Understanding the Epilepsy in POLG Related Disease

Abstract
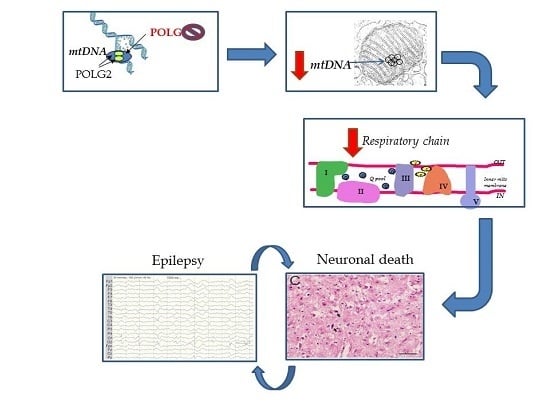
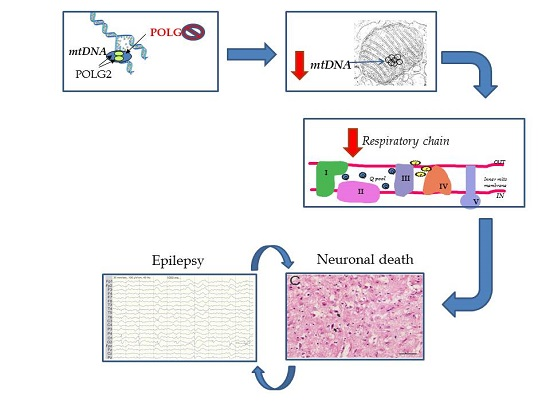{kind=link}
{kind=link}
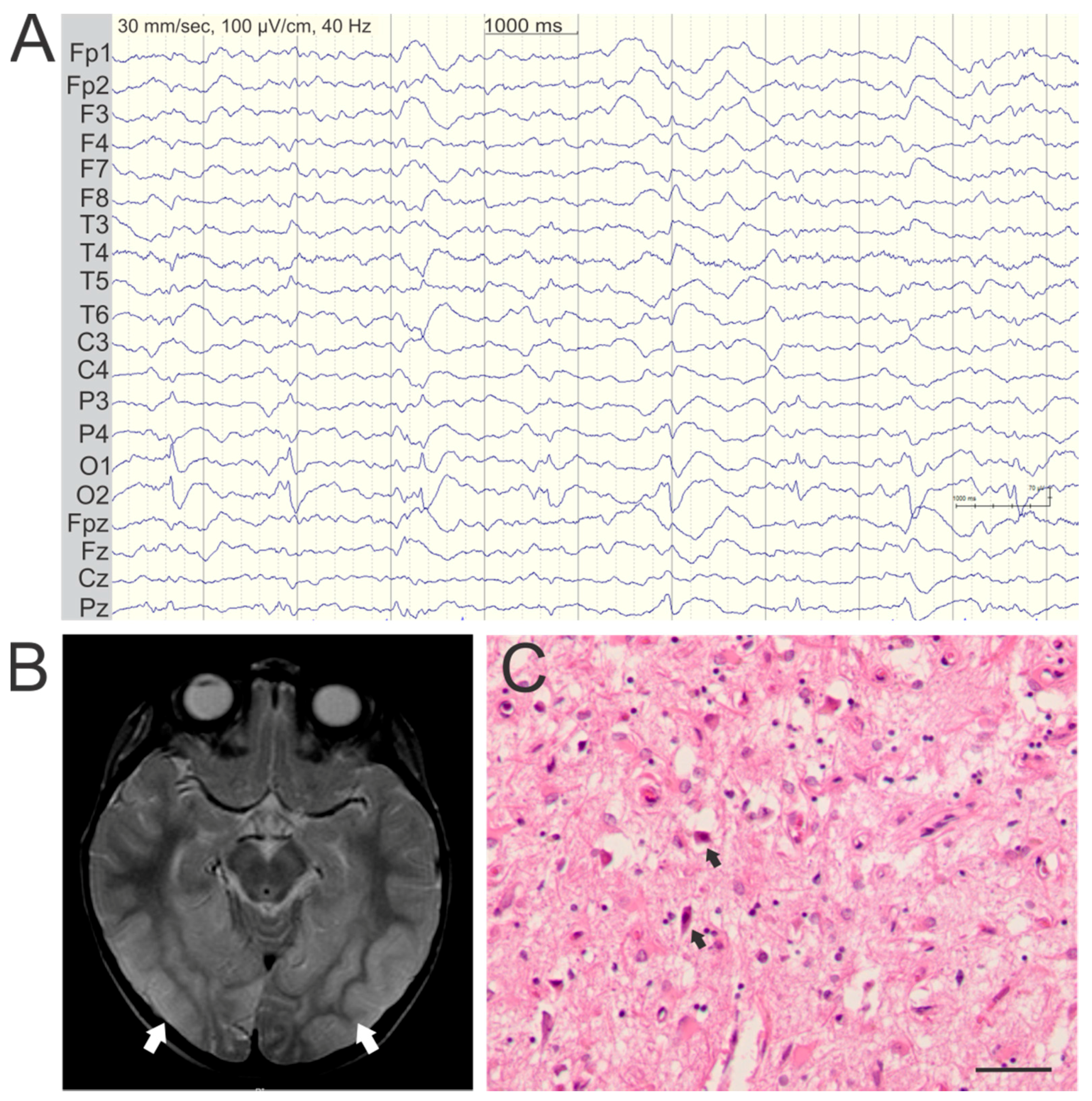{kind=link}
1. Introduction
2. Overview of Our Mechanistic Understanding
3. Seizure Semiology and EEG Findings
4. Epileptic Foci Correlate with Cortical Lesions on MRI
5. Pathology of Cortical Lesions Is Consistent with Energy Failure
6. Molecular Disease Mechanisms Underlying POLG Related Epilepsy
7. Treatment
Acknowledgments
Author Contributions
Conflicts of interest
References
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Zeviani, M. Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J. Intern. Med. 2009, 265, 174–192. [Google Scholar] [CrossRef] [PubMed]
- Longley, M.J.; Graziewicz, M.A.; Bienstock, R.J.; Copeland, W.C. Consequences of mutations in human DNA polymerase gamma. Gene 2005, 354, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Tran, G.T.; Coxhead, J.; Bertelsen, B.; Lilleng, P.K.; Balafkan, N.; Payne, B.; Miletic, H.; Chinnery, P.F.; Bindoff, L.A. Molecular pathogenesis of polymerase gamma-related neurodegeneration. Ann. Neurol. 2014, 76, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.H.; Chinnery, P.F.; Copeland, W.C. POLG-related disorders. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Fong, C.T., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chan, S.S.; Copeland, W.C. DNA polymerase gamma and mitochondrial disease: Understanding the consequence of POLG mutations. Biochim. Biophys. Acta 2009, 1787, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Debray, F.G.; Lambert, M.; Chevalier, I.; Robitaille, Y.; Decarie, J.C.; Shoubridge, E.A.; Robinson, B.H.; Mitchell, G.A. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 2007, 119, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Khurana, D.S.; Salganicoff, L.; Melvin, J.J.; Hobdell, E.F.; Valencia, I.; Hardison, H.H.; Marks, H.G.; Grover, W.D.; Legido, A. Epilepsy and respiratory chain defects in children with mitochondrial encephalopathies. Epilepsia 2008, 39, 8–13. [Google Scholar]
- Canafoglia, L.; Franceschetti, S.; Antozzi, C.; Carrara, F.; Farina, L.; Granata, T.; Lamantea, E.; Savoiardo, M.; Uziel, G.; Villani, F.; et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology 2001, 56, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S. Pathophysiology of mitochondrial disease causing epilepsy and status epilepticus. Epilepsy Behav. 2015, 49, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.G.; Devine, H.E.; Gorman, G.S.; Schaefer, A.M.; Horvath, R.; Ng, Y.; Nesbitt, V.; Lax, N.Z.; McFarland, R.; Cunningham, M.O.; et al. Epilepsy in adults with mitochondrial disease: A cohort study. Ann. Neurol. 