction
2. Clinical Context of Drug-Induced Liver Injury and Hepatic Injury due to Herbal and Dietary Supplements (DILI/HDS) and Autoimmune Hepatitis (AIH)
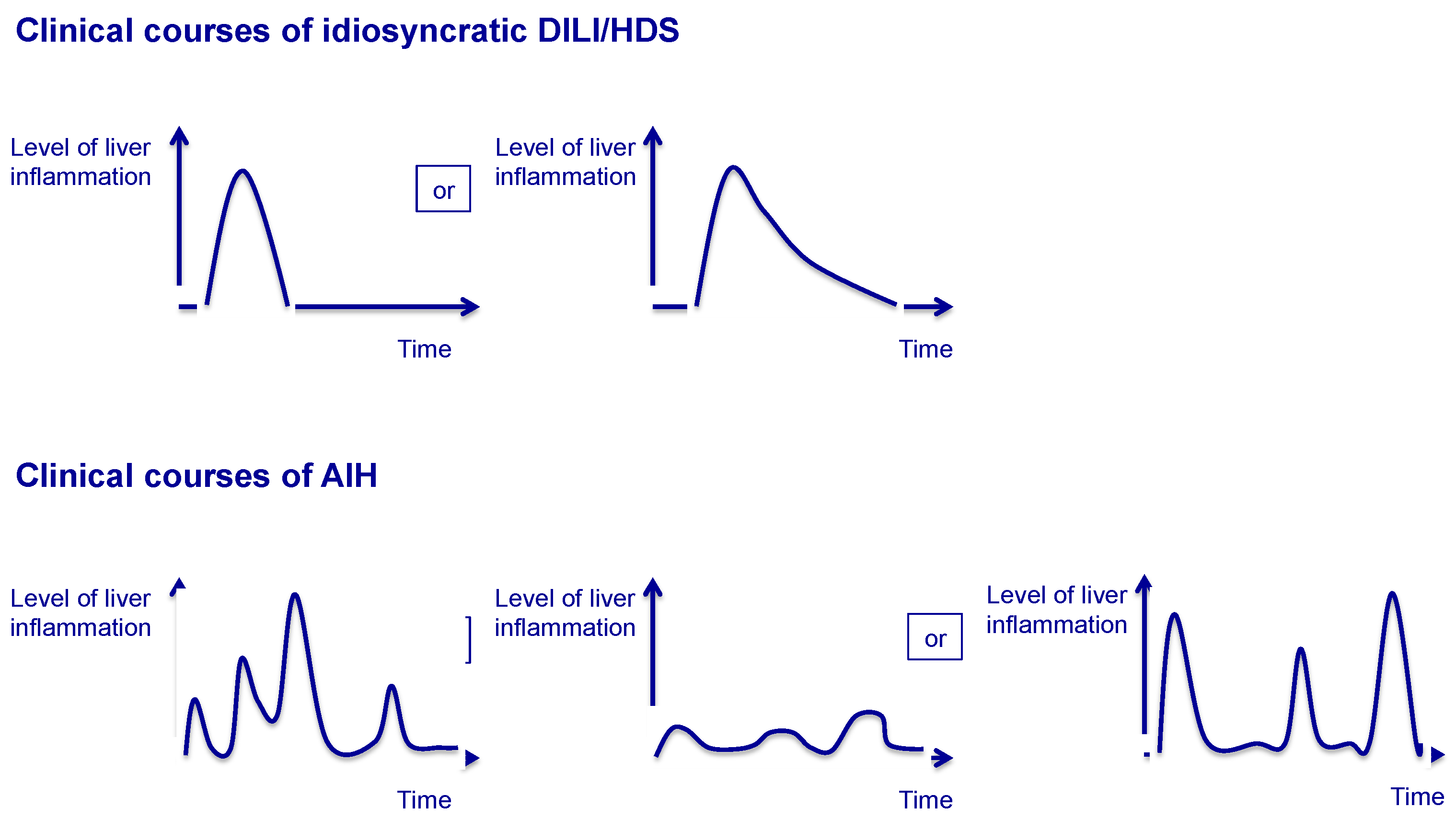
2.1. Drug-Induced Liver Injury and Hepatic Injury Due to Herbal and Dietary Supplements
2.2. Autoimmune Hepatitis
2.3. Clinical Scenarios Involving Both DILI/HDS and AIH
3. Molecular Mechanisms of DILI/HDS and AIH
3.1. Genetic Background
3.2. Neoantigens, Antigen Presentation and Triggering Events
3.3. Metabolism
3.4. Pro-Inflammatory Mechanisms
3.5. Regulatory Mechanisms
4. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AIH | Autoimmune hepatitis |
| ALH | Acute liver failure |
| ANA | Anti-nuclear antibodies |
| APAP | (N-) Acetyl-para-aminophenol, acetaminophen |
| APC | Antigen-presenting cells |
| CARD10 | Caspase recruitment domain 10 |
| Cbl-bCTLA-4 | Casitas B-cell lymphomaCytotoxic T lymphocyte antigen 4 |
| CYP | Cytochrome P450 |
| DAMPs | Damage associated molecular pattern molecules |
| DILI | Drug-induced liver injury |
| DC | Dendritic cells |
| DRESS | Drug rash with eosinophilia and systemic symptoms |
| ELISAFTCD | Enzyme-linked Immunosorbent AssayFormiminotransferase cyclodeaminase |
| FXR | Farnesoid X receptor |
| GWAS | Genome-wide association study |
| GST | Glutathione S-transferase |
| HDS | Herbal and dietary supplements |
| HLA | Human leucocyte antigen |
| IAIHG | International autoimmune hepatitis group |
| IL | Interleukin |
| IFNγ | Interferon gamma |
| LC1 | Liver cytosol 1 |
| LKM | Liver-kidney microsomes |
| LSEC | Liver sinusoidal endothelial cells |
| MELD | Model of end-stage liver disease |
| MDR3 | Multidrug resistance protein 3 |
| MHC | Major histocompatibility complex |
| MS | Multiple sclerosis |
| NAC | N-acetylcysteine |
| NAPQI | N-acetyl-p-benzoquinoneimine |
| NAT2 | N-acetyltransferase 2 |
| PBMC | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death-1 |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| RUCAM | Roussel uclaf causality assessment method |
| SEPSECS | O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase |
| SH2B3 | Scr homology 2 adaptor protein 3 |
| SLA/LP | Soluble liver antigen/Liver-pancreas antigen |
| SMA | Smooth muscle antigen |
| TGFβ | Transforming growth factor beta |
| TNFα | Tumor necrosis factor-alpha |
| Treg | Regulatory T cells |
References
- Aithal, G.P.; Watkins, P.B.; Andrade, R.J.; Larrey, D.; Molokhia, M.; Takikawa, H.; Hunt, C.M.; Wilke, R.A.; Avigan, M.; Kaplowitz, N.; et al. Case definition and phenotype standardization in drug-induced liver injury. Clin. Pharmacol. Ther. 2011, 89, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Chalasani, N.P.; Lee, W.M.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Hayashi, P.H.; Davern, T.J.; Navarro, V.; Reddy, R.; et al. Hepatic histological findings in suspected drug-induced liver injury: Systematic evaluation and clinical associations. Hepatology 2014, 59, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E. The histopathological evaluation of drug-induced liver injury. Histopathology 2017, 70, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and Outcomes of 899 Patients with Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 2015, 148, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.J.; Khan, I.; Björnsson, E.; Seeff, L.B.; Serrano, J.; Hoofnagle, J.H. Liver injury from herbal and dietary supplements. Hepatology 2017, 65, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Sgro, C.; Clinard, F.; Ouazir, K.; Chanay, H.; Allard, C.; Guilleminet, C.; Lenoir, C.; Lemoine, A.; Hillon, P. Incidence of drug-induced hepatic injuries: A French population-based study. Hepatology 2002, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S.; Bergmann, O.M.; Björnsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.J.; Barnhart, H.; Bonkovsky, H.L.; Davern, T.; Fontana, R.J.; Grant, L.; Reddy, K.R.; Seeff, L.B.; Serrano, J.; Sherker, A.H.; et al. Liver injury from herbals and dietary supplements in the U.S. Drug-Induced Liver Injury Network. Hepatology 2014, 60, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Koch, D.G.; Lee, W.M.; Acute Liver Failure Study Group. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 2010, 52, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.S.; Forde, K.A.; Carbonari, D.M.; Lewis, J.D.; Leidl, K.B.; Reddy, K.R.; Haynes, K.; Roy, J.; Sha, D.; Marks, A.R.; et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology 2015, 148, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug. Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.X.; Govindarajan, S.; Kaplowitz, N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 2004, 127, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.; Simonart, T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): An update. Dermatology 2003, 206, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Einarsson, S.; Saha, C.; Niklasson, A.; Bjornsson, E.; Chalasani, N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: Search for signals. Hepatology 2008, 47, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Benichou, C. Causality assessment of adverse reactions to drugs—I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol. 1993, 46, 1323–1330. [Google Scholar] [CrossRef]
