ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
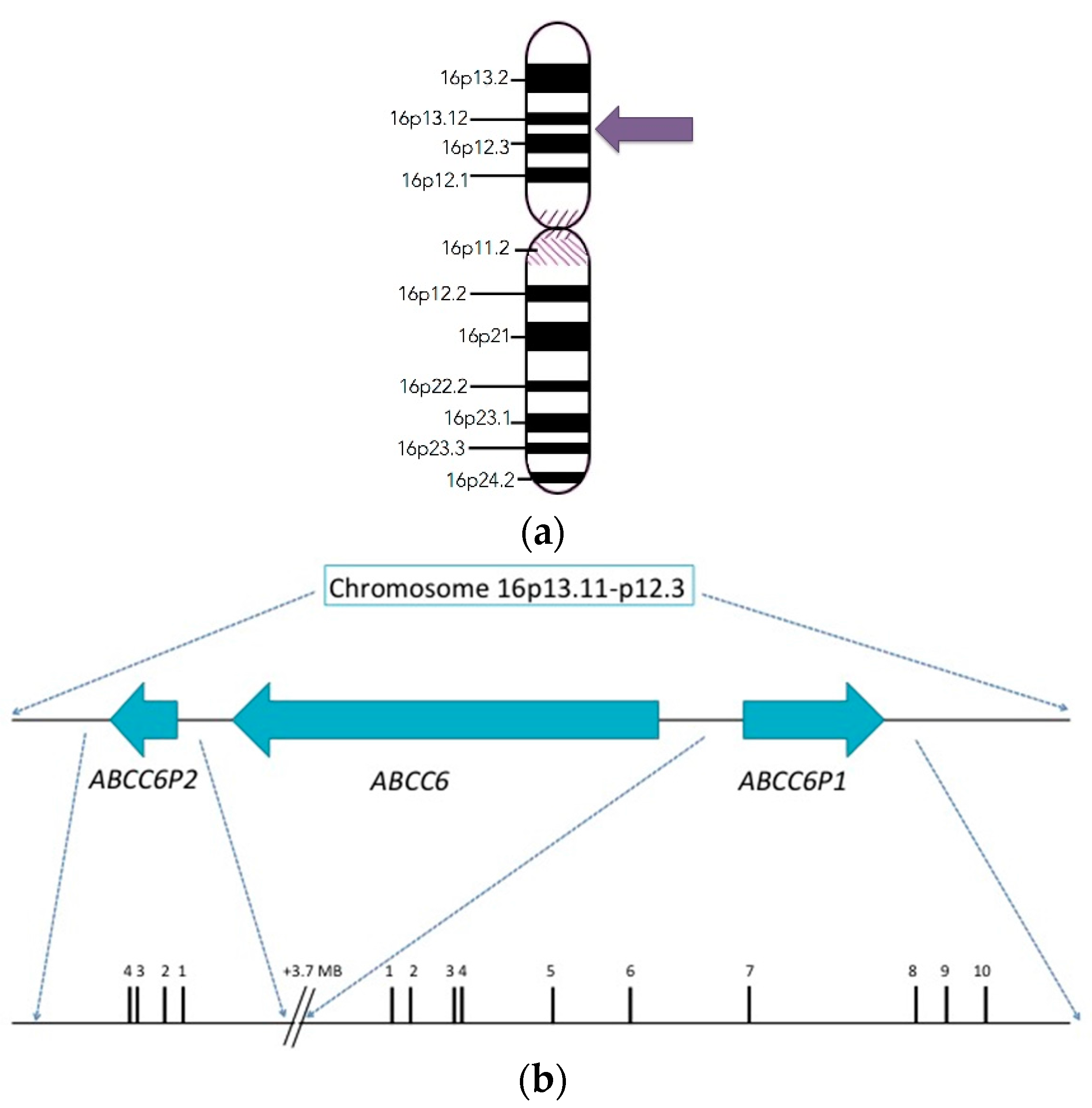