2015, 78, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Hudson, G.; Ferrari, G.; Futterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmuller, H.; McFarland, R.; Ramesh, V.; et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Engelsen, B.A.; Telstad, W.; Aasly, J.; Zeviani, M.; Winterthun, S.; Ferrari, G.; Aarseth, J.H.; Bindoff, L.A. The spectrum of clinical disease caused by the a467t and w748s POLG mutations: A study of 26 cases. Brain 2006, 129, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Winterthun, S.; Ferrari, G.; He, L.; Taylor, R.W.; Zeviani, M.; Turnbull, D.M.; Engelsen, B.A.; Moen, G.; Bindoff, L.A. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology 2005, 64, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, M.E.; Ng, Y.S.; Taylor, R.W.; McFarland, R. Epilepsy due to mutations in the mitochondrial polymerase gamma (POLG) gene: A clinical and molecular genetic review. Epilepsia 2016, 57, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Neckelmann, G.; Mork, S.J.; Engelsen, B.E.; Viscomi, C.; Moen, G.; Ersland, L.; Zeviani, M.; Bindoff, L.A. Localized cerebral energy failure in DNA polymerase gamma-associated encephalopathy syndromes. Brain 2010, 133, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Bindoff, L.A. Molecular genetics of DNA polymerase gamma-associated neurodegeneration. eLS 2016. [Google Scholar] [CrossRef]
- Engelsen, B.A.; Tzoulis, C.; Karlsen, B.; Lillebo, A.; Laegreid, L.M.; Aasly, J.; Zeviani, M.; Bindoff, L.A. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain 2008, 131, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Tzoulis, C.; Chong, W.K.; Chentouf, L.; Klingenberg, C.; Fratter, C.; Carr, L.J.; Prabhakar, P.; Kumaraguru, N.; Gissen, P.; et al. The clinical spectrum and natural history of early-onset diseases due to DNA polymerase gamma mutations. Genet. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Rahman, S.; Schmitt, B.; Taanman, J.W.; Duncan, A.J.; Harting, I.; Wohlrab, G.; Ebinger, F.; Rating, D.; Bast, T. Status epilepticus in children with alpers’ disease caused by POLG1 mutations: Eeg and mri features. Epilepsia 2009, 50, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Bindoff, L.A.; Engelsen, B.A. Mitochondrial diseases and epilepsy. Epilepsia 2012, 53, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.V.; Sharief, F.S.; Chan, S.S.; Copeland, W.C.; Naviaux, R.K. Molecular diagnosis of alpers syndrome. J. Hepatol. 2006, 45, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Nguyen, K.V. POLG mutations associated with alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004, 55, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Harding, B.N. Progressive neuronal degeneration of childhood with liver disease (alpers-huttenlocher syndrome): A personal review. J. Child Neurol. 