- Benichou, C.; Danan, G.; Flahault, A. Causality assessment of adverse reactions to drugs—II. An original model for validation of drug causality assessment methods: Case reports with positive rechallenge. J. Clin. Epidemiol. 1993, 46, 1331–1336. [Google Scholar] [CrossRef]
- Fontana, R.J.; Seeff, L.B.; Andrade, R.J.; Björnsson, E.; Day, C.P.; Serrano, J.; Hoofnagle, J.H. Standardization of nomenclature and causality assessment in drug-induced liver injury: Summary of a clinical research workshop. Hepatology 2010, 52, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Rakela, J.; Mosley, J.W.; Edwards, V.M.; Govindarajan, S.; Alpert, E.; The Acute Hepatic Failure Study Group. A double-blinded, randomized trial of hydrocortisone in acute hepatic failure. Dig. Dis. Sci. 1991, 36, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. Randomised trial of steroid therapy in acute liver failure. Gut 1979, 20, 620–623. [Google Scholar]
- Dalton, H.R.; Fellows, H.J.; Stableforth, W.; Joseph, M.; Thurairajah, P.H.; Warshow, U.; Hazeldine, S.; Remnarace, R.; Ijaz, S.; Hussaini, S.H.; et al. The role of hepatitis E virus testing in drug-induced liver injury. Aliment. Pharmacol. Ther. 2007, 15, 1429–1435. [Google Scholar]
- Davern, T.J.; Chalasani, N.; Fontana, R.J.; Hayashi, P.H.; Protiva, P.; Kleiner, D.E.; Engle, R.E.; Nguyen, H.; Emerson, S.U.; Purcell, R.H.; et al. Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology 2011, 141, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Karkhanis, J.; Verna, E.C.; Chang, M.S.; Stravitz, R.T.; Schilsky, M.; Lee, W.M.; Brown, R.S., Jr.; Acute Liver Failure Study Group. Steroid use in acute liver failure. Hepatology 2014, 59, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.; Talwalkar, J.; Treeprasertsuk, S.; Kamath, P.S.; Takahashi, N.; Sanderson, S.; Neuhauser, M.; Lindor, K. Drug-induced autoimmune hepatitis: Clinical characteristics and prognosis. Hepatology 2010, 51, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Dechêne, A.; Herzer, K.; Hilgard, P.; Syn, W.K.; Gerken, G.; Canbay, A. Steroid and ursodesoxycholic Acid combination therapy in severe drug-induced liver injury. Digestion 2011, 84, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M.; Hynan, L.S.; Rossaro, L.; Fontana, R.J.; Stravitz, R.T.; Larson, A.M.; Davern, T.J., 2nd; Murray, N.G.; McCashland, T.; Reisch, J.S.; et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009, 137, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Lohse, A.W.; Vergani, D. Autoimmune hepatitis—Update 2015. J. Hepatol. 2015, 62, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Parés, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.J.; McFarlane, I.G. Meeting report: International Autoimmune Hepatitis Group. Hepatology 1993, 18, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.; Berg, P.A.; Bianchi, F.B.; Bianchi, L.; Burroughs, A.K.; Cancado, E.L.; Chapman, R.W.; Cooksley, W.G.; Czaja, A.J.; Desmet, V.J.; et al. International Autoimmune Hepatitis Group Report: Review of criteria for diagnosis of autoimmune hepatitis. J. Hepatol. 1999, 31, 929–938. [Google Scholar]
- Suzuki, A.; Brunt, E.M.; Kleiner, D.E.; Miquel, R.; Smyrk, T.C.; Andrade, R.J.; Lucena, M.I.; Castiella, A.; Lindor, K.; Björnsson, E. The use of liver biopsy evaluation in discrimination of idiopathic autoimmune hepatitis versus drug-induced liver injury. Hepatology 2011, 54, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Vergani, D.; Alvarez, F.; Bianchi, F.B.; Cançado, E.L.; Mackay, I.R.; Manns, M.P.; Nishioka, M.; Penner, E.; International Autoimmune Hepatitis Group. Liver autoimmune serology: A consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J. Hepatol. 2004, 41, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Herkel, J.; Heidrich, B.; Nieraad, N.; Wies, I.; Rother, M.; Lohse, A.W. Fine specificity of autoantibodies to soluble liver antigen and liver/pancreas. Hepatology 2002, 35, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Stechemesser, E.; Klein, R.; Berg, P.A. Characterization and clinical relevance of liver-pancreas antibodies in autoimmune hepatitis. Hepatology 1993, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Homberg, J.C.; Abuaf, N.; Bernard, O.; Islam, S.; Alvarez, F.; Khalil, S.H.; Poupon, R.; Darnis, F.; Lévy, V.G.; Grippon, P. Chronic active hepatitis associated with antiliver/kidney microsome antibody type 1: A second type of "autoimmune" hepatitis. Hepatology 1987, 7, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Abuaf, N.; Cavalli, F.; Durand, V.; Johanet, C.; Homberg, J.C. Antibody to liver cytosol (anti-LC1) in patients with autoimmune chronic active hepatitis type 2. Hepatology 1988, 8, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, G.V.; Portmann, B.; Reid, F.; Donaldson, P.T.; Doherty, D.G.; McCartney, M.; Mowat, A.P.; Vergani, D.; Mieli-Vergani, G. Autoimmune hepatitis in childhood: A 20-year experience. Hepatology 1997, 25, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, K.H.; Melms, A.; Berg, P.A. Anti-actin antibodies of IgM and IgG class in chronic liver diseases detected by fluorometric immunoassay. Liver 1983, 3, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, C.; Herkel, J.; Lüth, S.; Galle, P.R.; Schramm, C.; Lohse, A.W. Evaluation of F-Actin ELISA for the diagnosis of autoimmune hepatitis. Am. J. Gastroenterol. 2006, 101, 2731–2736. [Google Scholar] [CrossRef] [PubMed]
- Hov, J.R.; Boberg, K.M.; Karlsen, T.H. Autoantibodies in primary sclerosing cholangitis. World. J. Gastroenterol. 2008, 14, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Ma, Y.; Smith, H.M.; Portmann, B.; Wendon, J.; Vergani, D. The significance of autoantibodies and immunoglobulins in acute liver failure: A cohort study. J. Hepatol. 2007, 47, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Gassert, D.J.; Garcia, H.; Tanaka, K.; Reinus, J.F. Corticosteroid-responsive cryptogenic chronic hepatitis: Evidence for seronegative autoimmune hepatitis. Dig. Dis. Sci. 2007, 52, 2433–2437. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Metzler, F.; Geiger, E.; Heinrich, E.; Hallensleben, M.; Manns, M.P.; Vogel, A. Prediction of short- and long-term outcome in patients with autoimmune hepatitis. Hepatology 2015, 62, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Couto, C.A.; Bittencourt, P.L.; Porta, G.; Abrantes-Lemos, C.P.; Carrilho, F.J.; Guardia, B.D.; Cançado, E.L. Antismooth muscle and antiactin antibodies are indirect markers of histological and biochemical activity of autoimmune hepatitis. Hepatology 2014, 59, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Wies, I.; Brunner, S.; Henninger, J.; Herkel, J.; Kanzler, S.; Meyer zum Büschenfelde, K.H.; Lohse, A.W. Identification of target antigen for SLA/LP autoantibodies in autoimmune hepatitis. Lancet 2000, 355, 1510–1515. [Google Scholar] [CrossRef]
- Mix, H.; Weiler-Normann, C.; Thimme, R.; Ahlenstiel, G.; Shin, E.C.; Herkel, J.; David, C.S.; Lohse, A.W.; Rehermann, B. Identification of CD4 T-cell epitopes in soluble liver antigen/liver pancreas autoantigen in autoimmune hepatitis. Gastroenterology 2008, 135, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, M.; Meunier-Rotival, M.; Bernard, O.; Alvarez, F. Anti-liver kidney microsome antibody recognizes a cytochrome P450 from the IID subfamily. J. Exp. Med. 1988, 168, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Johnson, E.F.; Griffin, K.J.; Tan, E.M.; Sullivan, K.F. Major antigen of liver kidney microsomal autoantibodies in idiopathic autoimmune hepatitis is cytochrome P450db1. J. Clin. Investig. 1989, 83, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Griffin, K.J.; Sullivan, K.F.; Johnson, E.F. LKM-1 autoantibodies recognize a short linear sequence in P450IID6, a cytochrome P-450 monooxygenase. J. Clin. Investig. 1991, 88, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, P.; Hajoui, O.; Homberg, J.C.; Alvarez, F. Formiminotransferase cyclodeaminase is an organ-specific autoantigen recognized by sera of patients with autoimmune hepatitis. Gastroenterology 1999, 116, 643–649. [Google Scholar] [CrossRef]
- Muratori, L.; Sztul, E.; Muratori, P.; Gao, Y.; Ripalti, A.; Ponti, C.; Lenzi, M.; Landini, M.P.; Bianchi, F.B. Distinct epitopes on formiminotransferase cyclodeaminase induce autoimmune liver cytosol antibody type 1. Hepatology 2001, 34, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Nishioka, M.; Morshed, S.A.; Hachiya, T. Patterns of nuclear immunofluorescence and reactivities to recombinant nuclear antigens in autoimmune hepatitis. Gastroenterology 1994, 107, 200–207. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Autoimmune hepatitis. J. Hepatol. 2015, 63, 971–1004. [Google Scholar]