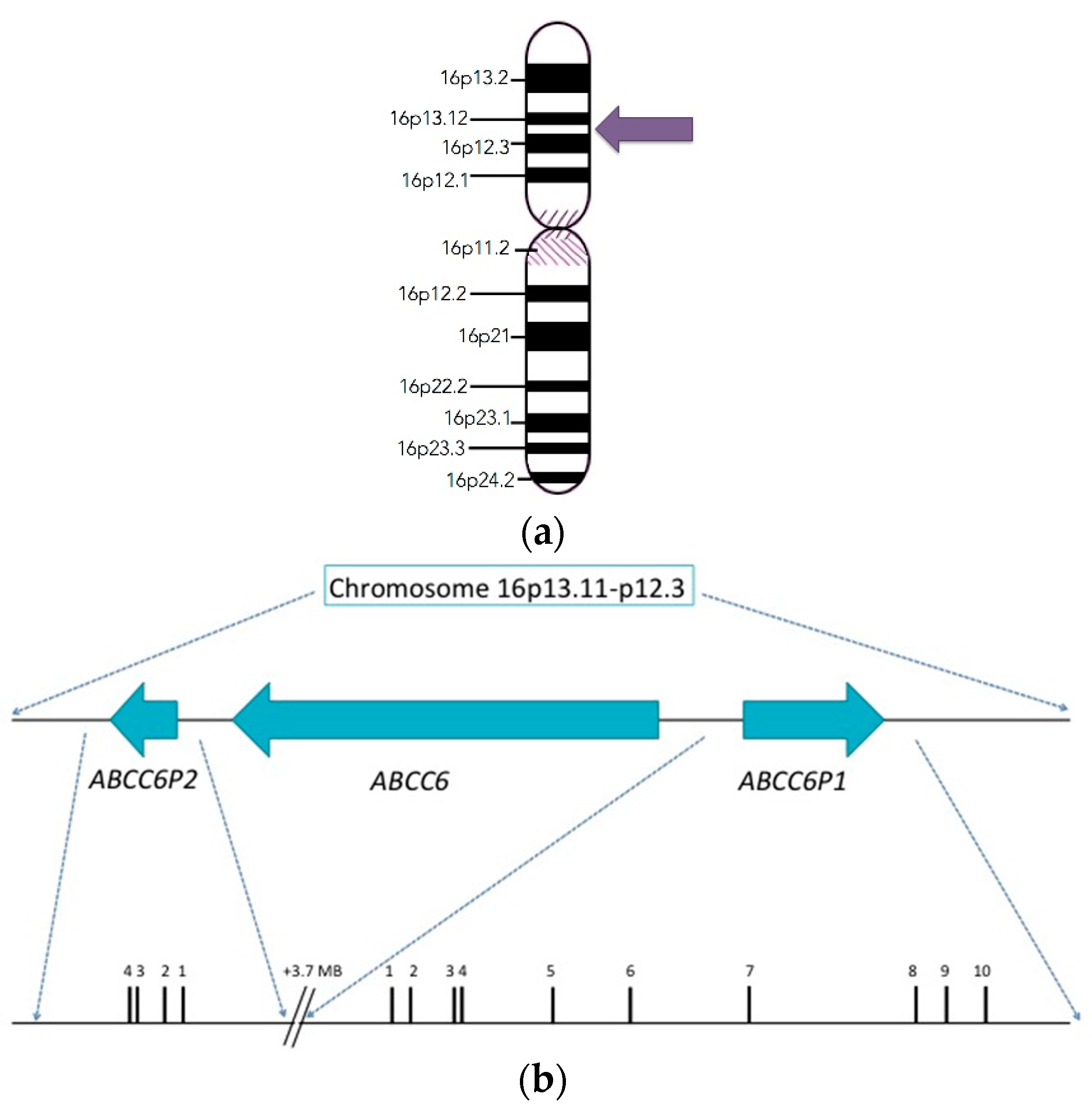
2. Discovery and Structure of the ABCC6 Gene
