Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
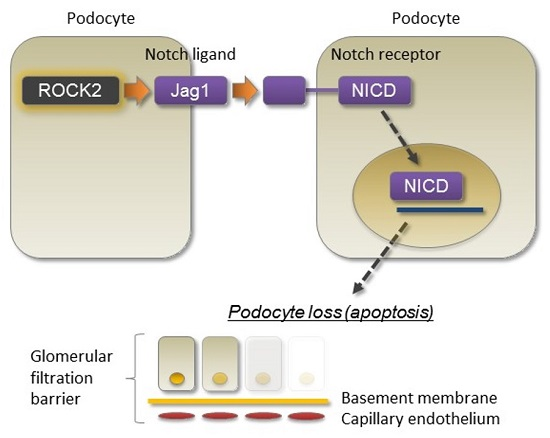
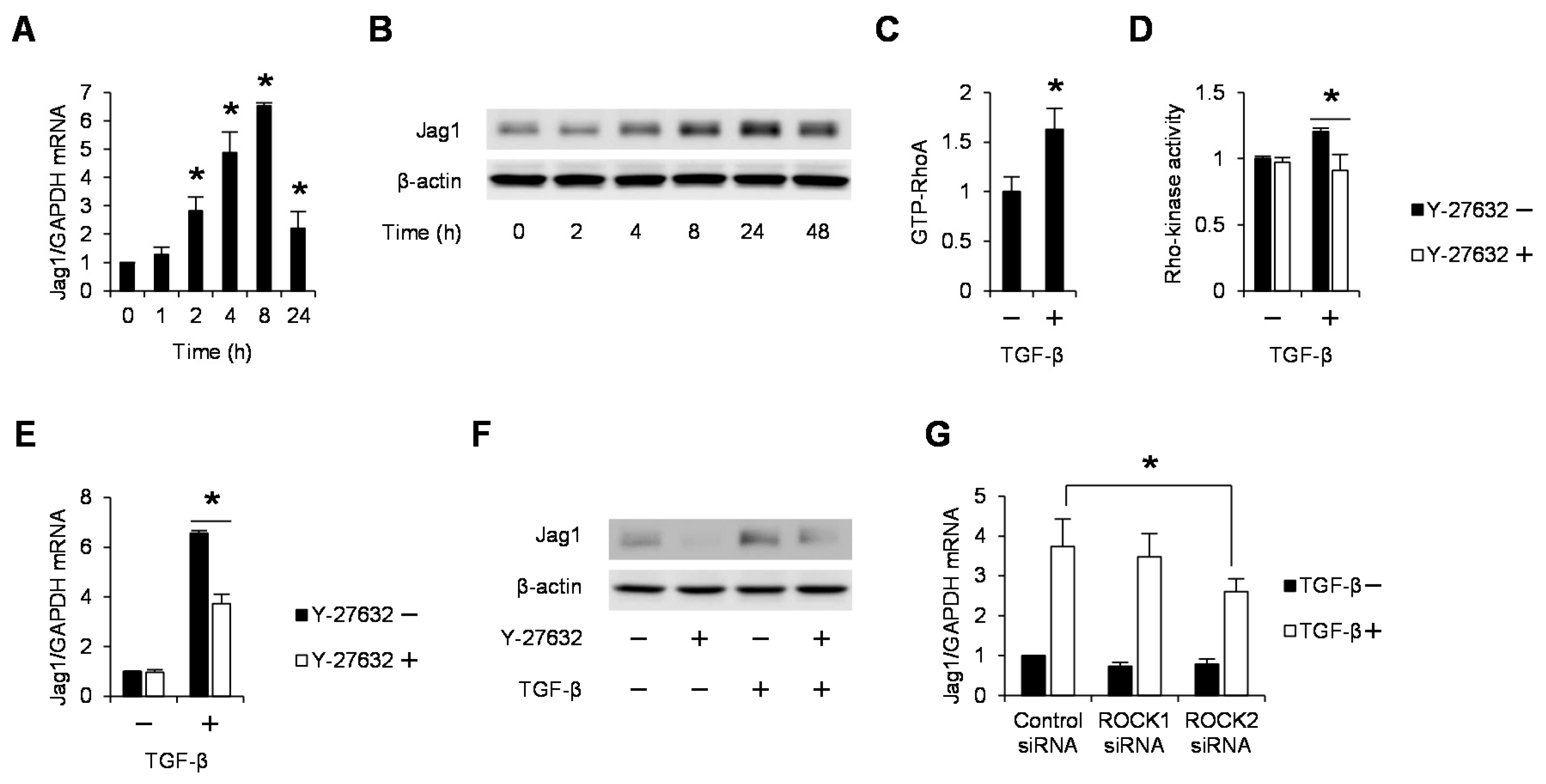
2.1. TGF-β Induces Jag1 Expression and Rho/Rho-Kinase Activation in Podocytes
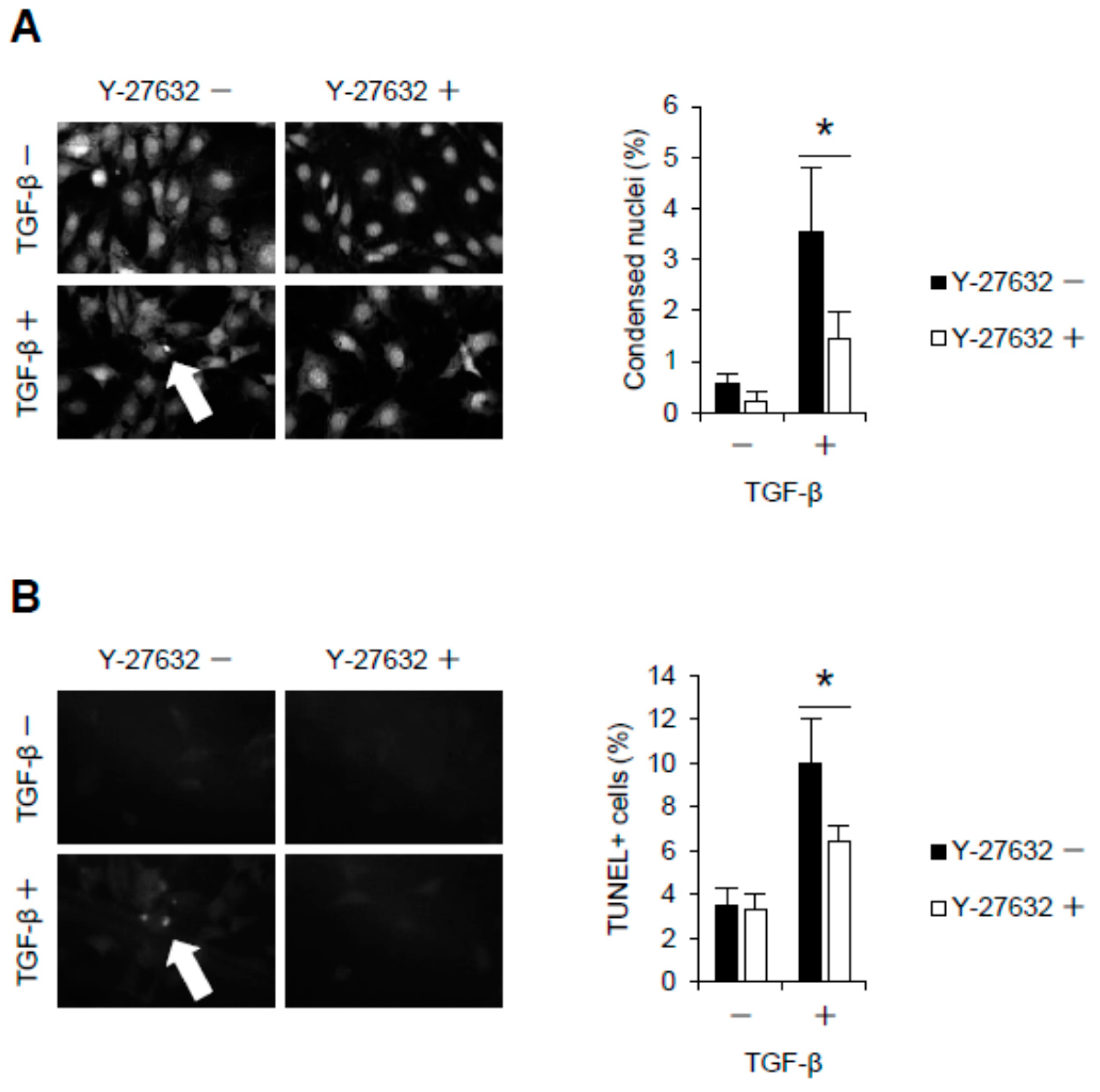
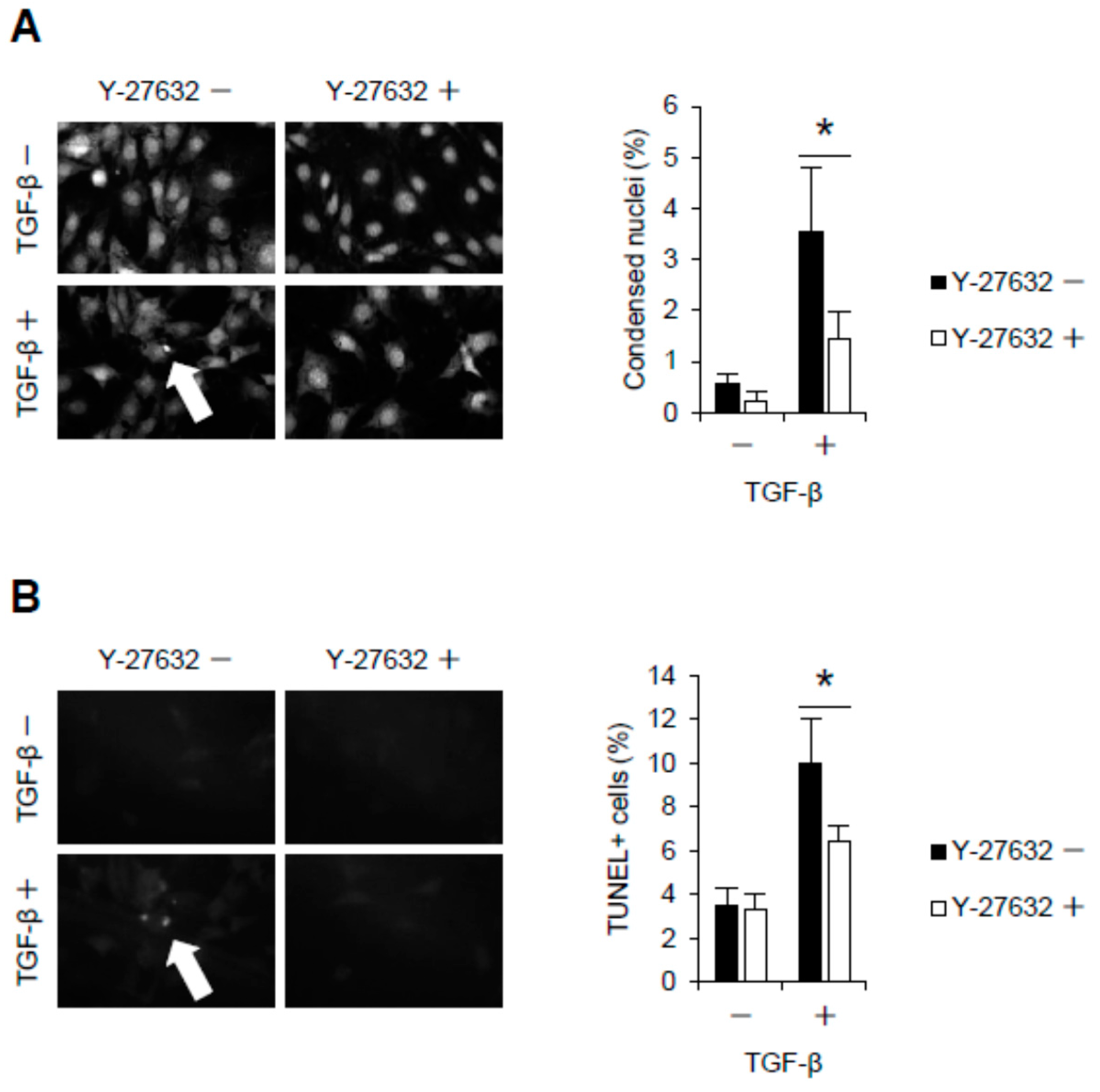
2.2. Rho-Kinase Inhibition Attenuates Podocyte Apoptosis
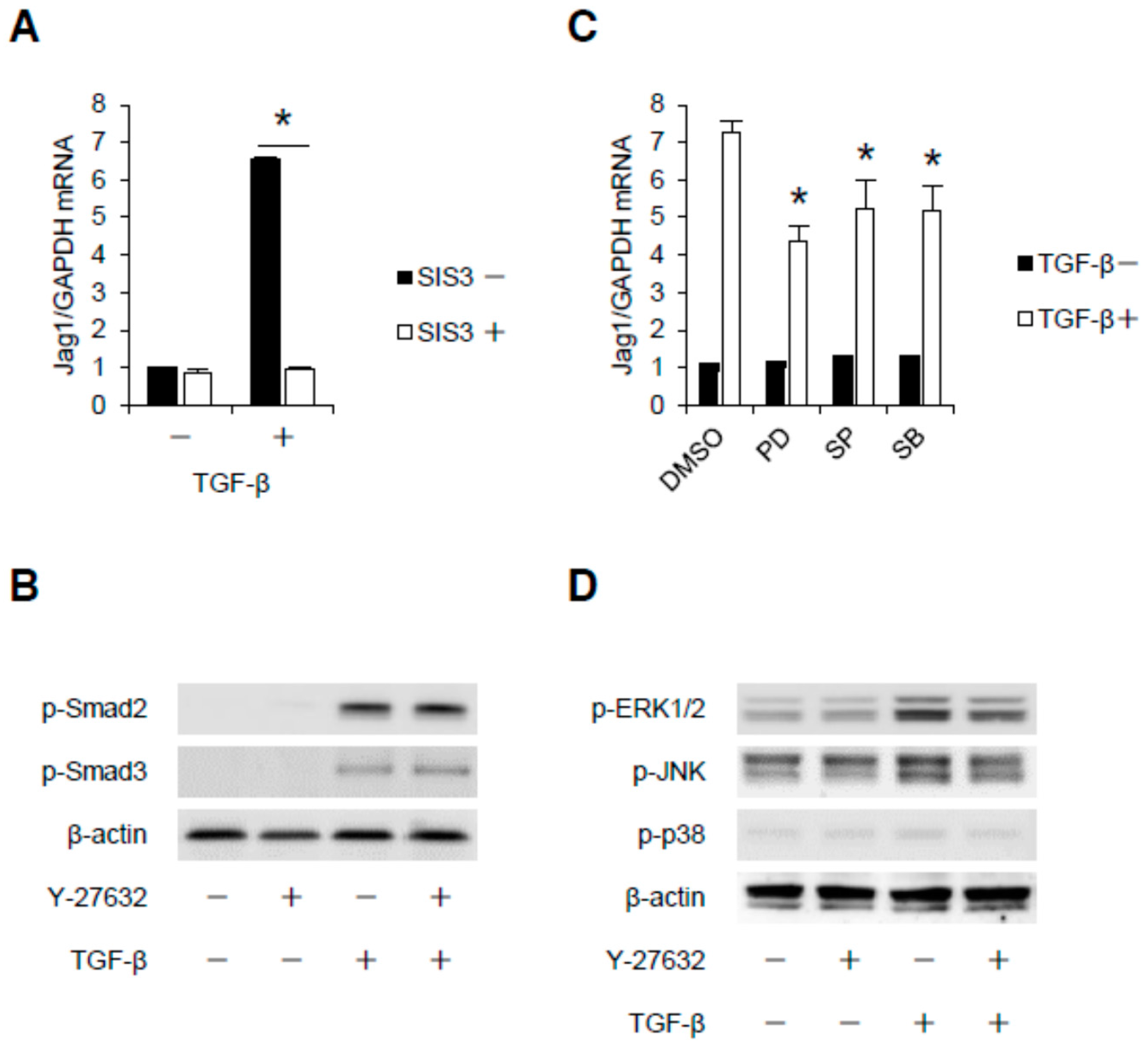
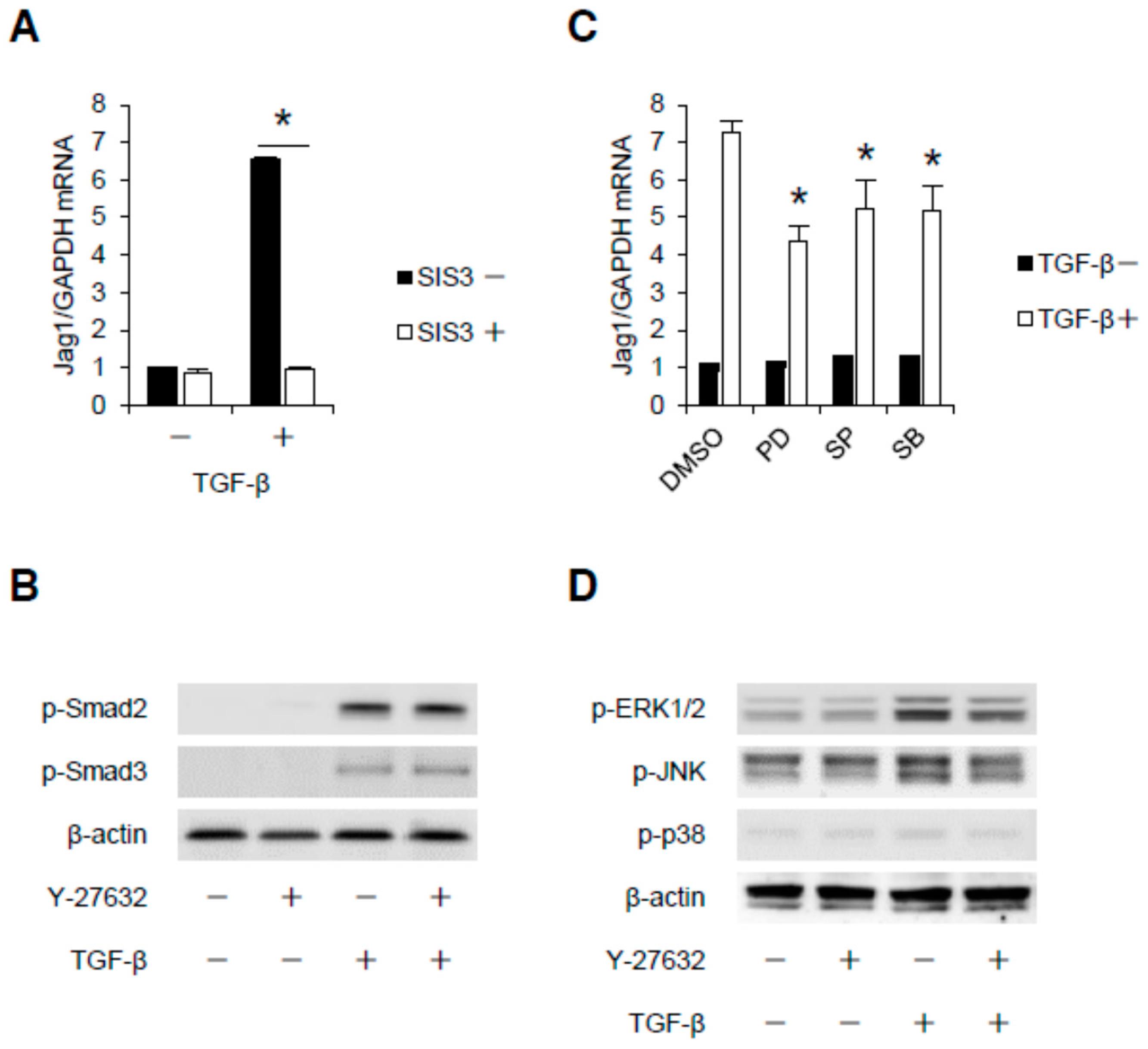
2.3. Rho-Kinase Promotes TGF-β-Induced Apoptosis in Podocytes via the Notch and Mitogen-activated Protein Kinase (MAPK) Signaling Pathways Independently of the Smads Cascade
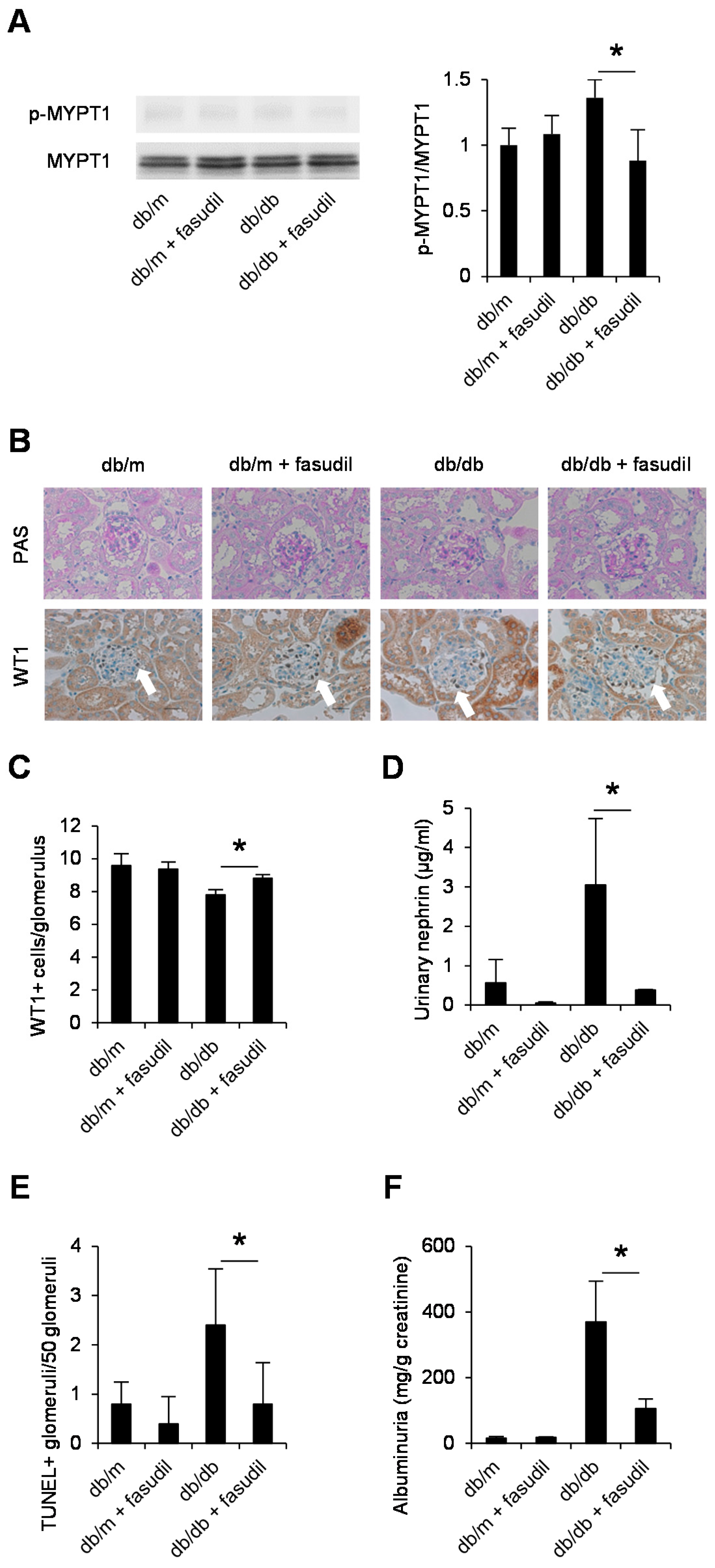
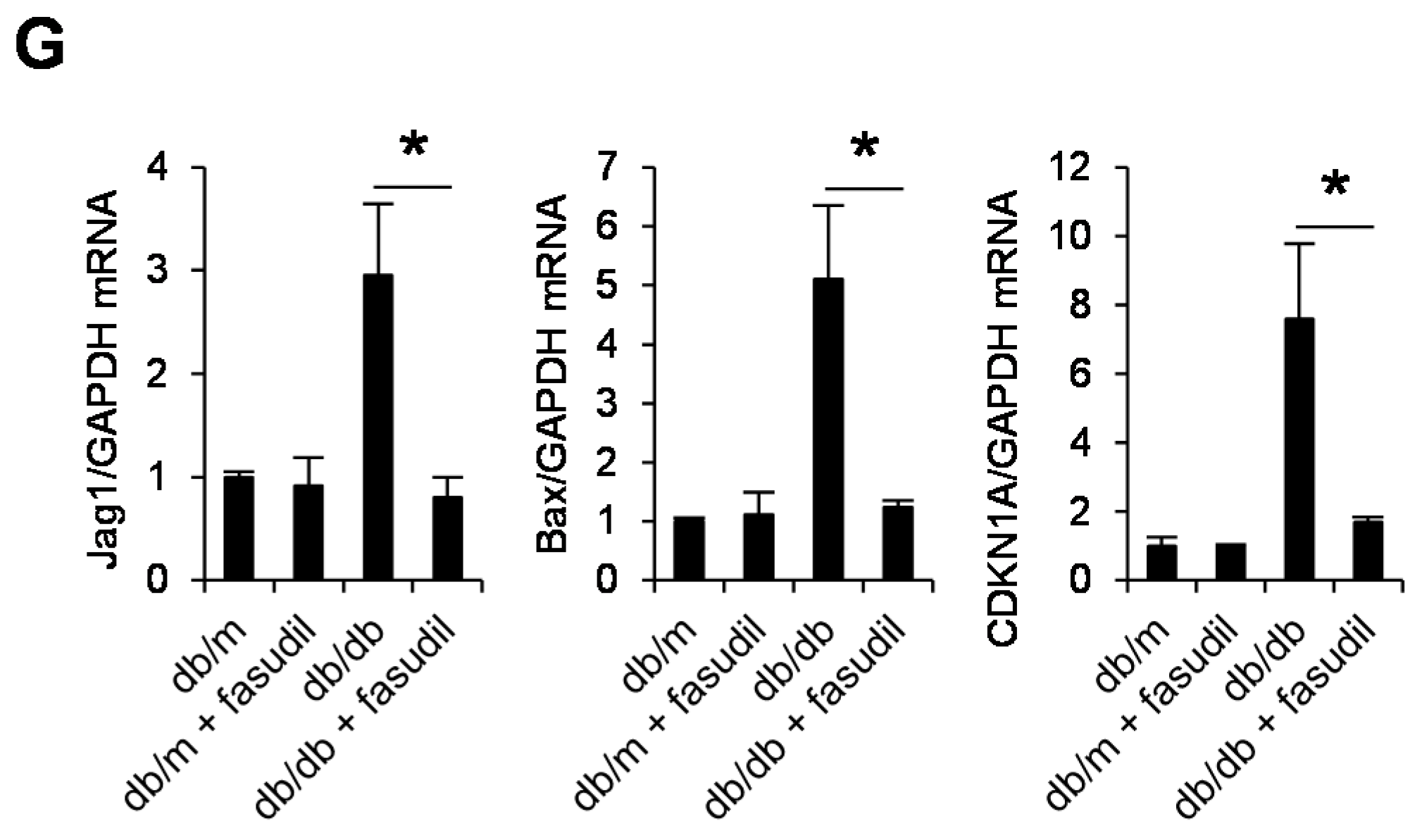
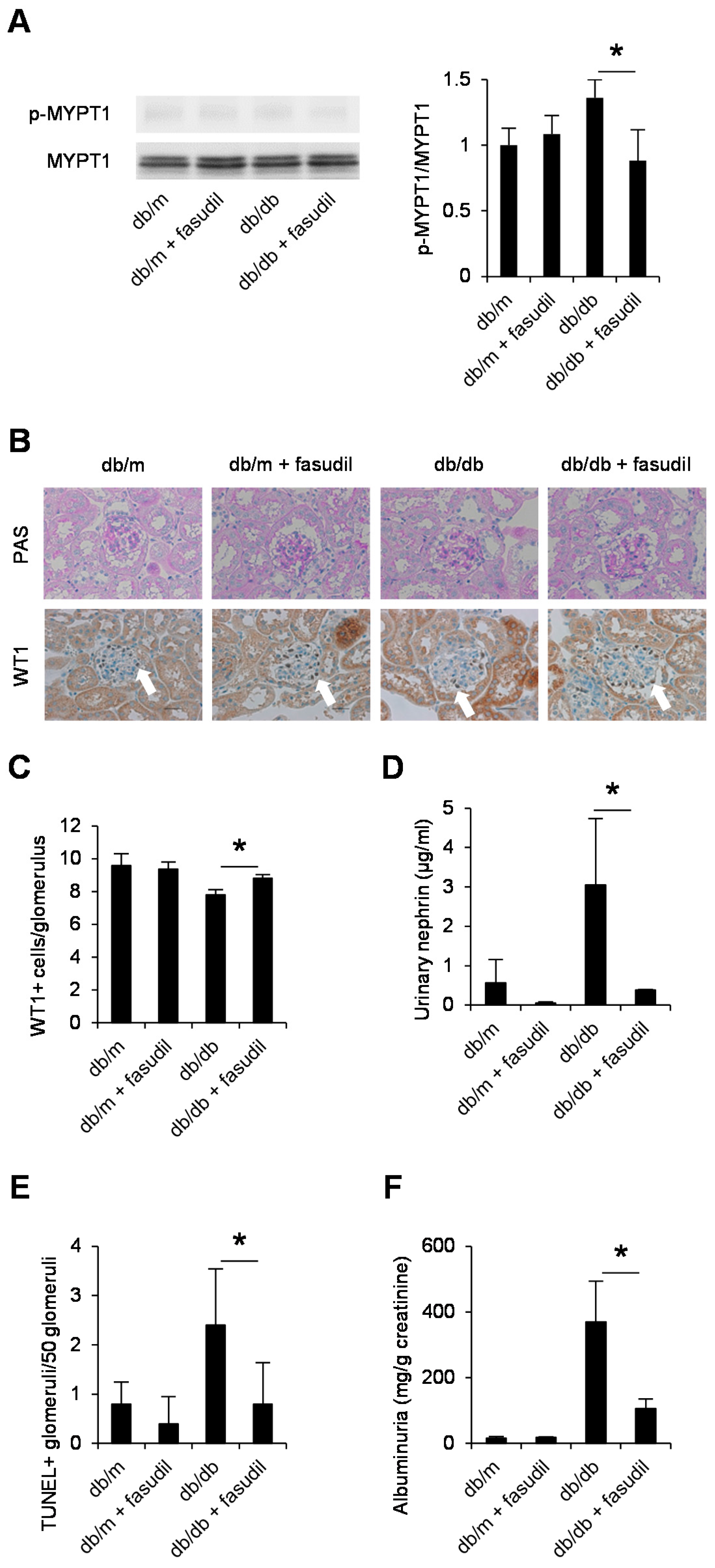
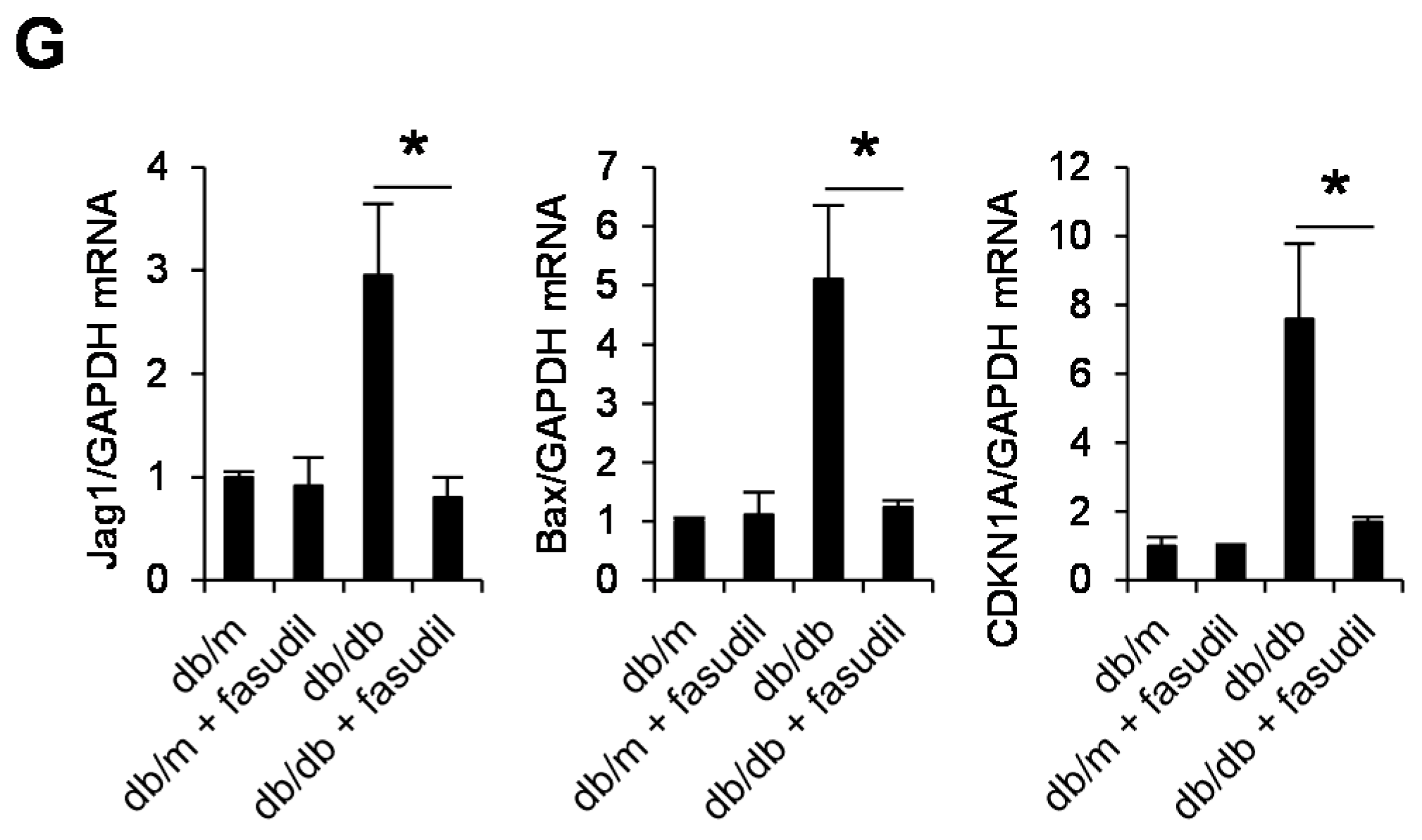
2.4. Rho-Kinase Inhibitor Fasudil Inhibits Podocyte Apoptosis in db/db Mice
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Animal Studies
4.4. Podocyte Isolation
4.5. Silencing of Rho-Kinase
4.6. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction
4.7. Western Blot Analyses
4.8. GTP-RhoA Activity Assay
4.9. Rho-Kinase Activity Assay
4.10. Detection of Condensed Nuclei and Apoptotic Podocytes
