Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes


Abstract
1. Introduction
2. Results
2.1. Blocking the Mechanistic Target of Rapamycin Complex (mTORC)1 Prevents Degradation of the Extracellular Matrix
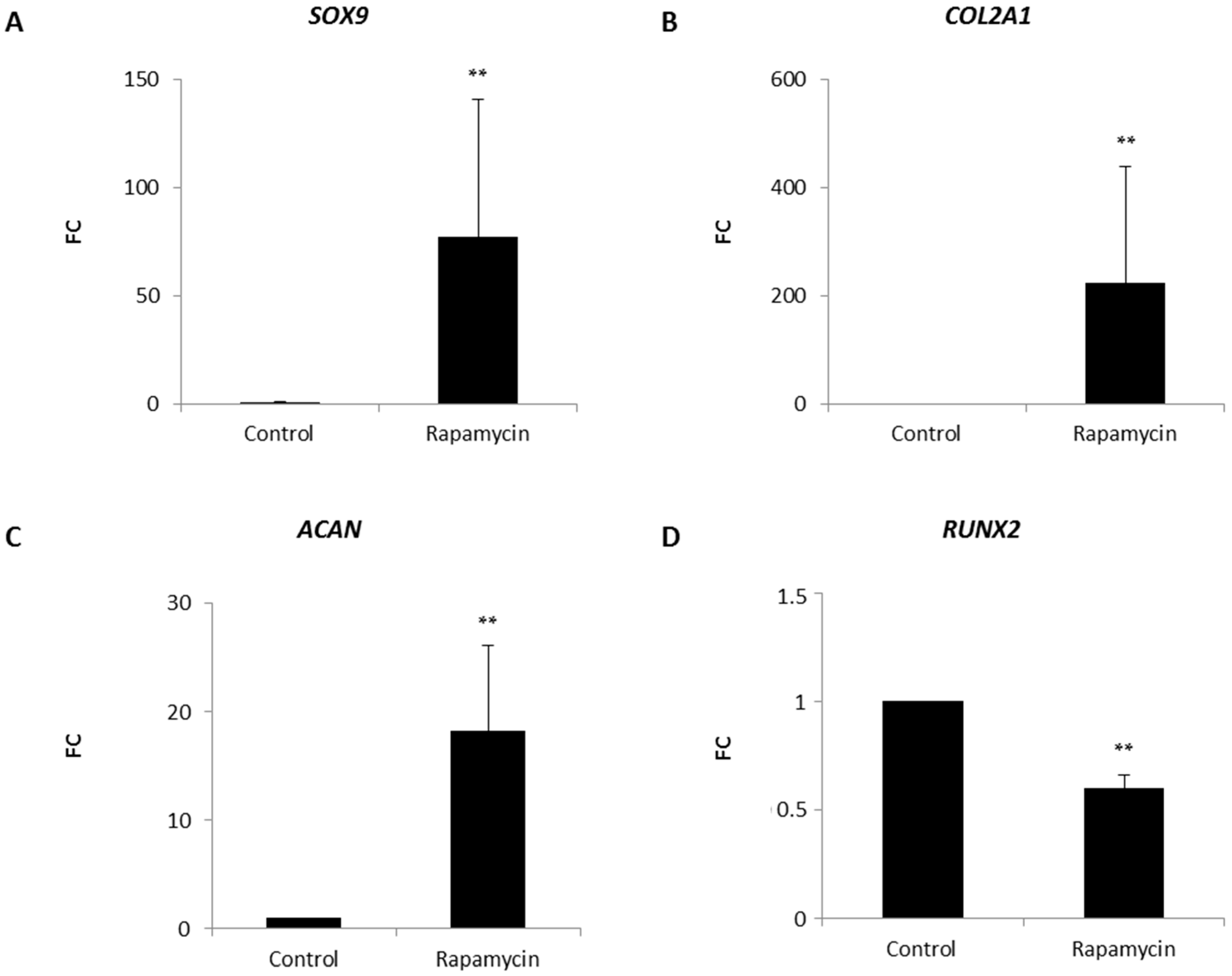
2.2. Effects of mTORC1 Inhibition on the Chondrogenic Phenotype of Patient-Derived Osteoarhritic (OA) Chondrocytes
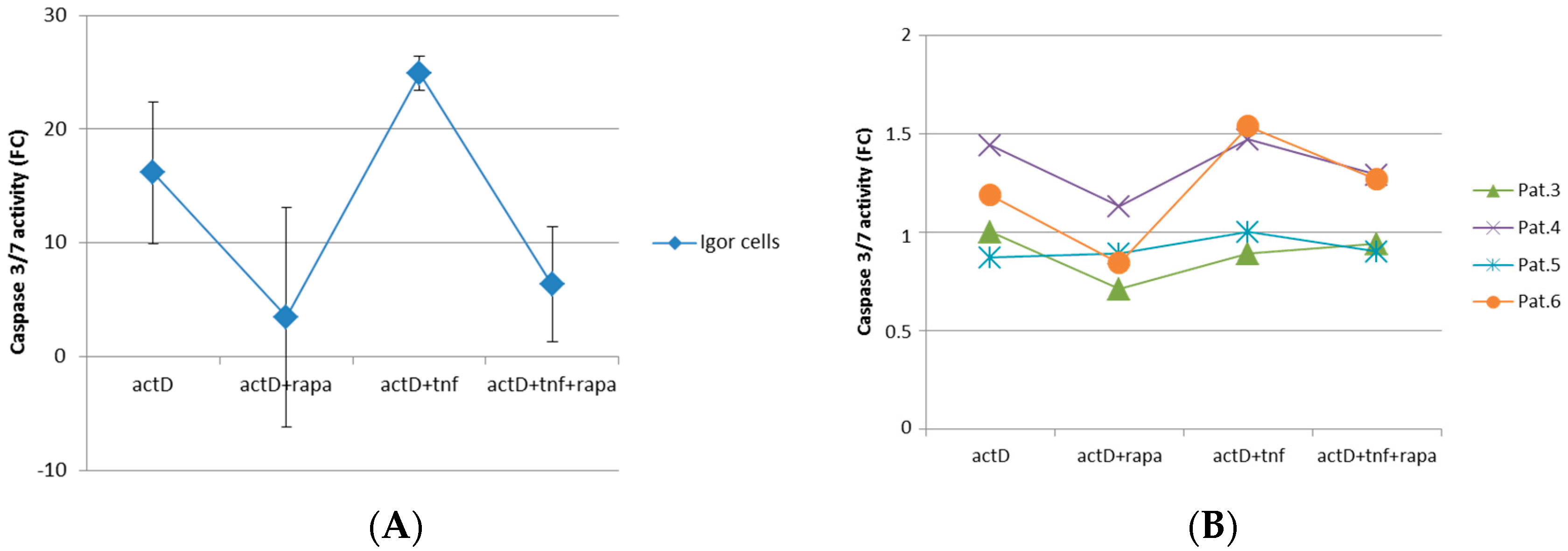
2.3. mTORC1 Inhibition Prevents Chondrocytes from Undergoing Apoptosis
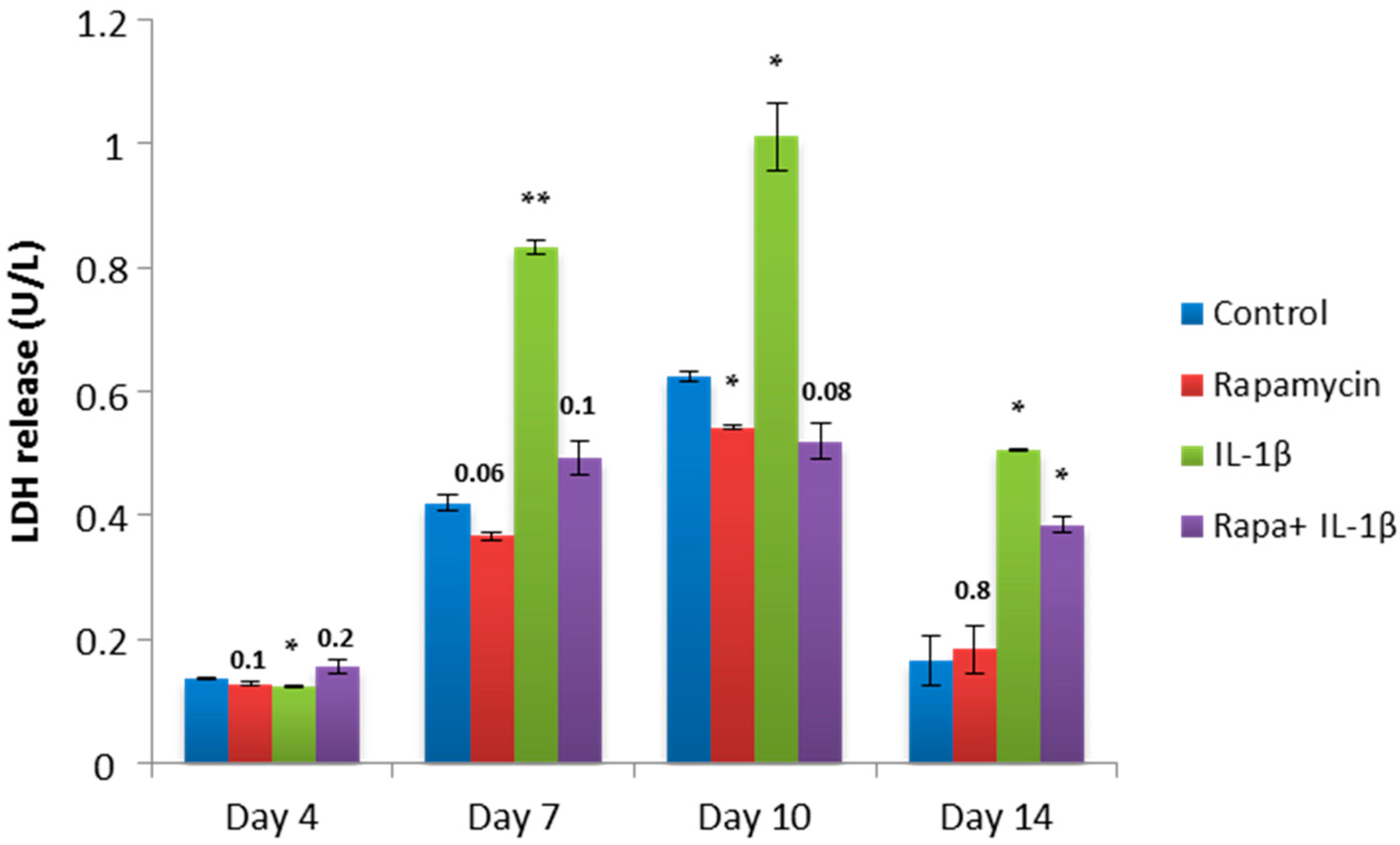
2.4. Block of mTORC1 Prohibits Cellular Cytotoxicity and Cytolysis of Patient-Derived OA Chondrocytes
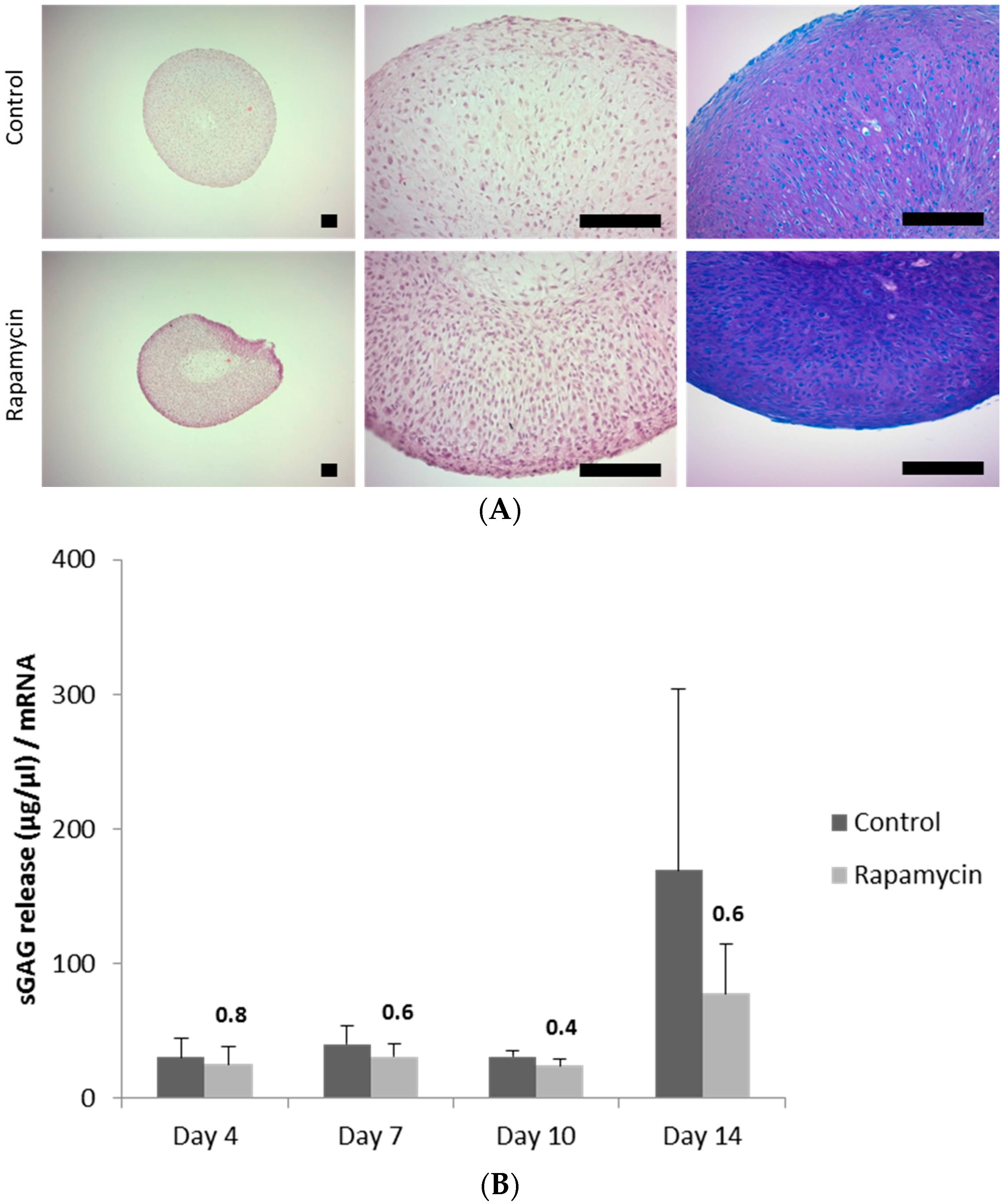
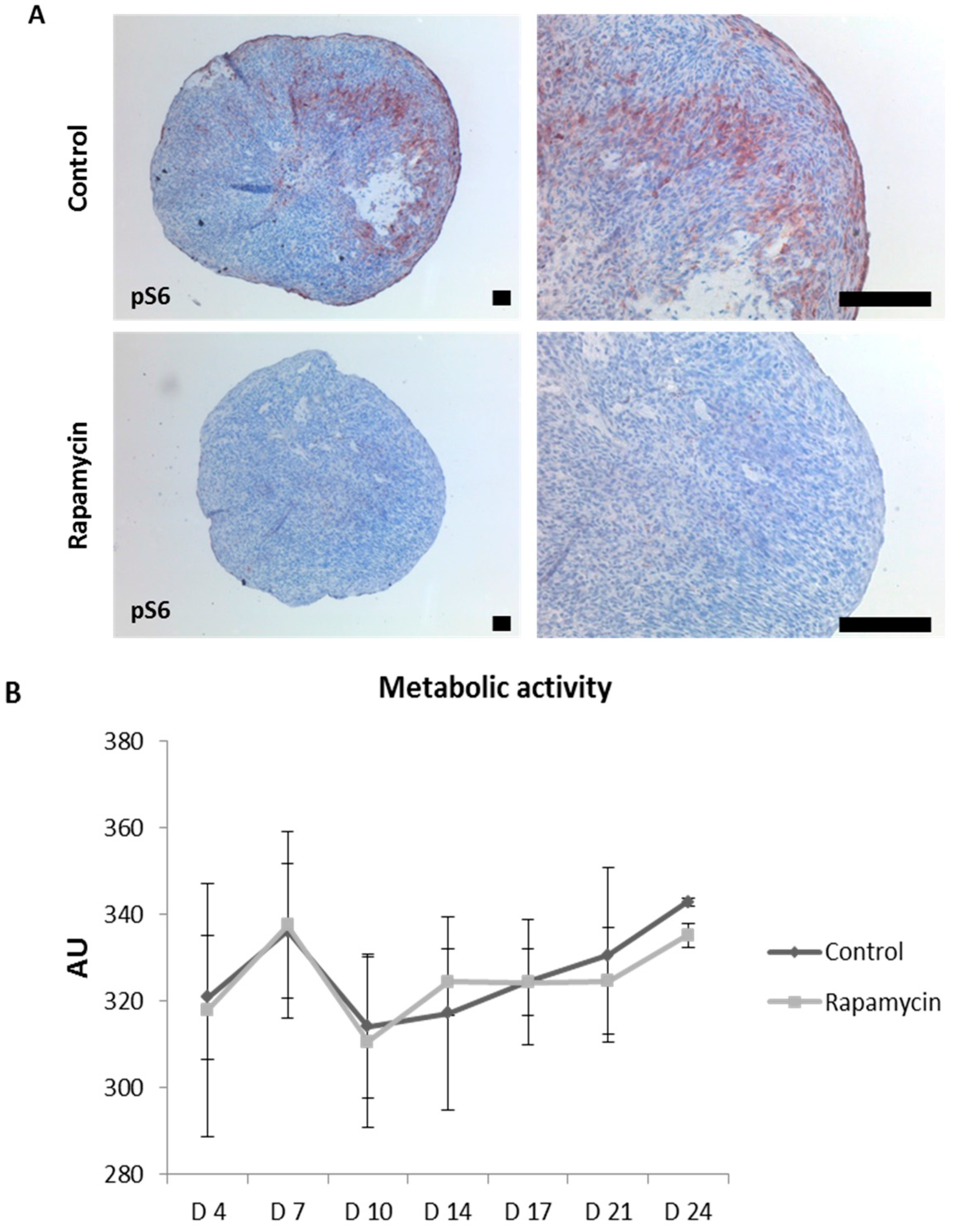
2.5. Blocking mTORC1 Promotes Chondrogenesis and Suppresses Cartilage Degrading and Inflammatory Processes in an OA-Model
3. Discussion
4. Materials and Methods
4.1. Isolation of Patient-Derived Osteoarthritic (OA) Chondrocytes
4.2. Cultivation of Patient-Derived OA Chondrocytes within a Collagen Type I Hydrogel
4.3. Cultivation of Patient-Derived OA Chondrocytes in a Three Dimensional Aggregate
4.4. IGOR Cells
4.5. Hematoxylin and Eosin Staining
4.6. Immunohistochemical Staining
4.7. 1,9 Dimethylmethylen Blue (DMMB) Staining
4.8. Sulfated Glycosaminoglycan (sGAG) Assay
4.9. Lactate Dehydrogenase (LDH) Assay
4.10. Caspase 3/7 Assay
4.11. Alamar Blue Assay
4.12. RNA Extraction and Polymerase Chain Reaction
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin-1 domains |
| Caspase | Cyseinyl-aspartate specific proteinase |
| CASY | Cell counter and analyser system |
| DMMB | 1,9 Dimethylmethylene blue |
| ECM | Extracellular matrix |