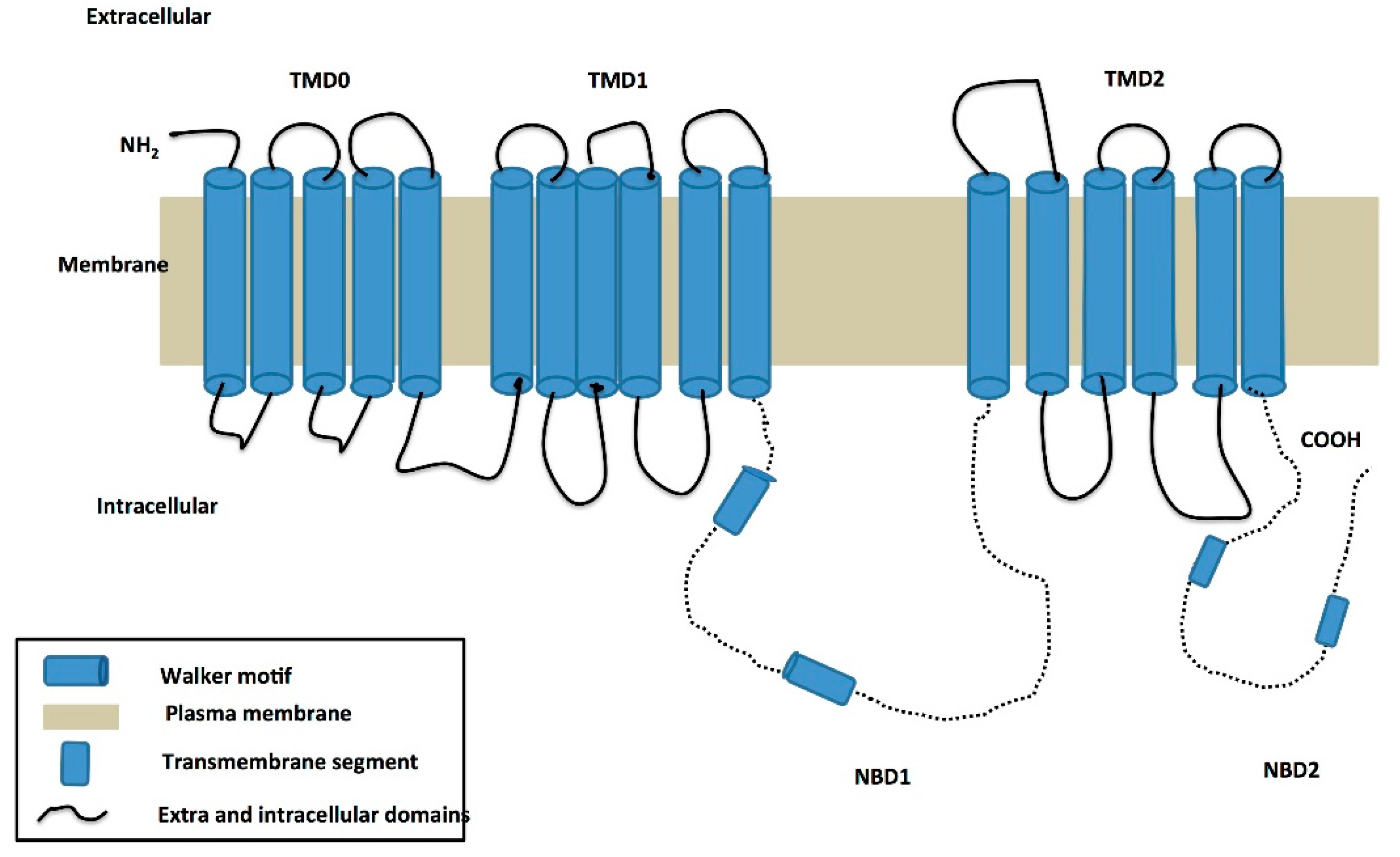
3. Structure of the ABCC6 Protein and