- Van Gerven, N.M.; Verwer, B.J.; Witte, B.I.; van Hoek, B.; Coenraad, M.J.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; de Man, R.A.; Drenth, J.P.; et al. Relapse is almost universal after withdrawal of immunosuppressive medication in patients with autoimmune hepatitis in remission. J. Hepatol. 2013, 58, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Weiler-Normann, C.; Schramm, C. Drug induced liver injury and its relationship to autoimmune hepatitis. J. Hepatol. 2011, 55, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, J.; Kanda, T.; Yasui, S.; Haga, Y.; Sasaki, R.; Nakamura, M.; Wu, S.; Nakamoto, S.; Arai, M.; Iino, Y.; et al. Autoimmune hepatitis following drug-induced liver injury in an elderly patient. Clin. J. Gastroenterol. 2016, 9, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Kaplowitz, N.; Hallal, H.; Castiella, A.; García-Bengoechea, M.; Otazua, P.; Berenguer, M.; Fernandez, M.C.; Planas, R.; Andrade, R.J. Recurrent drug-induced liver injury (DILI) with different drugs in the Spanish Registry: The dilemma of the relationship to autoimmune hepatitis. J. Hepatol. 2011, 55, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Aithal, P.G.; Day, C.P. The natural history of histologically proved drug induced liver disease. Gut 1999, 44, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.G.; Chalasani, N. Chronic liver injury induced by drugs: A systematic review. Liver Int. 2015, 35, 2343–2353. [Google Scholar] [CrossRef] [PubMed]
- Muratori, P.; Granito, A.; Quarneti, C.; Ferri, S.; Menichella, R.; Cassani, F.; Pappas, G.; Bianchi, F.B.; Lenzi, M.; Muratori, L. Autoimmune hepatitis in Italy: The Bologna experience. J. Hepatol. 2009, 50, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Dinh, H.; Arenovich, T.; Marcus, V.A.; Wanless, I.R.; Heathcote, E.J. Autoimmune hepatitis: Effect of symptoms and cirrhosis on natural history and outcome. Hepatology 2005, 42, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Ngu, J.H.; Gearry, R.B.; Frampton, C.M.; Stedman, C.A. Predictors of poor outcome in patients with autoimmune hepatitis: A population-based study. Hepatology 2013, 57, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- DeLemos, A.S.; Foureau, D.M.; Jacobs, C.; Ahrens, W.; Russo, M.W.; Bonkovsky, H.L. Drug-induced liver injury with autoimmune features. Semin. Liver Dis. 2014, 34, 194–204. [Google Scholar] [PubMed]
- Goldstein, N.S.; Bayati, N.; Silverman, A.L.; Gordon, S.C. Minocycline as a cause of drug-induced autoimmune hepatitis. Report of four cases and comparison with autoimmune hepatitis. Am. J. Clin. Pathol. 2000, 114, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P. Nonsteroidal anti-inflammatory drug-induced hepatotoxicity. Clin. Liver Dis. 2007, 11, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Strand, V.; Greenberg, J.D.; Curtis, J.R.; Kavanaugh, A.; Kremer, J.M.; Anofrei, A.; Reed, G.; Calabrese, L.; Hooper, M.; et al. Risk of elevated liver enzymes associated with TNF inhibitor utilisation in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Doherty, D.G.; Hayllar, K.M.; McFarlane, I.G.; Johnson, P.J.; Williams, R. Susceptibility to autoimmune chronic active hepatitis: Human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology 1991, 13, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Strettell, M.D.; Donaldson, P.T.; Thomson, L.J.; Santrach, P.J.; Moore, S.B.; Czaja, A.J.; Williams, R. Allelic basis for HLA-encoded susceptibility to type 1 autoimmune hepatitis. Gastroenterology 1997, 112, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- De Boer, Y.S.; van Gerven, N.M.; Zwiers, A.; Verwer, B.J.; van Hoek, B.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; Drenth, J.P.; den Ouden, J.W.; et al. Genome-wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology 2014, 147, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Donaldson, P.T. Genetic susceptibilities for immune expression and liver cell injury in autoimmune hepatitis. Immunol. Rev. 2000, 174, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.I.; Aithal, G.P. Human leukocyte antigen genetic risk factors of drug-induced liver toxicology. Expert Opin. Drug Metab. Toxicol. 2015, 11, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.T.; Daly, A.K.; Henderson, J.; Graham, J.; Pirmohamed, M.; Bernal, W.; Day, C.P.; Aithal, G.P. Human leucocyte antigen class II genotype in susceptibility and resistance to co-amoxiclav-induced liver injury. J. Hepatol. 2010, 53, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Ruiz-Cabello, F.; Donaldson, P.T.; Stephens, C.; et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Mallal, S.; Nolan, D.; Witt, C.; Masel, G.; Martin, A.M.; Moore, C.; Sayer, D.; Castley, A.; Mamotte, C.; Maxwell, D.; et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002, 359, 727–732. [Google Scholar] [CrossRef]
- Nicoletti, P.; Aithal, G.P.; Bjornsson, E.S.; Andrade, R.J.; Sawle, A.; Arrese, M.; Barnhart, H.X.; Bondon-Guitton, E.; Hayashi, P.H.; Bessone, F.; et al. Association of Liver Injury From Specific Drugs, or Groups of Drugs, With Polymorphisms in HLA and Other Genes in a Genome-Wide Association Study. Gastroenterology 2017, 152, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, K.; Czaja, A.J.; Jones, D.E.; Donaldson, P.T. Cytotoxic T lymphocyte antigen-4 (CTLA-4) gene polymorphisms and susceptibility to type 1 autoimmune hepatitis. Hepatology 2000, 31, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J.; Cookson, S.; Constantini, P.K.; Clare, M.; Underhill, J.A.; Donaldson, P.T. Cytokine polymorphisms associated with clinical features and treatment outcome in type 1 autoimmune hepatitis. Gastroenterology 1999, 117, 645–652. [Google Scholar] [CrossRef]