| EDTA | Ethylenediaminetetraacetic acid |
| ER | Endoplasmatic reticulum |
| FDA | Food and Drug Administration |
| FCS | Fetal calf serum |
| sGAG | Sulfated glycosaminoglycan |
| IL | Interleukin |
| ITS | Insulin-transferrin-selenium |
| LDH | Lactate dehydrogenase |
| MAPK | mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| mTOR | Mechanistic target of rapamycin |
| mTORC | Mechanistic target of rapamycin complex |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B-cells |
| NSAID | Nonsteroidal anti-inflammatory drugs |
| OA | Osteoarthritis |
| pS6 | Phospho-S6 |
| RT-PCR | Real time polymerase chain reaction |
| SOX | SRY-box |
| TNF | Tumor necrosis factor |
| TGF | Transforming growth factor |
Appendix A

References
- Jiang, L.; Tian, W.; Wang, Y.; Rong, J.; Bao, C.; Liu, Y.; Zhao, Y.; Wang, C. Body mass index and susceptibility to knee osteoarthritis: A systematic review and meta-analysis. Jt. Bone Spine 2012, 79, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Nehrer, S.; Minas, T. Treatment of articular cartilage defects. Investig. Radiol. 2000, 35, 639–646. [Google Scholar] [CrossRef]
- Chaganti, R.K.; Lane, N.E. Risk factors for incident osteoarthritis of the hip and knee. Curr. Rev. Musculoskelet. Med. 2011, 4, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Alhambra, D.; Judge, A.; Javaid, M.K.; Cooper, C.; Diez-Perez, A.; Arden, N.K. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: Influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis. 2014, 73, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Ono, K.; Miyazaki, J.; Olsen, B.R.; Muragaki, Y.; Adachi, E.; Yamamura, K.; Kimura, T. Osteoarthritis associated with mild chondrodysplasia in transgenic mice expressing α 1(IX) collagen chains with a central deletion. Proc. Natl. Acad. Sci. USA 1993, 90, 2870–2874. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- De Lange-Brokaar, B.J.; Ioan-Facsinay, A.; van Osch, G.J.; Zuurmond, A.M.; Schoones, J.; Toes, R.E.; Huizinga, T.W.; Kloppenburg, M. Synovial inflammation, immune cells and their cytokines in osteoarthritis: A review. Osteoarthr. Cartil. 2012, 20, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Melchiorri, C.; Meliconi, R.; Frizziero, L.; Silvestri, T.; Pulsatelli, L.; Mazzetti, I.; Borzi, R.M.; Uguccioni, M.; Facchini, A. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum. 1998, 41, 2165–2174. [Google Scholar] [CrossRef]
- Massicotte, F.; Lajeunesse, D.; Benderdour, M.; Pelletier, J.P.; Hilal, G.; Duval, N.; Martel-Pelletier, J. Can altered production of interleukin-1β, interleukin-6, transforming growth factor-β and prostaglandin E2 by isolated human subchondral osteoblasts identify two subgroups of osteoarthritic patients. Osteoarthr. Cartil. 2002, 10, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Farahat, M.N.; Yanni, G.; Poston, R.; Panayi, G.S. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann. Rheum. Dis. 1993, 52, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; van den Berg, W.B. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010, 62, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; McKenna, L.; Zien, A.; Fan, Z.; Gebhard, P.M.; Zimmer, R. Gene expression profiling of serum- and interleukin-1β-stimulated primary human adult articular chondrocytes—A molecular analysis based on chondrocytes isolated from one donor. Cytokine 2005, 31, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Guerne, P.A.; Carson, D.A.; Lotz, M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J. Immunol. 1990, 144, 499–505. [Google Scholar] [PubMed]