Its Functional Role in PXE
Predicted Functional Role of ABCC6 in PXE
4. Founder Effect in PXE
5. Clinical Manifestation of PXE
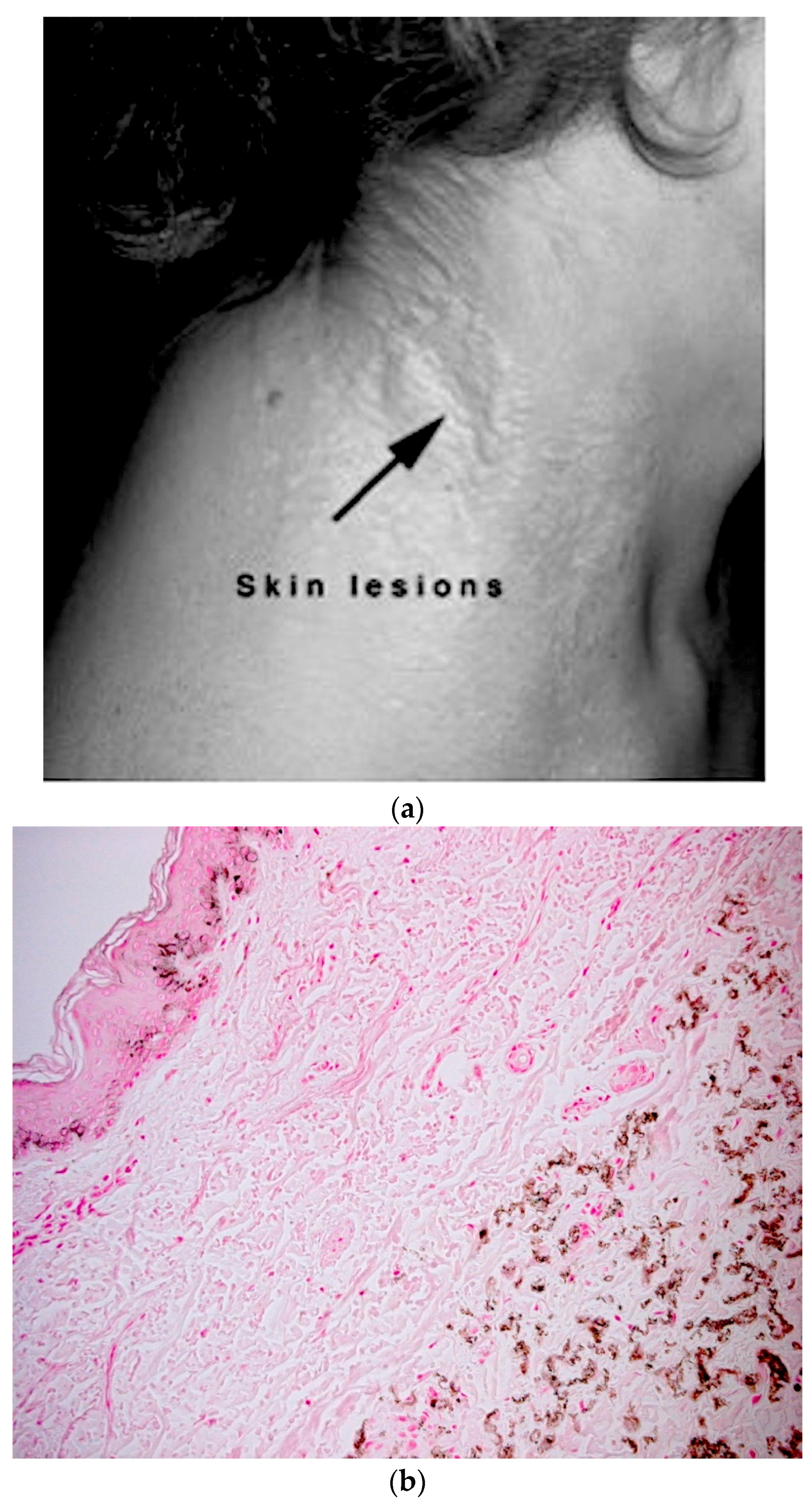