- Li, Y.; He, X.; Schembri-King, J.; Jakes, S.; Hayashi, J. Cloning and characterization of human Lnk, an adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J. Immunol. 2000, 164, 5199–5206. [Google Scholar] [CrossRef] [PubMed]
- Blonska, M.; Lin, X. NF-κB signaling pathways regulated by CARMA family of scaffold proteins. Cell. Res. 2011, 21, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Ramsay, L.; Daly, A.K.; Sonchit, N.; Leathart, J.B.; Alexander, G.; Kenna, J.G.; Caldwell, J.; Day, C.P. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology 2004, 39, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Urban, T.J.; Daly, A.K.; Aithal, G.P. Genetic basis of drug-induced liver injury: Present and future. Semin. Liver Dis. 2014, 34, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Vuilleumier, N.; Rossier, M.F.; Chiappe, A.; Degoumois, F.; Dayer, P.; Mermillod, B.; Nicod, L.; Desmeules, J.; Hochstrasser, D. CYP2E1 genotype and isoniazid-induced hepatotoxicity in patients treated for latent tuberculosis. Eur. J. Clin. Pharmacol. 2006, 62, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Chern, H.D.; Su, W.J.; Wu, J.C.; Chang, S.C.; Chiang, C.H.; Chang, F.Y.; Lee, S.D. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology 2003, 37, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Aithal, G.P.; Leathart, J.B.; Swainsbury, R.A.; Dang, T.S.; Day, C.P. Genetic susceptibility to diclofenac-induced hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 2007, 132, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Su, W.J.; Huang, Y.H.; Chen, C.Y.; Chang, F.Y.; Lin, H.C.; Lee, S.D. Genetic polymorphisms of manganese superoxide dismutase, NAD(P)H: Quinone oxidoreductase, glutathione S-transferase M1 and T1, and the susceptibility to drug-induced liver injury. J. Hepatol. 2007, 47, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Andrade, R.J.; Martínez, C.; Ulzurrun, E.; García-Martín, E.; Borraz, Y.; Fernández, M.C.; Romero-Gomez, M.; Castiella, A.; Planas, R.; et al. Glutathione S-transferase m1 and t1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology 2008, 48, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; García-Martín, E.; Andrade, R.J.; Martínez, C.; Stephens, C.; Ruiz, J.D.; Ulzurrun, E.; Fernandez, M.C.; Romero-Gomez, M.; Castiella, A.; et al. Mitochondrial superoxide dismutase and glutathione peroxidase in idiosyncratic drug-induced liver injury. Hepatology 2010, 52, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.E.; Meng, X.; Elliott, V.L.; Kitteringham, N.R.; Pirmohamed, M.; Park, B.K. Characterisation of flucloxacillin and 5-hydroxymethyl flucloxacillin haptenated HSA in vitro and in vivo. Proteom. Clin. Appl. 2009, 3, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, N.; Choudhuri, K.; Ma, Y.; Mahmoud, A.; Bogdanos, D.P.; Muratori, L.; Bianchi, F.; Williams, R.; Mieli-Vergani, G.; Vergani, D. Cytochrome P4502D6(193–212): A new immunodominant epitope and target of virus/self cross-reactivity in liver kidney microsomal autoantibody type 1-positive liver disease. J. Immunol. 2003, 170, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Lüth, S.; Huber, S.; Schramm, C.; Buch, T.; Zander, S.; Stadelmann, C.; Brück, W.; Wraith, D.C.; Herkel, J.; Lohse, A.W. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J. Clin. Investig. 2008, 118, 3403–3410. [Google Scholar] [PubMed]
- Carambia, A.; Frenzel, C.; Bruns, O.T.; Schwinge, D.; Reimer, R.; Hohenberg, H.; Huber, S.; Tiegs, G.; Schramm, C.; Lohse, A.W.; et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J. Hepatol. 2013, 58, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Muratori, L.; Parola, M.; Ripalti, A.; Robino, G.; Muratori, P.; Bellomo, G.; Carini, R.; Lenzi, M.; Landini, M.P.; Albano, E.; et al. Liver/kidney microsomal antibody type 1 targets CYP2D6 on hepatocyte plasma membrane. Gut 2000, 46, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Uetrecht, J. Evaluation of which reactive metabolite, if any, is responsible for a specific idiosyncratic reaction. Drug Metab. Rev. 2006, 38, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Meyer zum Büschenfelde, K.H.; Lohse, A.W.; Gerken, G.; Treichel, U.; Löhr, H.F.; Mohr, H.; Grosse, A.; Dienes, H.P. The role of autoimmunity in hepatitis C infection. J. Hepatol. 1995, 22, 93–96. [Google Scholar] [PubMed]