- Lotz, M.; Terkeltaub, R.; Villiger, P.M. Cartilage and joint inflammation. Regulation of IL-8 expression by human articular chondrocytes. J. Immunol. 1992, 148, 466–473. [Google Scholar] [PubMed]
- Verma, P.; Dalal, K. ADAMTS-4 and ADAMTS-5: Key enzymes in osteoarthritis. J. Cell. Biochem. 2011, 112, 3507–3514. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Peeters-Joris, C.; Vaes, G. Modulation by interleukin 1 and tumor necrosis factor α of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim. Biophys. Acta 1990, 1052, 366–378. [Google Scholar] [CrossRef]
- Xue, J.; Wang, J.; Liu, Q.; Luo, A. Tumor necrosis factor-α induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol. Med. Rep. 2013, 8, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182. [Google Scholar] [CrossRef]
- Siegel, N.; Valli, A.; Fuchs, C.; Rosner, M.; Hengstschlager, M. Expression of mTOR pathway proteins in human amniotic fluid stem cells. Int. J. Mol. Med. 2009, 23, 779–784. [Google Scholar] [PubMed]
- Rosner, M.; Hanneder, M.; Siegel, N.; Valli, A.; Fuchs, C.; Hengstschlager, M. The mTOR pathway and its role in human genetic diseases. Mutat. Res. 2008, 659, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Calne, R.Y.; Collier, D.S.; Lim, S.; Pollard, S.G.; Samaan, A.; White, D.J.; Thiru, S. Rapamycin for immunosuppression in organ allografting. Lancet 1989, 2, 227. [Google Scholar] [CrossRef]