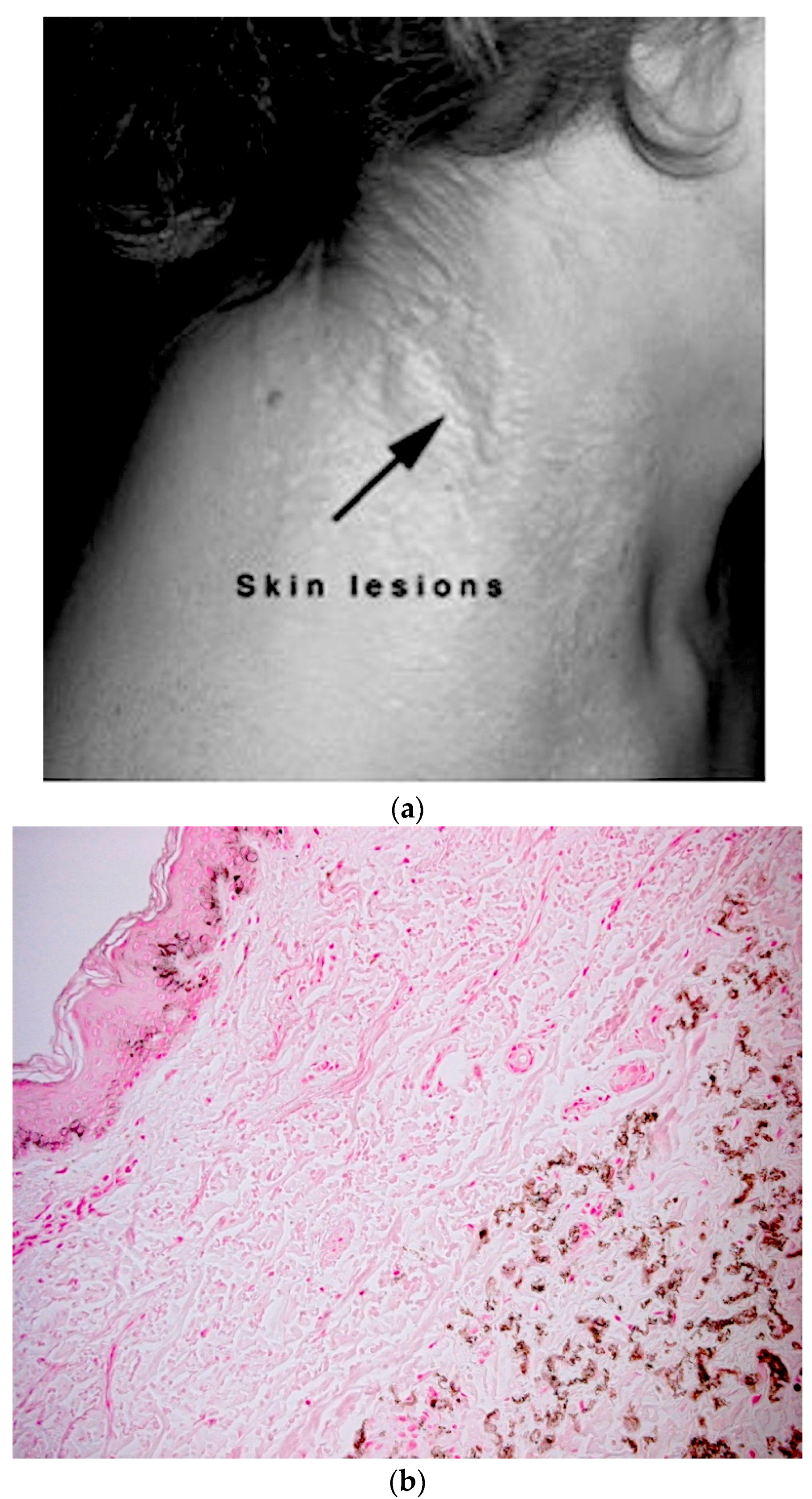
5.1. Skin Phenotype
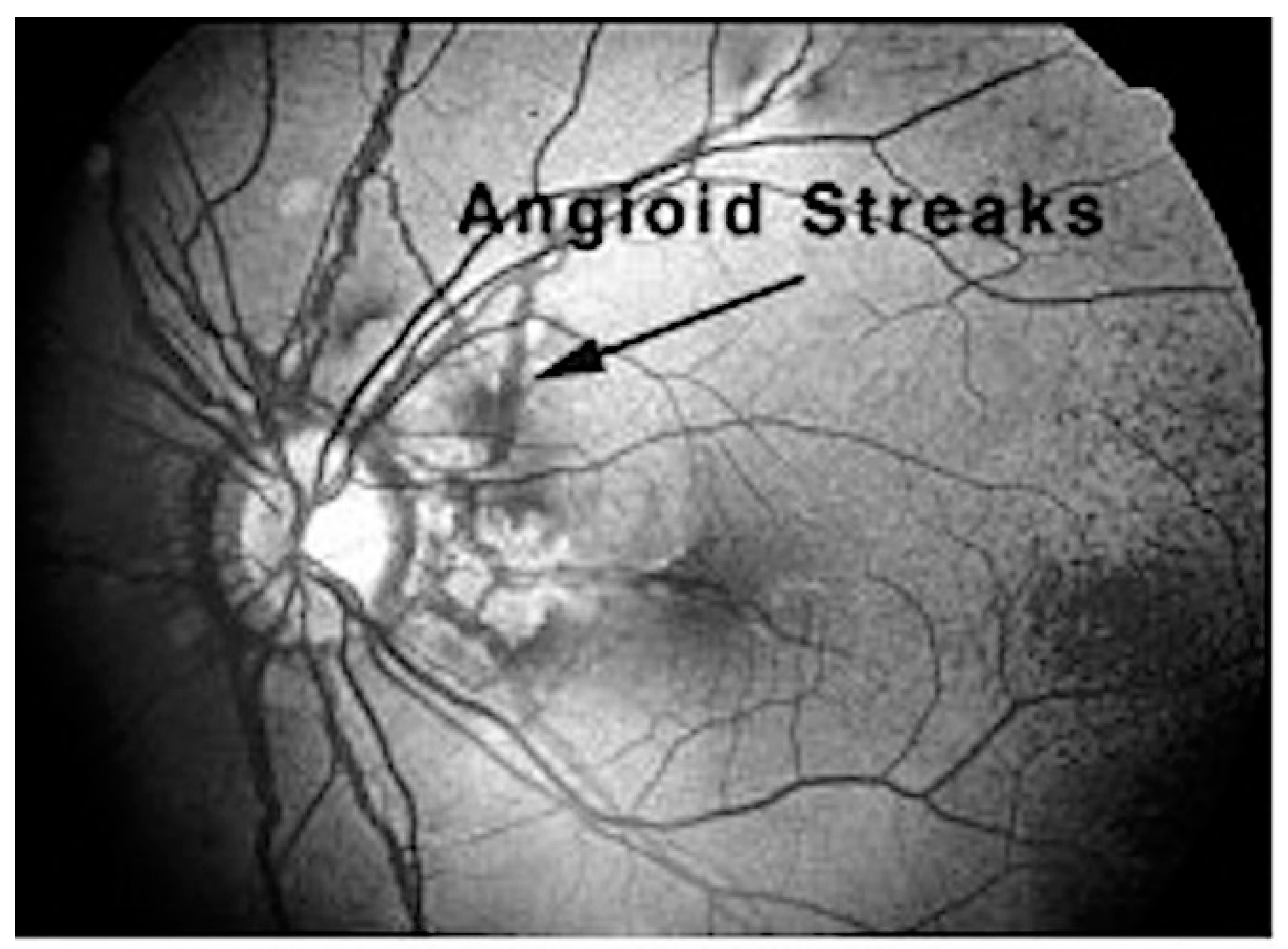