- Senaldi, G.; Portmann, B.; Mowat, A.P.; Mieli-Vergani, G.; Vergani, D. Immunohistochemical features of the portal tract mononuclear cell infiltrate in chronic aggressive hepatitis. Arch. Dis. Child. 1992, 67, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Jaeckel, E. Animal models of autoimmune hepatitis. Semin. Liver Dis. 2002, 22, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Bjornsson, E.; Niklasson, A.; Chalasani, N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology 2010, 51, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Senior, J.R. What is idiosyncratic hepatotoxicity? What is it not? Hepatology 2008, 47, 1813–1815. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Andrade, R.J.; Kaplowitz, N.; García-Cortes, M.; Fernández, M.C.; Romero-Gomez, M.; Bruguera, M.; Hallal, H.; Robles-Diaz, M.; Rodriguez-González, J.F.; et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: The influence of age and sex. Hepatology 2009, 49, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Björnsson, E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology 2010, 138, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Ingelman-Sundberg, M. The Importance of Patient-Specific Factors for Hepatic Drug Response and Toxicity. Int. J. Mol. Sci. 2016, 17, 1714. [Google Scholar] [CrossRef] [PubMed]
- Felker, D.; Lynn, A.; Wang, S.; Johnson, D.E. Evidence for a potential protective effect of carnitine-pantothenic acid co-treatment on valproic acid-induced hepatotoxicity. Expert Rev. Clin. Pharmacol. 2014, 7, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Kalhorn, T.F.; Slattery, J.T. Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology 2002, 36, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A.; Luyendyk, J.P.; Maddox, J.F.; Ganey, P.E. Inflammation and Drug Idiosyncrasy—Is There a Connection? J. Pharmacol. Exp. Ther. 2003, 307, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.; Harkema, J.; Pestka, J.; Ganey, P. Is Exposure to Bacterial Endotoxin a Determinant of Susceptibility to Intoxication from Xenobiotic Agents? Toxicol. Appl. Pharmacol. 1997, 147, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jones, A.D.; Harkema, J.R.; Roth, R.A.; Ganey, P.E. Amiodarone exposure during modest inflammation induces idiosyncrasy-like liver injury in rats: Role of tumor necrosis factor-α. Toxicol. Sci. 2012, 125, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Stachlewitz, R.F.; Liguori, M.J.; Blomme, E.A.G.; Waring, J.F.; Luyendyk, J.P.; Maddox, J.F.; Ganey, P.E.; Roth, R.A. Modest Inflammation Enhances Diclofenac Hepatotoxicity in Rats: Role of Neutrophils and Bacterial Translocation. J. Pharmacol. Exp. Ther. 2006, 319, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.R.; Uetrecht, J.; Shear, N.H. Idiosyncratic drug reactions: The reactive metabolite syndromes. Lancet 2000, 356, 1587–1591. [Google Scholar] [CrossRef]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Azuma, J.; Ohno, M.; Kubota, R.; Yokota, S.; Nagai, T.; Tsuyuguchi, K.; Okuda, Y.; Takashima, T.; Kamimura, S.; Fujio, Y.; et al. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: A randomized controlled trial for pharmacogenetics-based therapy. Eur. J. Clin. Pharmacol. 2013, 69, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018. [Google Scholar] [CrossRef] [PubMed]
- Martin-Murphy, B.V.; Holt, M.P.; Ju, C. The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett. 2010, 192, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.E.; Trauner, M.; van Staden, C.J.; Lee, P.H.; Ramachandran, B.; Eschenberg, M.; Afshari, C.A.; Qualls, C.W., Jr.; Lightfoot-Dunn, R.; Hamadeh, H.K. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol. Sci. 2010, 118, 485–500. [Google Scholar] [CrossRef] [PubMed]
- Mosedale, M.; Watkins, P.B. Drug-induced liver injury: Advances in mechanistic understanding that will inform risk management. Clin. Pharmacol. Ther. 2017, 101, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; McDermott, W.V. Comfrey herb tea and hepatic veno-occlusive disease. Lancet 1989, 1, 657–658. [Google Scholar] [CrossRef]
- Lin, G.; Wang, J.Y.; Li, N.; Li, M.; Gao, H.; Ji, Y.; Zhang, F.; Wang, H.; Zhou, Y.; Ye, Y.; et al. Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J. Hepatol. 2011, 54, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Lekehal, M.; Pessayre, D.; Lereau, J.M.; Moulis, C.; Fouraste, I.; Fau, D. Hepatotoxicity of the herbal medicine germander: Metabolic activation of its furano diterpenoids by cytochrome P450 3A Depletes cytoskeleton-associated protein thiols and forms plasma membrane blebs in rat hepatocytes. Hepatology 1996, 24, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Fau, D.; Lekehal, M.; Farrell, G.; Moreau, A.; Moulis, C.; Feldmann, G.; Haouzi, D.; Pessayre, D. Diterpenoids from germander, an herbal medicine, induce apoptosis in isolated rat hepatocytes. Gastroenterology 1997, 113, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Navarro, V.J.; Bonkovsky, H.L.; Hwang, S.I.; Vega, M.; Barnhart, H.; Serrano, J. Catechins in dietary supplements and hepatotoxicity. Dig. Dis. Sci. 2013, 58, 2682–2690. [Google Scholar] [CrossRef] [PubMed]