4.11. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adler, A.I.; Stevens, R.J.; Manley, S.E.; Bilous, R.W.; Cull, C.A.; Holman, R.R.; Ukpds, G. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 2003, 63, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, P.W. The podocyte as a target for therapies-new and old. Nat. Rev. Nephrol. 2011, 8, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 1), i37–i45. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Bonegio, R.; Susztak, K. Notch signaling in diabetic nephropathy. Exp. Cell Res. 2012, 318, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Signaling pathways in diabetic nephropathy. Histol. Histopathol. 2016, 31, 1059–1067. [Google Scholar] [PubMed]
- Matoba, K.; Kawanami, D.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-kinase mediates TNF-α-induced MCP-1 expression via p38 MAPK signaling pathway in mesangial cells. Biochem. Biophys. Res. Commun. 2010, 402, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Kawanami, D.; Okada, R.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; et al. Rho-kinase inhibition prevents the progression of diabetic nephropathy by downregulating hypoxia-inducible factor 1α. Kidney Int. 2013, 84, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Kawanami, D.; Tsukamoto, M.; Kinoshita, J.; Ito, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Murai, N.; Matsufuji, S.; et al. Rho-kinase regulation of TNF-α-induced nuclear translocation of NF-κB RelA/p65 and M-CSF expression via p38 MAPK in mesangial cells. Am. J. Physiol. Ren. Physiol. 2014, 307, F571–F580. [Google Scholar] [CrossRef] [PubMed]
- Gojo, A.; Utsunomiya, K.; Taniguchi, K.; Yokota, T.; Ishizawa, S.; Kanazawa, Y.; Kurata, H.; Tajima, N. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2007, 568, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Uehata, M.; Tamechika, I.; Keel, J.; Nonomura, K.; Maekawa, M.; Narumiya, S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 2000, 57, 976–983. [Google Scholar] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Kovall, R.A. Structures of CSL, Notch and Mastermind proteins: Piecing together an active transcription complex. Curr. Opin. Struct. Biol. 2007, 17, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Bonegio, R.G.; Beck, L.H.; Kahlon, R.K.; Lu, W.; Salant, D.J. The fate of Notch-deficient nephrogenic progenitor cells during metanephric kidney development. Kidney Int. 2011, 79, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Miner, J.H.; Lin, M.; Tansey, M.G.; Roth, K.; Kopan, R. γ-Secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development 2003, 130, 5031–5042. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.T.; Kim, M.; Valerius, M.T.; Surendran, K.; Schuster-Gossler, K.; Gossler, A.; McMahon, A.P.; Kopan, R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 2007, 134, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Pereira, F.A.; Beasley, D.; Zheng, H. Presenilins are required for the formation of comma- and S-shaped bodies during nephrogenesis. Development 2003, 130, 5019–5029. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Al-Awqati, Q. Segmental expression of Notch and Hairy genes in nephrogenesis. Am. J. Physiol. Ren. Physiol. 2005, 288, F939–F952. [Google Scholar] [CrossRef] [PubMed]
- Leimeister, C.; Schumacher, N.; Gessler, M. Expression of Notch pathway genes in the embryonic mouse metanephros suggests a role in proximal tubule development. Gene Expr. Patterns 2003, 3, 595–598. [Google Scholar] [CrossRef]
- Piscione, T.D.; Wu, M.Y.; Quaggin, S.E. Expression of Hairy/Enhancer of Split genes, Hes1 and Hes5, during murine nephron morphogenesis. Gene Expr. Patterns 2004, 4, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, T.; Bielesz, B.; Gruenwald, A.; Ponda, M.P.; Kopp, J.B.; Thomas, D.B.; Susztak, K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 2008, 14, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.; Guo, G.; Moridaira, K.; Fitzgerald, M.; McCracken, R.; Tolley, T.; Klahr, S. Transforming growth factor-β induces renal epithelial jagged-1 expression in fibrotic disease. J. Am. Soc. Nephrol. 2002, 13, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zhang, J.; Peng, X.; Dong, Y.; Jia, L.; Li, H.; Du, J. The Notch γ-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation. Int. J. Biochem. Cell Biol. 2014, 55, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Zhang, B.; Wu, D.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. TGF-β-induced RhoA activation and fibronectin production in mesangial cells require caveolae. Am. J. Physiol. Ren. Physiol. 2008, 295, F153–F164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, W.; Yang, Q.; Ren, Z.; Chen, X.; Zha, D.; Singhal, P.C.; Ding, G. IQGAP1 mediates angiotensin II-induced apoptosis of podocytes via the ERK1/2 MAPK signaling pathway. Am. J. Nephrol. 2013, 38, 430–444. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Xu, S.; Nguyen, T.T.; Quan, X.; Choi, S.K.; Kim, S.J.; Lee, E.Y.; Cha, S.K.; Park, K.S. Transforming growth factor-β1-induced apoptosis in podocytes via extracellular-signal-regulated kinase-mammalian target of rapamycin complex 1-nadph oxidase 4 axis. J. Biol. Chem. 2015, 290, 30830–30842. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lin, Q.; Liao, H.; Feng, J.; Dong, X.; Ye, J. TGF-β1 induces podocyte injury through Smad3-ERK-NF-κB pathway and Fyn-dependent TRPC6 phosphorylation. Cell Physiol. Biochem. 2010, 26, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Simeone, D.M.; Zhang, L.; Graziano, K.; Nicke, B.; Pham, T.; Schaefer, C.; Logsdon, C.D. Smad4 mediates activation of mitogen-activated protein kinases by TGF-β in pancreatic acinar cells. Am. J. Physiol. Cell Physiol. 2001, 281, C311–C319. [Google Scholar] [PubMed]
- Mertens, P.R.; Raffetseder, U.; Rauen, T. Notch receptors: A new target in glomerular diseases. Nephrol. Dial. Transplant. 2008, 23, 2743–2745. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Vilchis-Landeros, M.M.; Ponce-Coria, J.; Mendoza, V.; Hernandez-Pando, R.; Bobadilla, N.A.; Lopez-Casillas, F. Soluble β-glycan reduces renal damage progression in db/db mice. Am. J. Physiol. Renal Physiol. 2007, 292, F321–F329. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Ziyadeh, F.N. The emerging role of transforming growth factor-β in kidney diseases. Am. J. Physiol. 1994, 266, F829–F842. [Google Scholar] [PubMed]