- Lopez de Figueroa, P.; Lotz, M.K.; Blanco, F.J.; Carames, B. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheum. 2015, 67, 966–976. [Google Scholar] [CrossRef]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Matsushita, T.; Tabata, Y.; Saito, T.; Matsumoto, T.; Nagai, K.; Kuroda, R.; Kurosaka, M. Intra-articular administration of gelatin hydrogels incorporating rapamycin-micelles reduces the development of experimental osteoarthritis in a murine model. Biomaterials 2014, 35, 9904–9911. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawakami, Y.; Kobayashi, M.; Greco, N.; Cummins, J.H.; Matsushita, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Huard, J. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 2014, 16, 482. [Google Scholar] [CrossRef] [PubMed]
- Facchini, A.; Borzi, R.M.; Olivotto, E.; Platano, D.; Pagani, S.; Cetrullo, S.; Flamigni, F. Role of polyamines in hypertrophy and terminal differentiation of osteoarthritic chondrocytes. Amino Acids 2012, 42, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Vitellozzi, R.; Fernandez, P.; Falcieri, E.; Battistelli, M.; Burattini, S.; Facchini, A.; Flamigni, F.; Santi, S.; Borzi, R.M. Chondrocyte hypertrophy and apoptosis induced by GROα require three-dimensional interaction with the extracellular matrix and a co-receptor role of chondroitin sulfate and are associated with the mitochondrial splicing variant of cathepsin B. J. Cell. Physiol. 2007, 210, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Huey, D.J.; Hu, J.C.; Athanasiou, K.A. Unlike bone, cartilage regeneration remains elusive. Science 2012, 338, 917–921. [Google Scholar] [CrossRef]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Lopez de Figueroa, P.; Blanco, F.J.; Mendes, A.F.; Carames, B. Insulin decreases autophagy and leads to cartilage degradation. Osteoarthr. Cartil. 