- Monshi, M.M.; Faulkner, L.; Gibson, A.; Jenkins, R.E.; Farrell, J.; Earnshaw, C.J.; Alfirevic, A.; Cederbrant, K.; Daly, A.K.; French, N.; et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology 2013, 57, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Saide, K.; Farrell, J.; Faulkner, L.; Tailor, A.; Ogese, M.; Daly, A.K.; Pirmohamed, M.; Park, B.K.; Naisbitt, D.J. Characterization of amoxicillin- and clavulanic acid-specific T cells in patients with amoxicillin-clavulanate-induced liver injury. Hepatology 2015, 62, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Maria, V.A.; Victorino, R.M. Diagnostic value of specific T cell reactivity to drugs in 95 cases of drug induced liver injury. Gut 1997, 41, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Matsumaru, K.; Feng, G.; Kaplowitz, N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor-α-induced apoptosis in cultured mouse hepatocytes. Hepatology 2002, 36, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, J.; Liu, F.; Uetrecht, J.P. Involvement of T helper 17 cells in d-penicillamine-induced autoimmune disease in Brown Norway rats. Toxicol. Sci. 2011, 120, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Steuerwald, N.M.; Foureau, D.M.; Norton, H.J.; Zhou, J.; Parsons, J.C.; Chalasani, N.; Fontana, R.J.; Watkins, P.B.; Lee, W.M.; Reddy, K.R.; et al. Profiles of serum cytokines in acute drug-induced liver injury and their prognostic significance. PLoS ONE 2013, 8, e81974. [Google Scholar] [CrossRef] [PubMed]
- Wuillemin, N.; Terracciano, L.; Beltraminelli, H.; Schlapbach, C.; Fontana, S.; Krähenbühl, S.; Pichler, W.J.; Yerly, D. T cells infiltrate the liver and kill hepatocytes in HLA-B(∗)57:01-associated floxacillin-induced liver injury. Am. J. Pathol. 2014, 184, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S.; Gunnarsson, B.I.; Gröndal, G.; Jonasson, J.G.; Einarsdottir, R.; Ludviksson, B.R.; Gudbjörnsson, B.; Olafsson, S. Risk of drug-induced liver injury from tumor necrosis factor antagonists. Clin. Gastroenterol. Hepatol. 2015, 13, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.F.; Proverbs-Singh, T.A.; Postow, M.A. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol. 2016, 2, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Weiler-Normann, C.; Schramm, C.; Quaas, A.; Wiegard, C.; Glaubke, C.; Pannicke, N.; Möller, S.; Lohse, A.W. Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J. Hepatol. 2013, 58, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Burak, K.W.; Swain, M.G.; Santodomingo-Garzon, T.; Lee, S.S.; Urbanski, S.J.; Aspinall, A.I.; Coffin, C.S.; Myers, R.P. Rituximab for the treatment of patients with autoimmune hepatitis who are refractory or intolerant to standard therapy. Can. J. Gastroenterol. 2013, 27, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Schlaak, J.F.; Löhr, H.; Gallati, H.; Meyer zum Büschenfelde, K.H.; Fleischer, B. Analysis of the in vitro cytokine production by liver-infiltrating T cells of patients with autoimmune hepatitis. Clin. Exp. Immunol. 1993, 94, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Löhr, H.F.; Schlaak, J.F.; Lohse, A.W.; Böcher, W.O.; Arenz, M.; Gerken, G.; Meyer Zum Büschenfelde, K.H. Autoreactive CD4+ LKM-specific and anticlonotypic T-cell responses in LKM-1 antibody-positive autoimmune hepatitis. Hepatology 1996, 24, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kobayashi, M.; Hosaka, T.; Someya, T.; Akuta, N.; Kobayashi, M.; Suzuki, F.; Tsubota, A.; Saitoh, S.; Arase, Y.; et al. Peripheral CD8+/CD25+ lymphocytes may be implicated in hepatocellular injuries in patients with acute-onset autoimmune hepatitis. J. Gastroenterol. 2004, 39, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Ferri, S.; Longhi, M.S.; de Molo, C.; Lalanne, C.; Muratori, P.; Granito, A.; Hussain, M.J.; Ma, Y.; Lenzi, M.; et al. A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology 2010, 52, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Gorham, J.D.; Lin, J.T.; Sung, J.L.; Rudner, L.A.; French, M.A. Genetic regulation of autoimmune disease: BALB/c background TGF-β 1-deficient mice develop necroinflammatory IFN-gamma-dependent hepatitis. J. Immunol. 2001, 166, 6413–6422. [Google Scholar] [CrossRef] [PubMed]
