Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer

Abstract
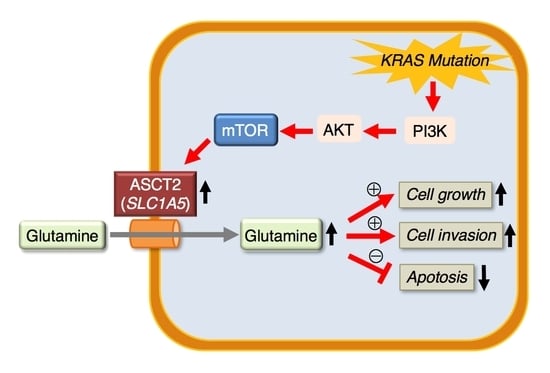
:
1. Introduction
2. Results
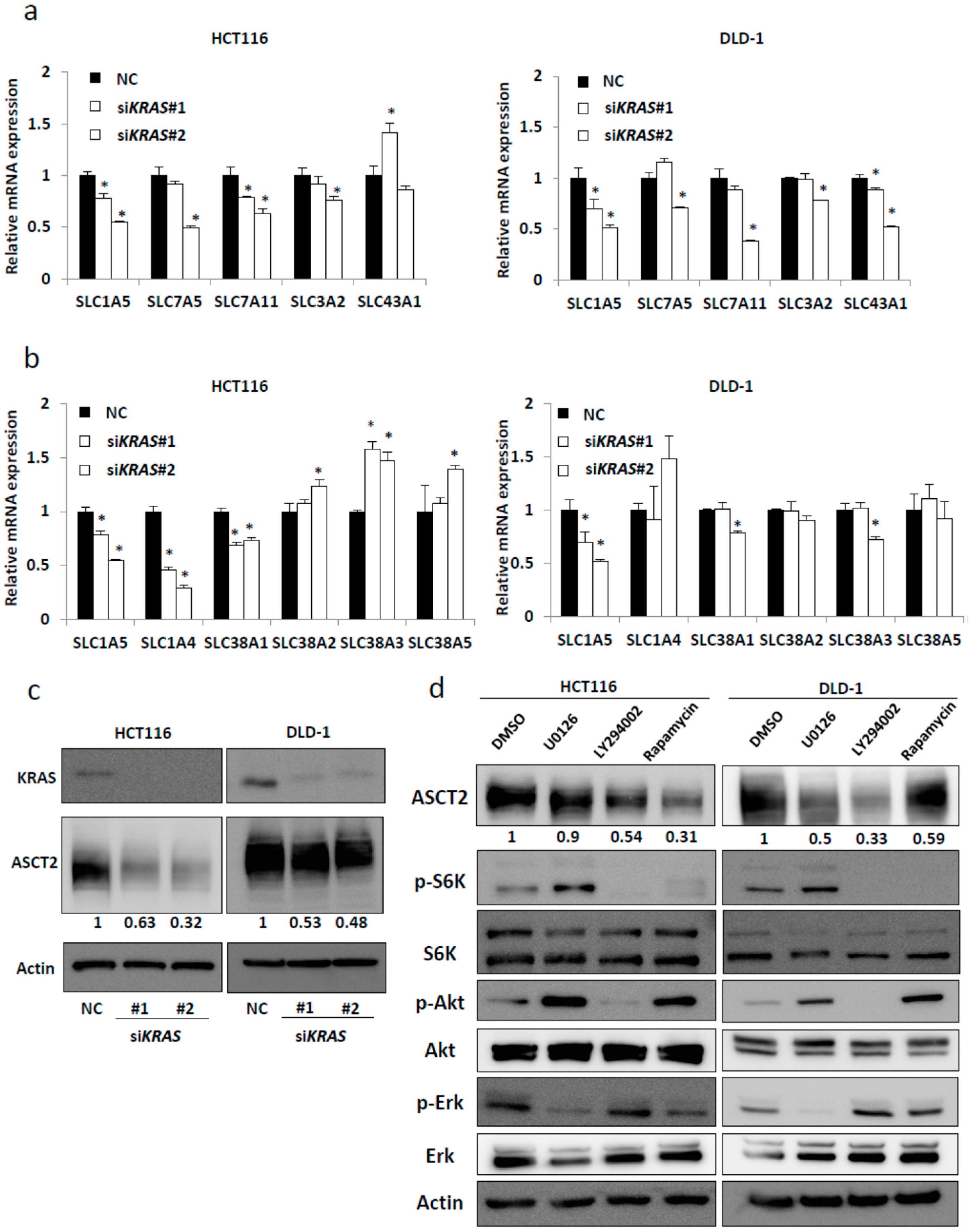
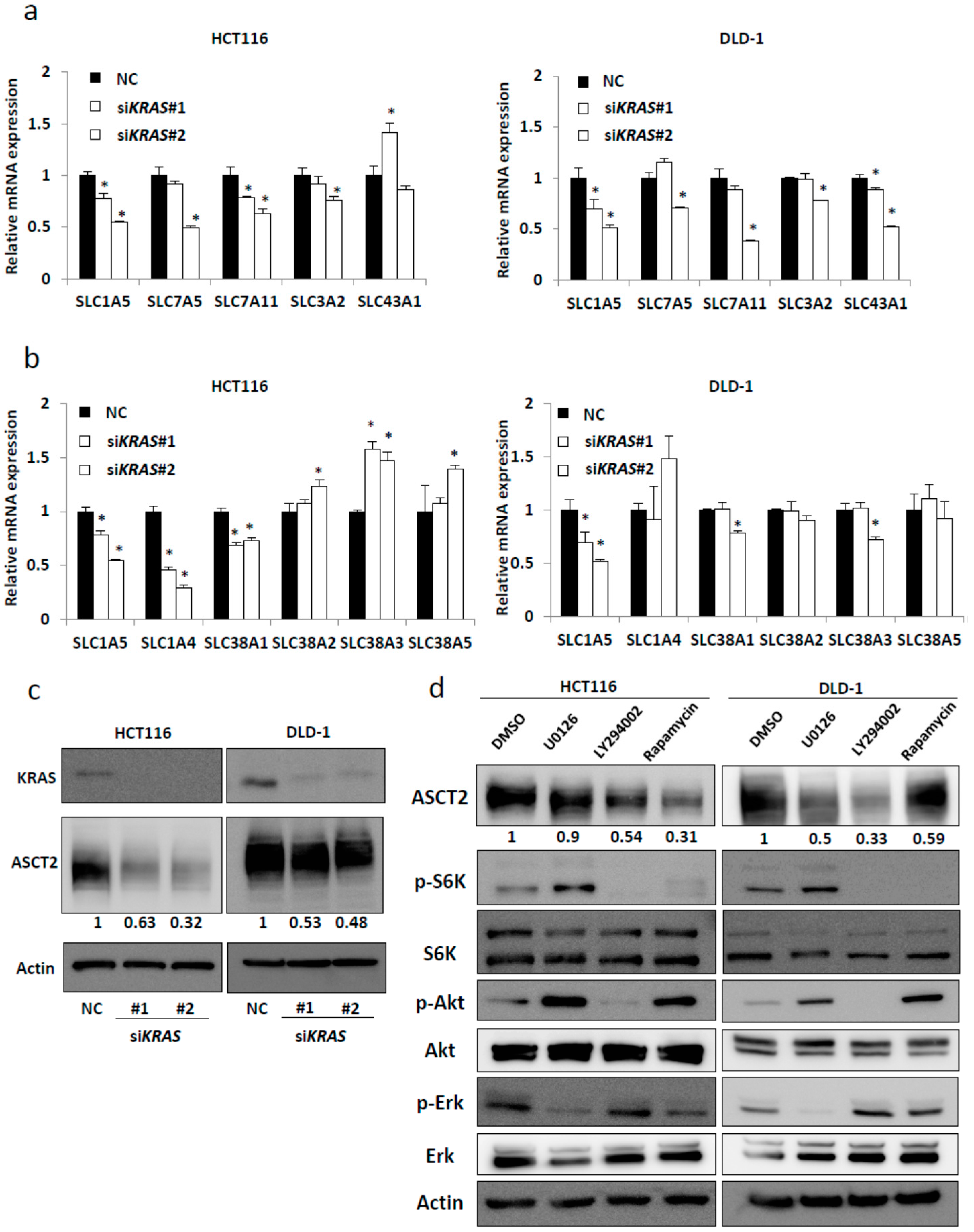
2.1. SLC1A5 (ASCT2) Is Regulated through KRAS Signaling in KRAS-Mutant CRC Cells
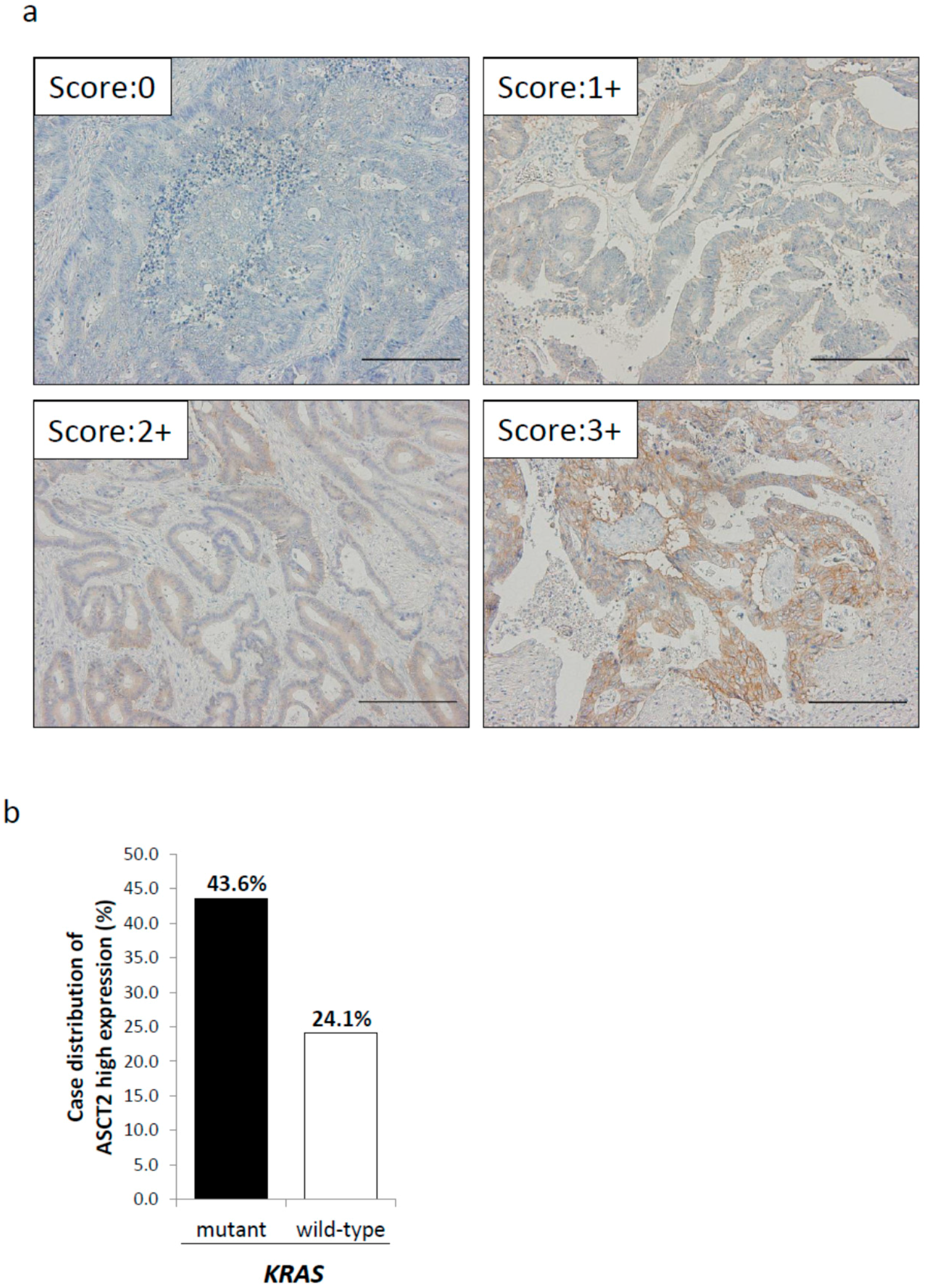
2.2. Relationship between ASCT2 Expression and KRAS Mutational Status in CRC Clinical Samples
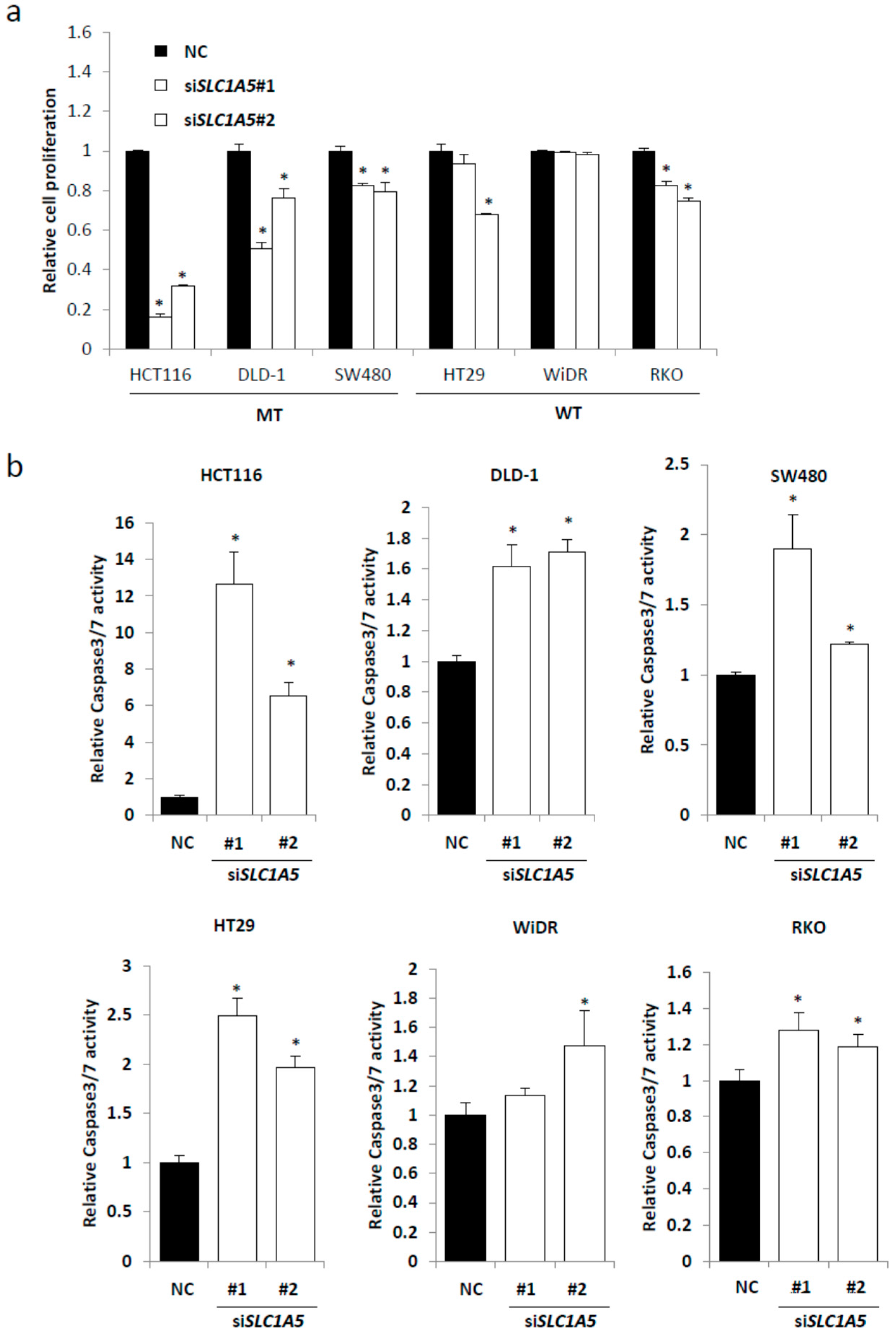
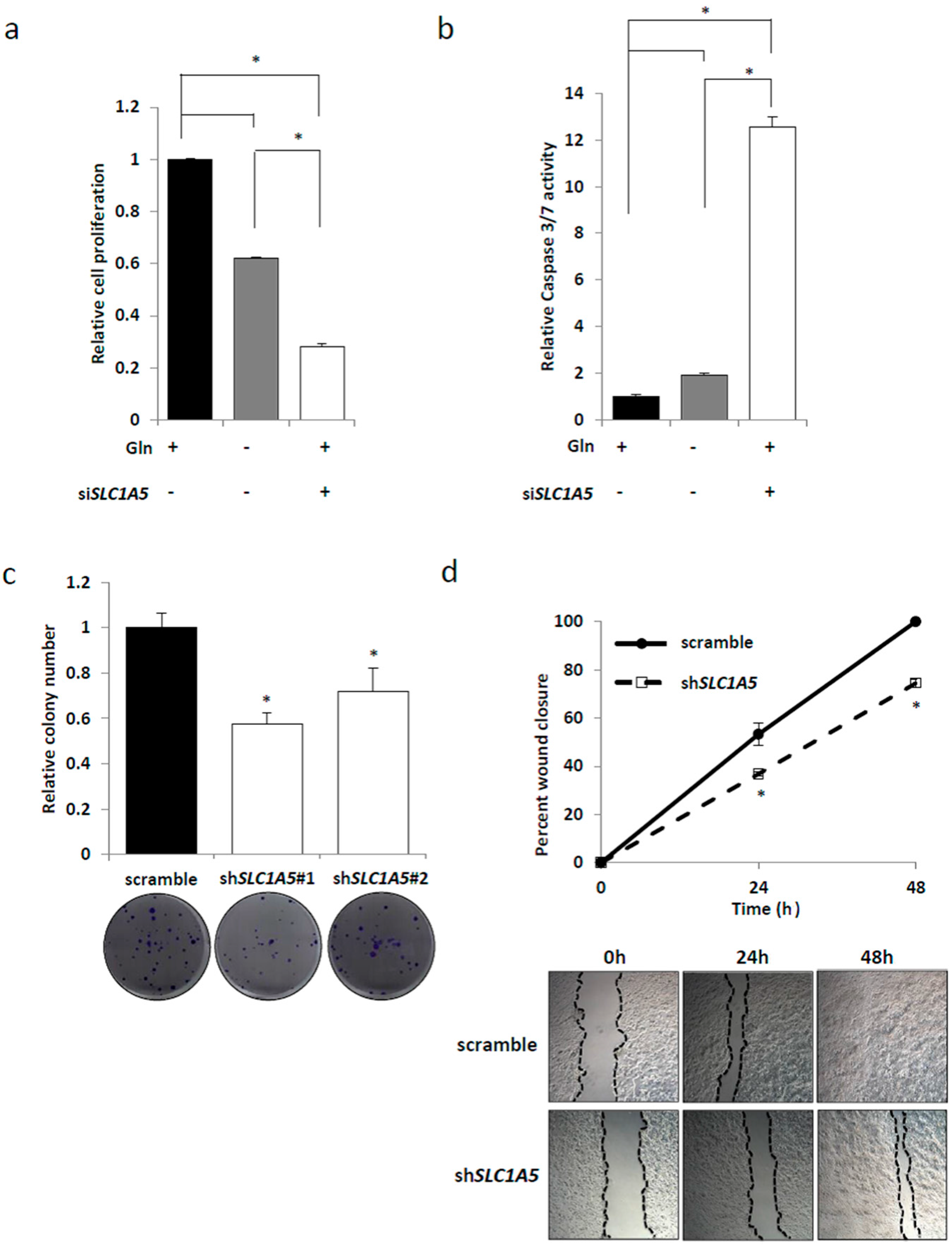
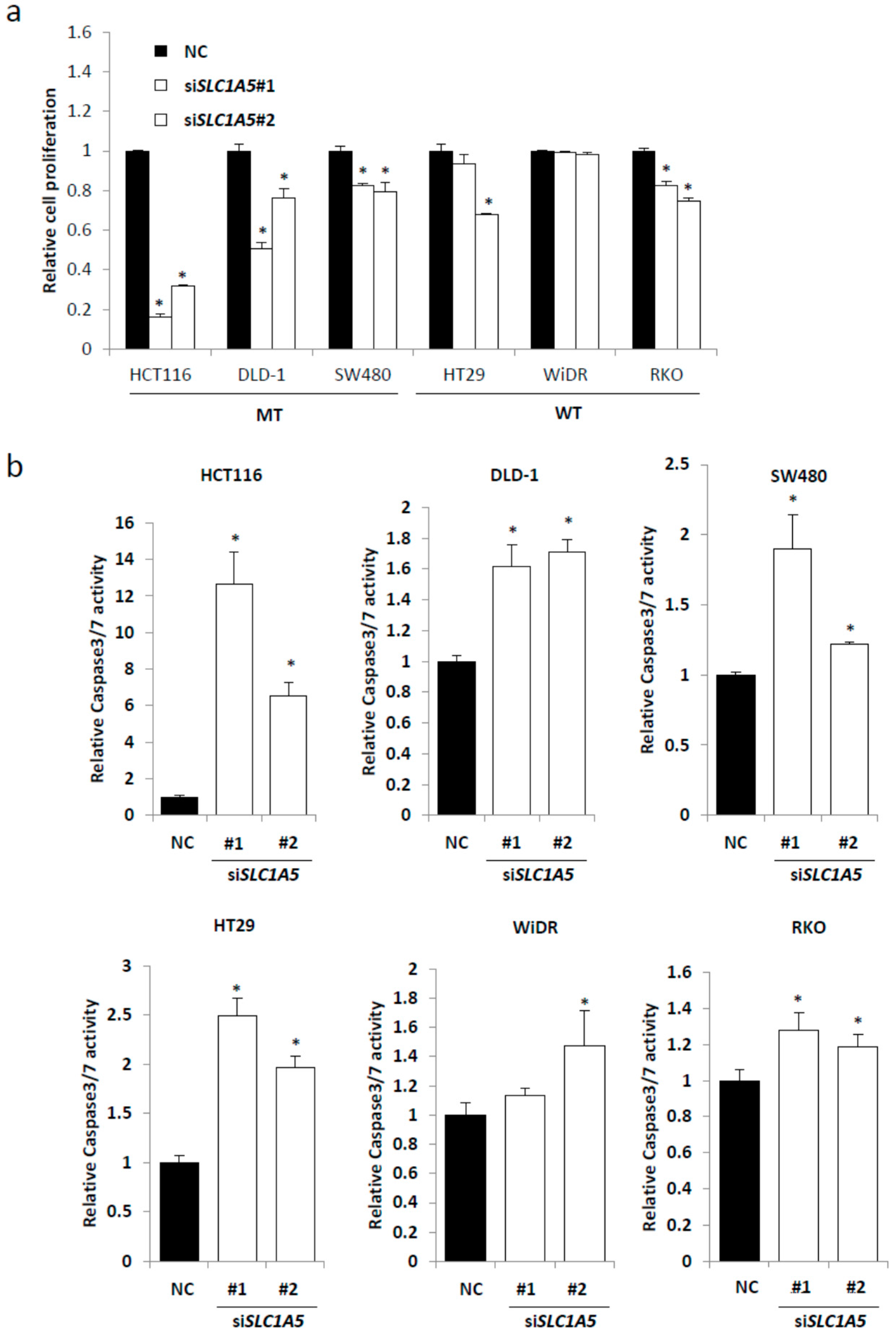
2.3. Knockdown of SLC1A5 (ASCT2) Results in Suppression of Cell Growth
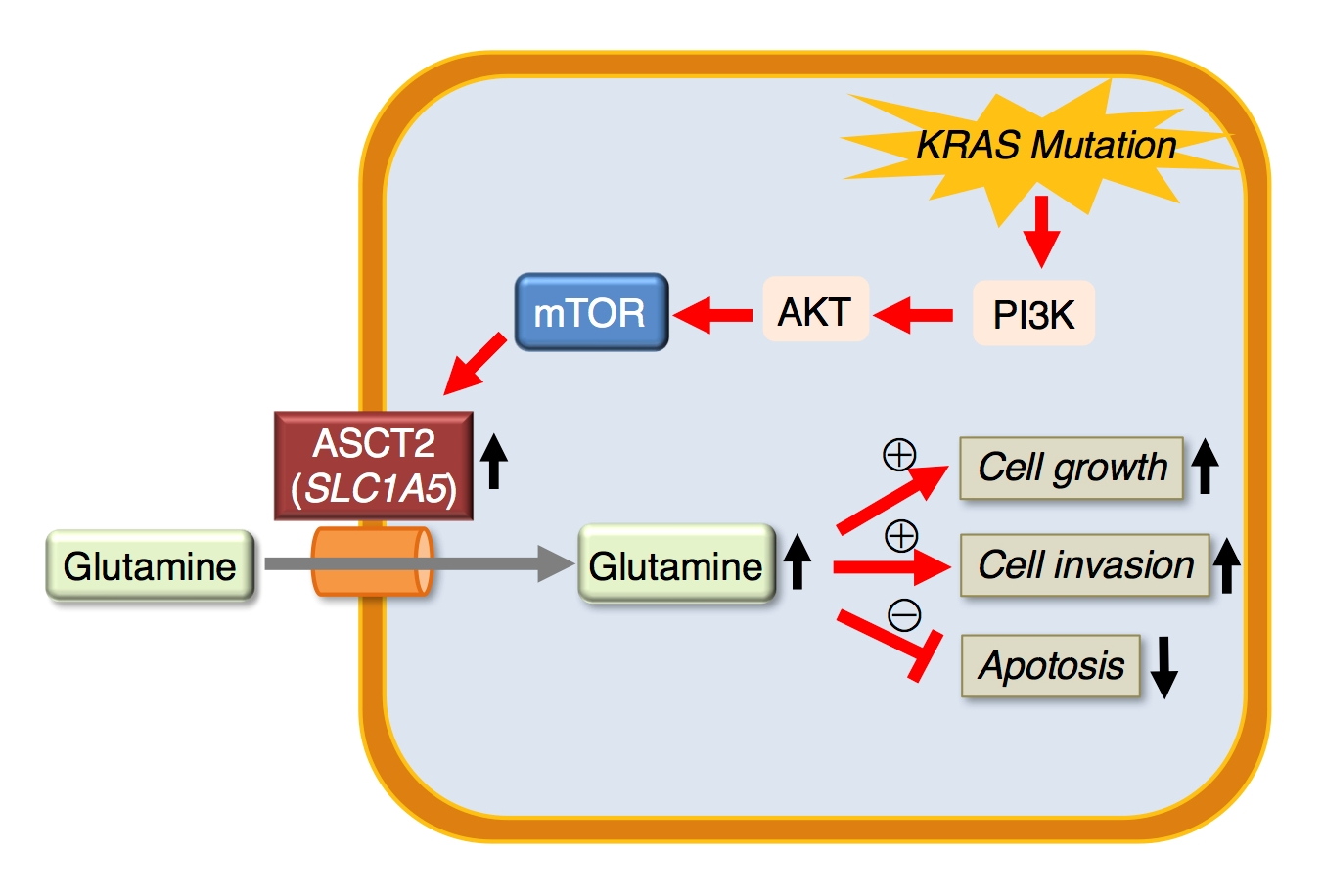
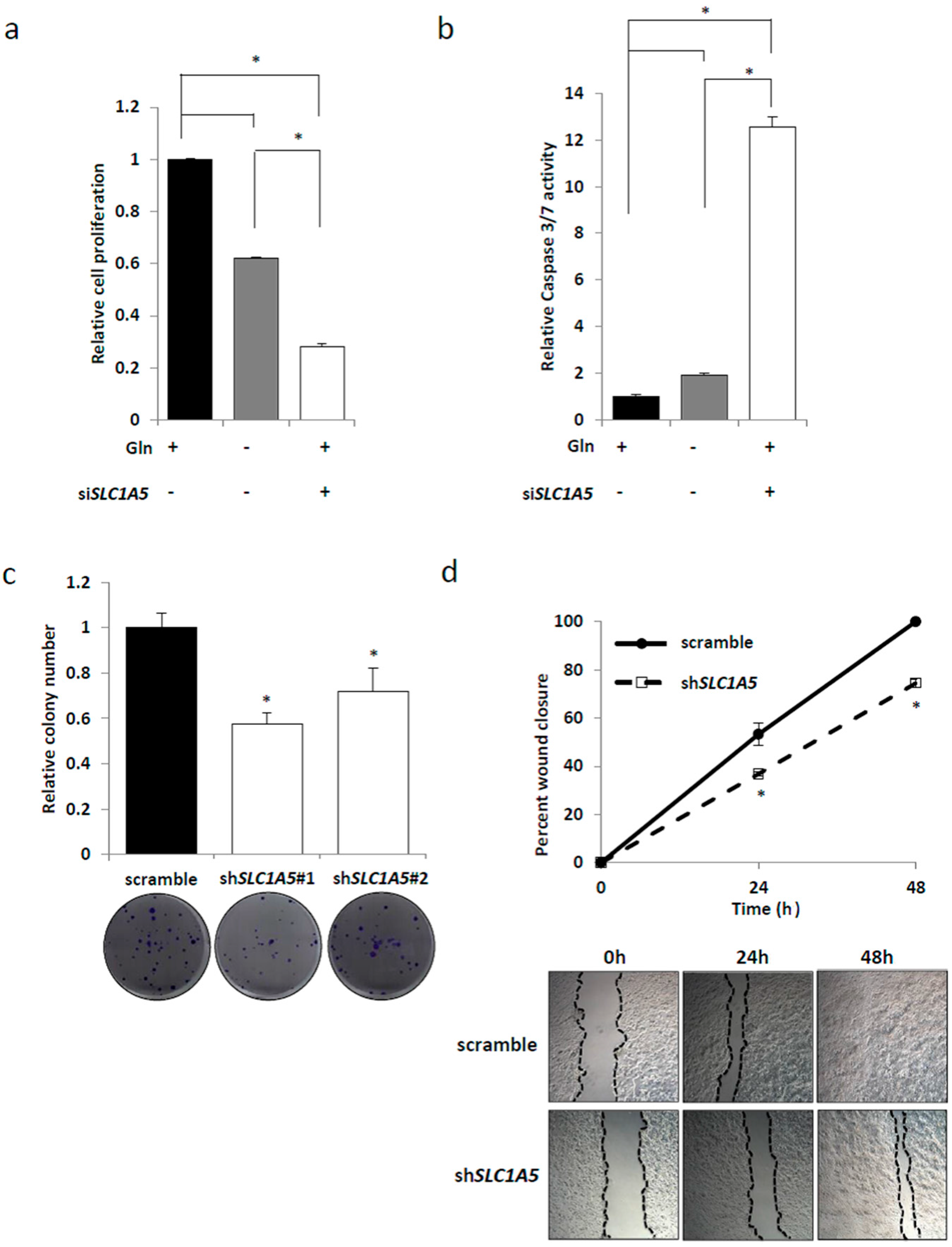
2.4. Role of SLC1A5 (ASCT2) in KRAS-Mutant CRC Cells
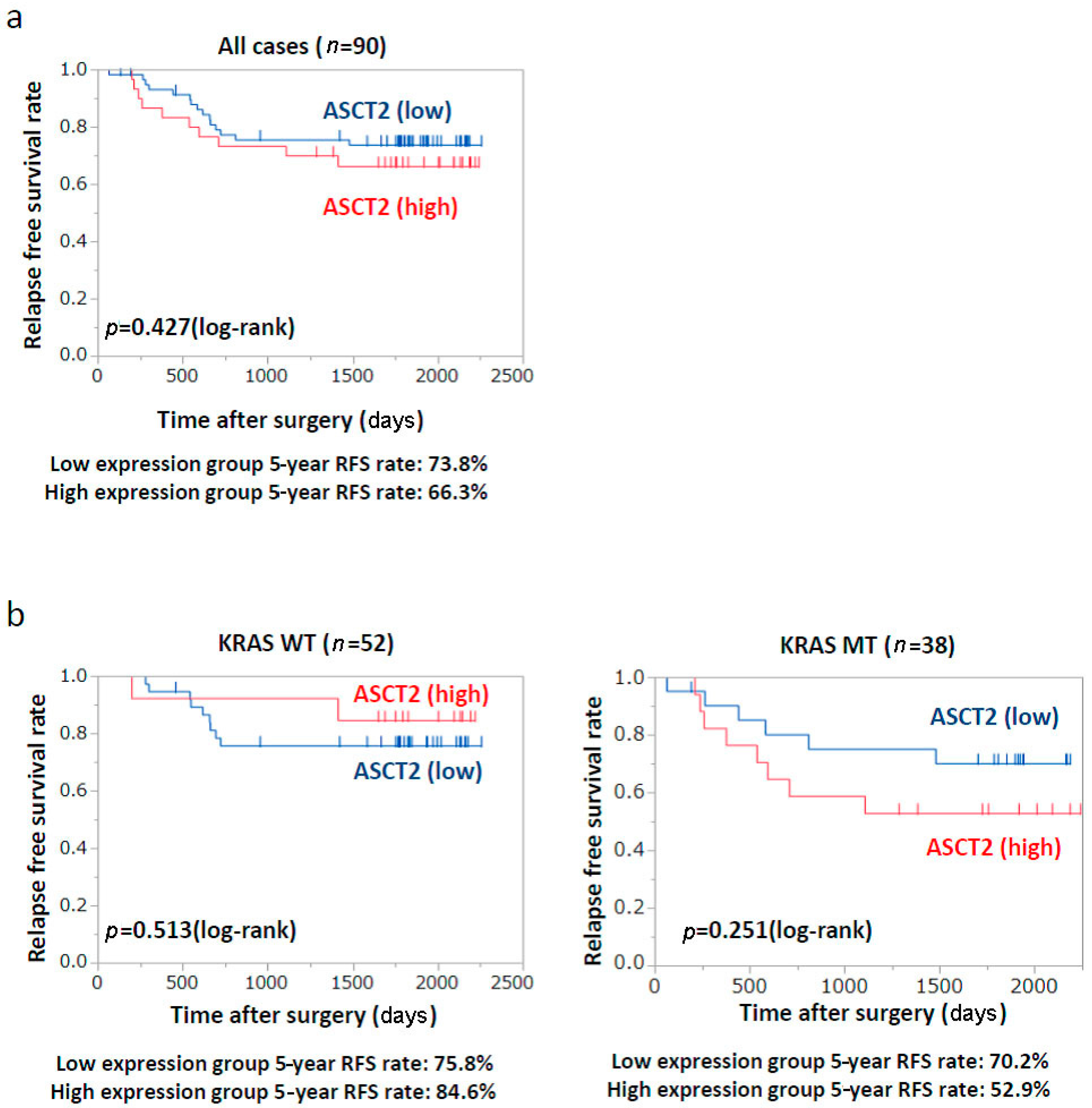
2.5. Tumor Characteristics and ASCT2 Expression in CRC Clinical Samples
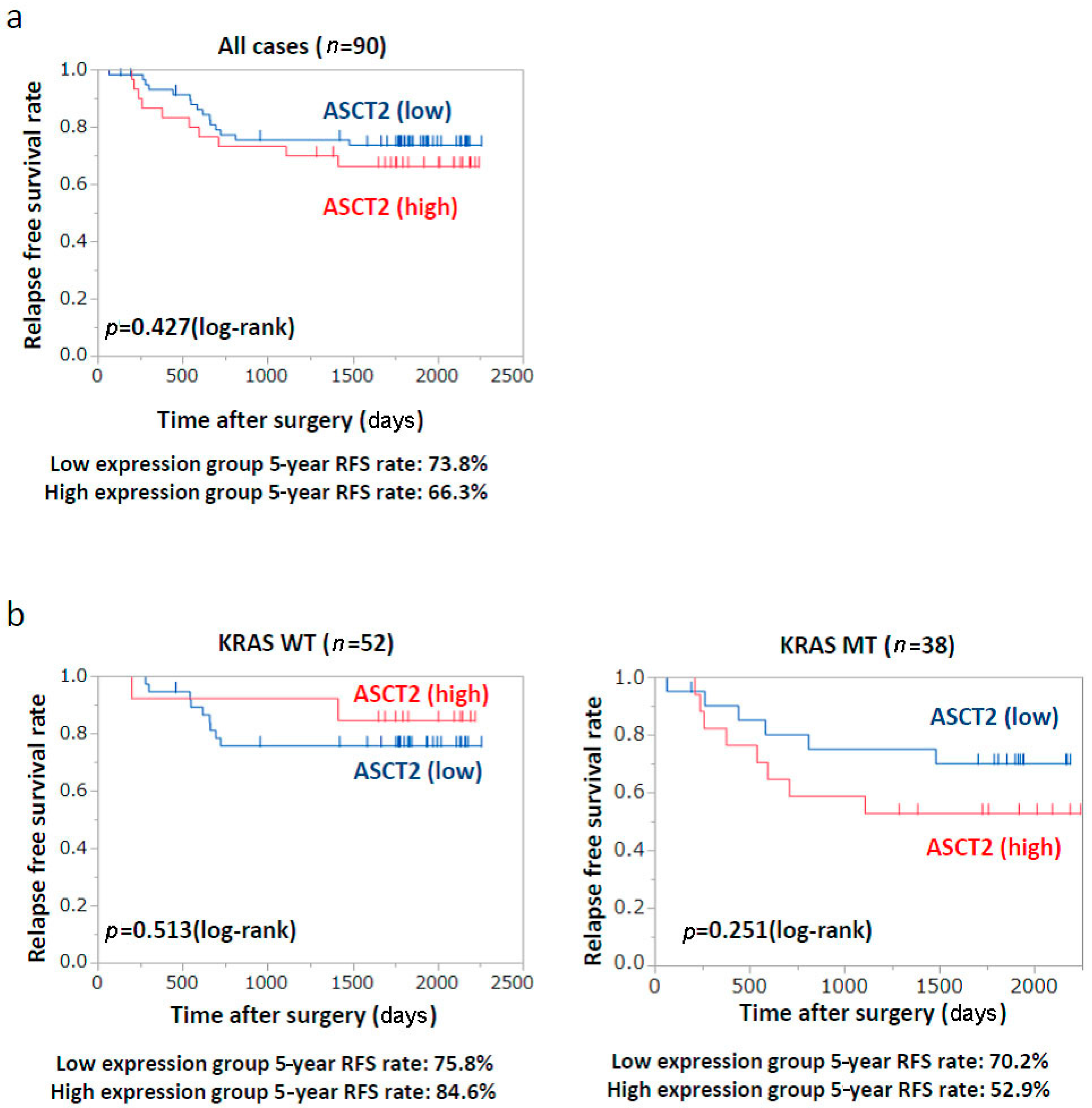
2.6. Patients’ Prognosis
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
4.3. Western Blot Analysis
4.4. Small Interfering RNA and Short Hairpin RNA
4.5. Cell Proliferation Assay
4.6. Clonogenic Assay