2016, 24, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Preitschopf, A.; Schorghofer, D.; Kinslechner, K.; Schutz, B.; Zwickl, H.; Rosner, M.; Joo, J.G.; Nehrer, S.; Hengstschlager, M.; Mikula, M. Rapamycin-induced hypoxia inducible factor 2A is essential for chondrogenic differentiation of amniotic fluid stem cells. Stem Cells Transl. Med. 2016, 5, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Vlad, S.C.; Neogi, T.; Aliabadi, P.; Fontes, J.D.; Felson, D.T. No association between markers of inflammation and osteoarthritis of the hands and knees. J. Rheumatol. 2011, 38, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, A.; Kamrava, S.K.; Joghataei, M.T.; Darabi, R.; Shakeri-Zadeh, A.; Shahriari, M.; Reiter, R.J.; Ghaznavi, H.; Mehrzadi, S. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J. Pineal Res. 2016, 61, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Calich, A.L.; Domiciano, D.S.; Fuller, R. Osteoarthritis: Can anti-cytokine therapy play a role in treatment? Clin. Rheumatol. 2010, 29, 451–455. [Google Scholar] [CrossRef]
- Pulsatelli, L.; Addimanda, O.; Brusi, V.; Pavloska, B.; Meliconi, R. New findings in osteoarthritis pathogenesis: Therapeutic implications. Ther. Adv. Chron. Dis. 2013, 4, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Messier, S.P. Osteoarthritis of the knee and associated factors of age and obesity: Effects on gait. Med. Sci. Sports Exerc. 