- Holdener, M.; Hintermann, E.; Bayer, M.; Rhode, A.; Rodrigo, E.; Hintereder, G.; Johnson, E.F.; Gonzalez, F.J.; Pfeilschifter, J.; Manns, M.P.; et al. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J. Exp. Med. 2008, 205, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.; Qian, P.; Allen, P.M.; Peters, M.G. Hepatitis resulting from liver-specific expression and recognition of self-antigen. J. Autoimmun. 2008, 31, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Bogdanos, D.P.; Hussain, M.J.; Underhill, J.; Bansal, S.; Longhi, M.S.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology 2006, 130, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Sebode, M.; Hartl, J.; Vergani, D.; Lohse, A.W.; International Autoimmune Hepatitis Group (IAIHG). Autoimmune hepatitis: From current knowledge and clinical practice to future research agenda. Liver Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Liu, G.; Yang, R. MicroRNAs: Novel Regulators during the Immune Response. J. Cell. Physiol. 2009, 218, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. MicroRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Mkrtchian, S.; Ingelman-Sundberg, M. The role of microRNAs in liver injury at the crossroad between hepatic cell death and regeneration. Biochem. Biophys. Res. Commun. 2017, 482, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Hamada, Y.; Yamada, H.; Horii, I. Changes of micro-RNA expression in rat liver treated by acetaminophen or carbon tetrachloride--regulating role of micro-RNA for RNA expression. J. Toxicol. Sci. 2007, 32, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Kido, M.; Watanabe, N.; Okazaki, T.; Akamatsu, T.; Tanaka, J.; Saga, K.; Nishio, A.; Honjo, T.; Chiba, T. Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD-1-mediated signaling. Gastroenterology 2008, 135, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Ma, Y.; Bogdanos, D.P.; Cheeseman, P.; Mieli-Vergani, G.; Vergani, D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J. Hepatol. 2004, 41, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.R.; Liberal, R.; Holder, B.S.; Cardone, J.; Ma, Y.; Robson, S.C.; Mieli-Vergani, G.; Vergani, D.; Longhi, M.S. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology 2014, 59, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Sebode, M.; Franke, B.; Wortmann, F.; Schwinge, D.; Quaas, A.; Baron, U.; Olek, S.; Wiegard, C.; Lohse, A.W.; et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J. Hepatol. 2012, 57, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Taubert, R.; Hardtke-Wolenski, M.; Noyan, F.; Wilms, A.; Baumann, A.K.; Schlue, J.; Olek, S.; Falk, C.S.; Manns, M.P.; Jaeckel, E. Intrahepatic regulatory T cells in autoimmune hepatitis are associated with treatment response and depleted with current therapies. J. Hepatol. 2014, 61, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Metushi, I.G.; Hayes, M.A.; Uetrecht, J. Treatment of PD-1(-/-) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015, 61, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Pachkoria, K.; Lucena, M.I.; Crespo, E.; Ruiz-Cabello, F.; Lopez-Ortega, S.; Fernandez, M.A.; Romero-Gomez, M.; Madrazo, A.; Durán, J.A.; de Dios, A.M.; et al. Analysis of IL-10, IL-4 and TNF-α polymorphisms in drug-induced liver injury (DILI) and its outcome. J. Hepatol. 2008, 49, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Xiang, X.; Mo, R.; Bao, R.; Wang, P.; Guo, S.; Zhao, G.; Gui, H.; Wang, H.; Bao, S.; et al. Protective effect of Th22 cells and intrahepatic IL-22 in drug induced hepatocellular injury. J. Hepatol. 2015, 63, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.B.; Seeff, L.B. Drug-induced liver injury: Summary of a single topic clinical research conference. Hepatology 2006, 43, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Singla, R.; Sarda, P.; Mohan, A.; Makharia, G.; Jayaswal, A.; Sreenivas, V.; Singh, S. Safety of 3 different reintroduction regimens of antituberculosis drugs after development of antituberculosis treatment-induced hepatotoxicity. Clin. Infect. Dis. 2010, 50, 833–839. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Scenario | Characteristics |
|---|---|
| DILI/HDS on top of AIH |
|
| Drug-induced AIH |
|
| Second episode of DILI mimics relapsing course of AIH |
|
| Chronic DILI mimics AIH |
|
| DILI/HDS with characteristics of AIH (“autoimmune(-like)” DILI/HDS, “immune-mediated” DILI/HDS) |
|
| Mechanism | Characteristics |
|---|---|
| Antigen presentation |
|
| Metabolism |
|
| Pro-inflammatory mechanisms |
|
| Regulatory mechanisms |
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sebode, M.; Schulz, L.; Lohse, A.W. “Autoimmune(-Like)” Drug and Herb Induced Liver Injury: New Insights into Molecular Pathogenesis. Int. J. Mol. Sci. 2017, 18, 1954. https://doi.org/10.3390/ijms18091954
Sebode M, Schulz L, Lohse AW. “Autoimmune(-Like)” Drug and Herb Induced Liver Injury: New Insights into Molecular Pathogenesis. International Journal of Molecular Sciences. 2017; 18(9):1954. https://doi.org/10.3390/ijms18091954
Chicago/Turabian StyleSebode, Marcial, Lisa Schulz, and Ansgar W. Lohse. 2017. "“Autoimmune(-Like)” Drug and Herb Induced Liver Injury: New Insights into Molecular Pathogenesis" International Journal of Molecular Sciences 18, no. 9: 1954. https://doi.org/10.3390/ijms18091954
APA StyleSebode, M., Schulz, L., & Lohse, A. W. (2017). “Autoimmune(-Like)” Drug and Herb Induced Liver Injury: New Insights into Molecular Pathogenesis. International Journal of Molecular Sciences, 18(9), 1954. https://doi.org/10.3390/ijms18091954