4.7. Apoptosis Assay
4.8. Wound Healing Assay
4.9. Immunohistochemistry
4.10. Patients, Clinicopathological Data
4.11. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ASCT2 | Alanine-serine-cysteine amino acid transporter |
| ASNS | Asparagine synthetase |
| CRC | Colorectal cancer |
| EGFR | Epidermal growth factor receptor |
| GLUD1 | Glutamine dehydrogenase 1 |
| GLUT1 | Glucose transporter 1 |
| PPP | Pentose phosphate pathway |
| RFS | Recurrence-free survival (RFS) |
| RT-PCR | Reverse-transcription polymerase chain reaction |
| siRNA | Small interfering RNA |
| TCA cycle | Tricarboxylic acid cycle |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-Ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C. Metabolic dependencies in RAS-driven cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Li, X.; Xu, J.; Kang, W.; Tong, J.H.; To, K.F.; Jin, Y.; Li, W.; Chen, H.; et al. SLC25A22 Promotes proliferation and survival of colorectal cancer cells with KRAS mutations and xenograft tumor progression in mice via intracellular synthesis of aspartate. Gastroenterology 2016, 151, 945–960. [Google Scholar] [CrossRef] [PubMed]
- Toda, K.; Kawada, K.; Iwamoto, M.; Inamoto, S.; Sasazuki, T.; Shirasawa, S.; Hasegawa, S.; Sakai, Y. Metabolic alterations caused by KRAS mutations in colorectal cancer contribute to cell adaptation to glutamine depletion by upregulation of asparagine synthetase. Neoplasia 2016, 18, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Miyo, M.; Konno, M.; Nishida, N.; Sueda, T.; Noguchi, K.; Matsui, H.; Colvin, H.; Kawamoto, K.; Koseki, J.; Haraguchi, N.; et al. Metabolic adaptation to nutritional stress in human colorectal cancer. Sci. Rep. 2016, 6, 38415. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Kawada, K.; Toda, K.; Nakamoto, Y.; Iwamoto, M.; Hatano, E.; Chen, F.; Hasegawa, S.; Togashi, K.; Date, H.; Uemoto, S.; et al. Relationship between 18F-FDG PET/CT scans and KRAS mutations in metastatic colorectal cancer. J. Nucl. Med. 2015, 56, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Kawada, K.; Nakamoto, Y.; Itatani, Y.; Inamoto, S.; Toda, K.; Kimura, H.; Sasazuki, T.; Shirasawa, S.; Okuyama, H.; et al. Regulation of 18F-FDG accumulation in colorectal cancer cells with mutated KRAS. J. Nucl. Med. 2014, 55, 2038–2044. [Google Scholar] [CrossRef] [PubMed]