1994, 26, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Roos, E.M.; Dahlberg, L. Positive effects of moderate exercise on glycosaminoglycan content in knee cartilage: A four-month, randomized, controlled trial in patients at risk of osteoarthritis. Arthritis Rheumatol. 2005, 52, 3507–3514. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, G.B.; Press, A.N.; Lindsley, H.B.; Hinton, R.; Cagle, P.E.; Carlson, K.; Scott, S.; Dahl, J.; Kramer, B. Effects of exercise on fatigue, aerobic fitness, and disease activity measures in persons with rheumatoid arthritis. Res. Nurs. Health 1997, 20, 195–204. [Google Scholar] [CrossRef]
- Hoff, P.; Buttgereit, F.; Burmester, G.R.; Jakstadt, M.; Gaber, T.; Andreas, K.; Matziolis, G.; Perka, C.; Rohner, E. Osteoarthritis synovial fluid activates pro-inflammatory cytokines in primary human chondrocytes. Int. Orthop. 2013, 37, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Moskowitz, N.K.; Gibofsky, A. The post-NSAID era: What to use now for the pharmacologic treatment of pain and inflammation in osteoarthritis. Curr. Pain Headache Rep. 2007, 11, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Burch, F.; Codding, C.; Patel, N.; Sheldon, E. Lidocaine patch 5% improves pain, stiffness, and physical function in osteoarthritis pain patients. A prospective, multicenter, open-label effectiveness trial. Osteoarthr. Cartil. 2004, 12, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Preitschopf, A.; Li, K.; Schorghofer, D.; Kinslechner, K.; Schutz, B.; Thi Thanh Pham, H.; Rosner, M.; Joo, G.J.; Rohrl, C.; Weichhart, T.; et al. mTORC1 is essential for early steps during Schwann cell differentiation of amniotic fluid stem cells and regulates lipogenic gene expression. PLoS ONE 2014, 9, e107004. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | F-Primer | R-Primer |