1990, 5, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Saneto, R.P.; Naviaux, R.K. Polymerase γ disease through the ages. Dev. Disabil. Res. Rev. 2010, 16, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Janssen, W.; Quaegebeur, A.; van Goethem, G.; Ann, L.; Smets, K.; Vandenberghe, R.; van Paesschen, W. The spectrum of epilepsy caused by POLG mutations. Acta Neurol. Belg. 2016, 116, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, C.; Henriksen, E.; Miletic, H.; Bindoff, L.A. No evidence of ischemia in stroke-like lesions of mitochondrial POLG encephalopathy. Mitochondrion 2017, 32, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Sakai, F. Pathogenesis of stroke-like episodes in melas: Analysis of neurovascular cellular mechanisms. Curr. Neurovasc. Res. 2005, 2, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Bathla, G.; Policeni, B.; Agarwal, A. Neuroimaging in patients with abnormal blood glucose levels. AJNR Am. J. Neuroradiol. 2014, 35, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S. The role of mitochondria in epileptogenesis. Curr. Opin. Neurol. 2002, 15, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Kilbride, S.M.; Telford, J.E.; Tipton, K.F.; Davey, G.P. Partial inhibition of complex I activity increases ca-independent glutamate release rates from depolarized synaptosomes. J. Neurochem. 2008, 106, 826–834. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C. The glutamate-glutamine cycle is not stoichiometric: Fates of glutamate in brain. J. Neurosci. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Allen Hauser, W.; Mathern, G.; Moshe, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc task force of the ilae commission on therapeutic strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Pruss, H.; Holtkamp, M. Ketamine successfully terminates malignant status epilepticus. Epilepsy Res. 2008, 82, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hikmat, O.; Eichele, T.; Tzoulis, C.; Bindoff, L.A. Understanding the Epilepsy in POLG Related Disease. Int. J. Mol. Sci. 2017, 18, 1845. https://doi.org/10.3390/ijms18091845
Hikmat O, Eichele T, Tzoulis C, Bindoff LA. Understanding the Epilepsy in POLG Related Disease. International Journal of Molecular Sciences. 2017; 18(9):1845. https://doi.org/10.3390/ijms18091845
Chicago/Turabian StyleHikmat, Omar, Tom Eichele, Charalampos Tzoulis, and Laurence A. Bindoff. 2017. "Understanding the Epilepsy in POLG Related Disease" International Journal of Molecular Sciences 18, no. 9: 1845. https://doi.org/10.3390/ijms18091845
APA StyleHikmat, O., Eichele, T., Tzoulis, C., & Bindoff, L. A. (2017). Understanding the Epilepsy in POLG Related Disease. International Journal of Molecular Sciences, 18(9), 1845. https://doi.org/10.3390/ijms18091845