- Kawada, K.; Nakamoto, Y.; Kawada, M.; Hida, K.; Matsumoto, T.; Murakami, T.; Hasegawa, S.; Togashi, K.; Sakai, Y. Relationship between 18F-fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin. Cancer Res. 2012, 18, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.W.; Yu, X.J.; Wu, W.C.; Chen, J.; Shi, M.; Zheng, L.; Xu, J. GLUT1 and ASCT2 as predictors for prognosis of hepatocellular carcinoma. PLoS ONE 2016, 11, e0168907. [Google Scholar] [CrossRef] [PubMed]
- Honjo, H.; Kaira, K.; Miyazaki, T.; Yokobori, T.; Kanai, Y.; Nagamori, S.; Oyama, T.; Asao, T.; Kuwano, H. Clinicopathological significance of LAT1 and ASCT2 in patients with surgically resected esophageal squamous cell carcinoma. J. Surg. Oncol. 2016, 113, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, T.; Shimizu, K.; Kaira, K.; Nagashima, T.; Ohtaki, Y.; Atsumi, J.; Obayashi, K.; Nagamori, S.; Kanai, Y.; Oyama, T.; et al. Clinical significance of coexpression of l-type amino acid transporter 1 (LAT1) and ASC amino acid transporter 2 (ASCT2) in lung adenocarcinoma. Am. J. Transl. Res. 2015, 7, 1126–1139. [Google Scholar] [PubMed]