|---|---|---|
| SOX9 | 5′-AGCGCCCCCACTTTTGCT-3′ | 5′-TGGCCGGGAAAGGCGAG-3′ |
| COL2A1 | 5′- GATAAGGATGTGTGGAAGCCGGAGC-3′ | 5′-TCCTTTCTGTCCCTTTGGTCCTGGT-3′ |
| ACAN | 5′-TACACGCTACACCCTCGACTTTGA-3′ | 5′-TACGTCCTCACACCAGGAAACTCA-3′ |
| RUNX2 | 5′-GTTACTGTCATGGCGGGTAACGAT-3′ | 5′-TCAAGCTTCTGTCTGTGCCTTCTG-3′ |
| MMP13 | 5′-CCAGAAGTGCGGGGTAGGGG-3′ | 5′-TGTGTCCCATTTGTGGTGTGGGA-3′ |
| CASP3 | 5′-CTCCTAGCGGATGGGTGCTATTGT-3′ | 5′-AGACCGAGATGTCATTCCAGTGCTT–3′ |
| IL6 | 5′-GGCTGCTCCTGGTGTTTGCCT-3′ | 5′-TGCCAGTGCCTCTTTGCTGCT-3′ |
| ACTB | 5′-CTATCCAGGCTGTGCTATCCCTGT-3′ | 5′-CCTTAATGTCACGCACGATTTCC-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna-Preitschopf, A.D.; Zwickl, H.; Nehrer, S.; Hengstschläger, M.; Mikula, M. Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes. Int. J. Mol. Sci. 2017, 18, 1494. https://doi.org/10.3390/ijms18071494
Luna-Preitschopf AD, Zwickl H, Nehrer S, Hengstschläger M, Mikula M. Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes. International Journal of Molecular Sciences. 2017; 18(7):1494. https://doi.org/10.3390/ijms18071494
Chicago/Turabian StyleLuna-Preitschopf, Andrea De, Hannes Zwickl, Stefan Nehrer, Markus Hengstschläger, and Mario Mikula. 2017. "Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes" International Journal of Molecular Sciences 18, no. 7: 1494. https://doi.org/10.3390/ijms18071494
APA StyleLuna-Preitschopf, A. D., Zwickl, H., Nehrer, S., Hengstschläger, M., & Mikula, M. (2017). Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes. International Journal of Molecular Sciences, 18(7), 1494. https://doi.org/10.3390/ijms18071494