- Zhou, Q.; Gensch, C.; Liao, J.K. Rho-associated coiled-coil-forming kinases (ROCKs): Potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol. Sci. 2011, 32, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Rajagopalan, L.E. Rho kinase-2 activation in human endothelial cells drives lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-κB p65. J. Biol. Chem. 2010, 285, 12536–12542. [Google Scholar] [CrossRef] [PubMed]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.P.; Liao, J.K.; D’Amore, P.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3895. [Google Scholar] [CrossRef] [PubMed]
- Noma, K.; Rikitake, Y.; Oyama, N.; Yan, G.; Alcaide, P.; Liu, P.Y.; Wang, H.; Ahl, D.; Sawada, N.; Okamoto, R.; et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J. Clin. Investig. 2008, 118, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Oshiro, H.; Kanzaki, S.; Yamaguchi, A.; Yamanaka, S.; Furuya, M.; Miura, S.; Kanno, H.; Nagashima, Y.; Aoki, I.; et al. A novel method for isolating podocytes using magnetic activated cell sorting. Nephrol. Dial. Transplant. 2010, 25, 3884–3890. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matoba, K.; Kawanami, D.; Nagai, Y.; Takeda, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K. Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy. Int. J. Mol. Sci. 2017, 18, 1795. https://doi.org/10.3390/ijms18081795
Matoba K, Kawanami D, Nagai Y, Takeda Y, Akamine T, Ishizawa S, Kanazawa Y, Yokota T, Utsunomiya K. Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy. International Journal of Molecular Sciences. 2017; 18(8):1795. https://doi.org/10.3390/ijms18081795
Chicago/Turabian StyleMatoba, Keiichiro, Daiji Kawanami, Yosuke Nagai, Yusuke Takeda, Tomoyo Akamine, Sho Ishizawa, Yasushi Kanazawa, Tamotsu Yokota, and Kazunori Utsunomiya. 2017. "Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy" International Journal of Molecular Sciences 18, no. 8: 1795. https://doi.org/10.3390/ijms18081795
APA StyleMatoba, K., Kawanami, D., Nagai, Y., Takeda, Y., Akamine, T., Ishizawa, S., Kanazawa, Y., Yokota, T., & Utsunomiya, K. (2017). Rho-Kinase Blockade Attenuates Podocyte Apoptosis by Inhibiting the Notch Signaling Pathway in Diabetic Nephropathy. International Journal of Molecular Sciences, 18(8), 1795. https://doi.org/10.3390/ijms18081795