- Ren, P.; Yue, M.; Xiao, D.; Xiu, R.; Gan, L.; Liu, H.; Qing, G. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 2015, 235, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Nikkuni, O.; Kaira, K.; Toyoda, M.; Shino, M.; Sakakura, K.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; Suzuki, M.; Iijima, M.; et al. Expression of amino acid transporters (LAT1 and ASCT2) in patients with stage III/IV laryngeal squamous cell carcinoma. Pathol. Oncol. Res. 2015, 21, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, L.; An, H.; Chang, Y.; Zhang, W.; Zhu, Y.; Xu, L.; Xu, J. High expression of solute carrier family 1, member 5 (SLC1A5) is associated with poor prognosis in clear-cell renal cell carcinoma. Sci. Rep. 2015, 5, 16954. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; Clark, J.E.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Kaira, K.; Ohshima, Y.; Ishioka, N.S.; Shino, M.; Sakakura, K.; Takayasu, Y.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; et al. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer 2014, 110, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kaira, K.; Tomizawa, Y.; Sunaga, N.; Kawashima, O.; Oriuchi, N.; Tominaga, H.; Nagamori, S.; Kanai, Y.; Yamada, M.; et al. ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br. J. Cancer 2014, 110, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Panni, S.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Transport mechanism and regulatory properties of the human amino acid transporter ASCT2 (SLC1A5). Amino Acids 2014, 46, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhao, Y.; Zhao, J.; Wu, S.; Jiang, Y.; Ma, H.; Zhang, T. Upregulated SLC1A5 promotes cell growth and survival in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6006–6014. [Google Scholar] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total | KRAS Wild-Type | KRAS Mutant | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ASCT2 | ASCT2 | ASCT2 | |||||||
| High (n = 30) | Low (n = 63) | p-value | High (n = 13) | Low (n = 41) | p-value | High (n = 17) | Low (n = 22) | p-value | |
| Age, mean ± SD (y) | 71.2 ± 9.4 | 68.0 ± 10.7 | 0.16 | 69.8 ± 10.5 | 67.2 ± 10.6 | 0.42 | 72.2 ± 9.8 | 69.4 ± 11.1 | 0.41 |
| Sex | |||||||||
| Male | 16 | 39 | 0.43 | 8 | 28 | 0.65 | 8 | 11 | 0.86 |
| Female | 14 | 24 | 5 | 13 | 9 | 11 | |||
| Location | |||||||||
| Left | 18 | 50 | 0.049 * | 9 | 33 | 0.45 | 9 | 17 | 0.11 |
| Right | 12 | 13 | 4 | 8 | 8 | 5 | |||
| Tumor size (mm) | |||||||||
| ≥50 | 12 | 25 | 0.98 | 5 | 15 | 0.9 | 12 | 25 | 0.98 |
| <50 | 18 | 38 | 8 | 26 | 18 | 38 | |||
| UICC-TMN stage | |||||||||
| I/II | 14 | 33 | 0.61 | 7 | 23 | 0.89 | 7 | 10 | 0.79 |
| III/IV | 16 | 30 | 6 | 18 | 10 | 12 | |||
| T-category | |||||||||
| 1/2 | 5 | 25 | 0.16 | 4 | 10 | 0.72 | 1 | 9 | 0.024 * |
| 3/4 | 25 | 44 | 9 | 31 | 16 | 13 | |||
| M-category | |||||||||
| Negative | 25 | 53 | 0.92 | 10 | 35 | 0.67 | 15 | 18 | 0.75 |
| Positive | 5 | 10 | 3 | 6 | 2 | 4 | |||
| N-category | |||||||||
| Negative | 16 | 35 | 0.84 | 8 | 23 | 0.73 | 8 | 12 | 0.64 |
| Positive | 14 | 28 | 5 | 18 | 9 | 10 | |||
| Lymphatic invasion | |||||||||
| Negative | 18 | 39 | 0.86 | 6 | 25 | 0.35 | 12 | 14 | 0.65 |
| Positive | 12 | 24 | 7 | 16 | 5 | 8 | |||
| Vascular invasion | |||||||||
| Negative | 6 | 19 | 0.3 | 3 | 8 | 1 | 3 | 11 | 0.049 * |
| Positive | 24 | 44 | 10 | 33 | 14 | 11 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toda, K.; Nishikawa, G.; Iwamoto, M.; Itatani, Y.; Takahashi, R.; Sakai, Y.; Kawada, K. Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 1632. https://doi.org/10.3390/ijms18081632
Toda K, Nishikawa G, Iwamoto M, Itatani Y, Takahashi R, Sakai Y, Kawada K. Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer. International Journal of Molecular Sciences. 2017; 18(8):1632. https://doi.org/10.3390/ijms18081632
Chicago/Turabian StyleToda, Kosuke, Gen Nishikawa, Masayoshi Iwamoto, Yoshiro Itatani, Ryo Takahashi, Yoshiharu Sakai, and Kenji Kawada. 2017. "Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer" International Journal of Molecular Sciences 18, no. 8: 1632. https://doi.org/10.3390/ijms18081632
APA StyleToda, K., Nishikawa, G., Iwamoto, M., Itatani, Y., Takahashi, R., Sakai, Y., & Kawada, K. (2017). Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer. International Journal of Molecular Sciences, 18(8), 1632. https://doi.org/10.3390/ijms18081632