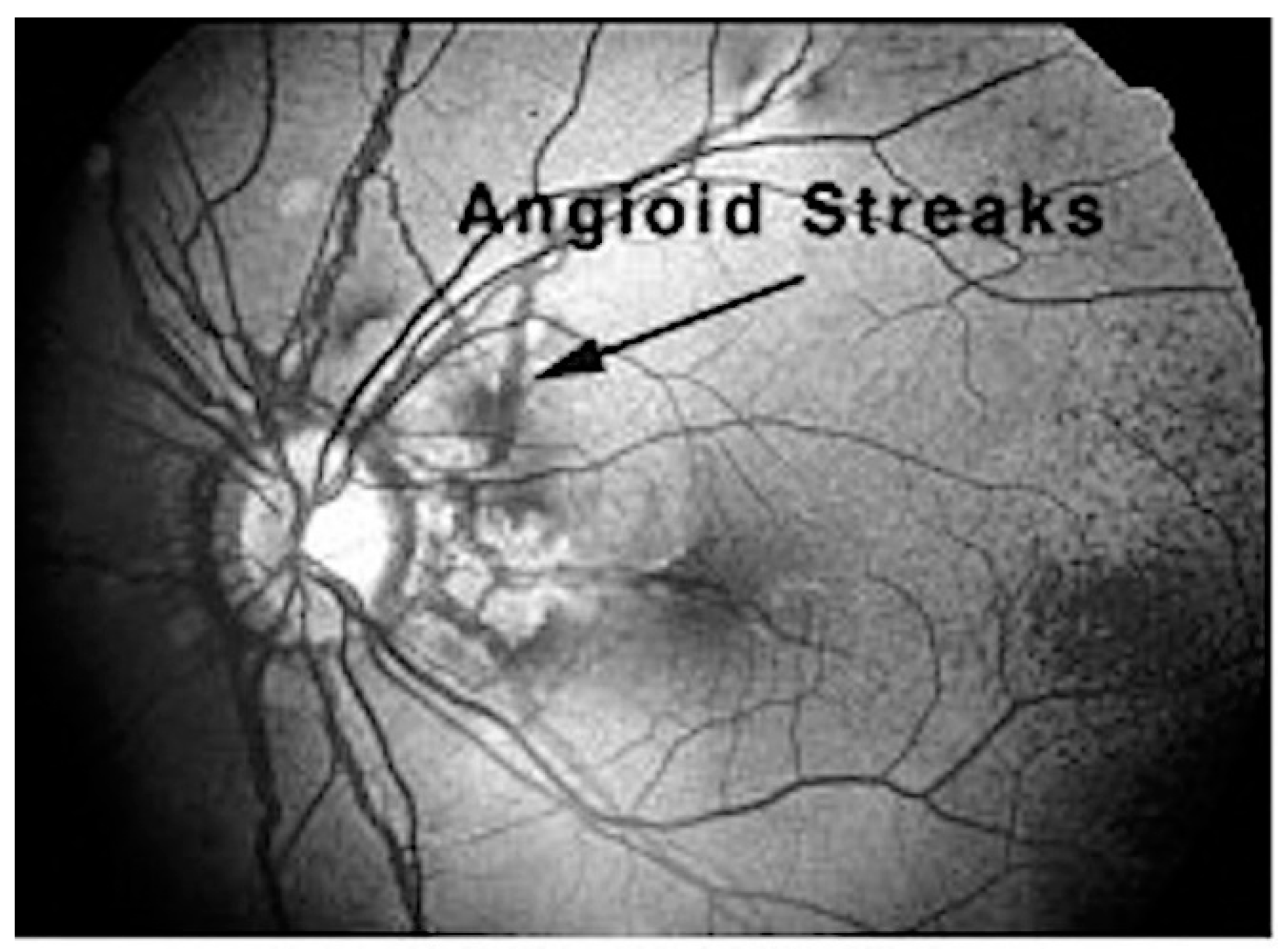
5.2. Ocular Manifestations
5.3. Cardiovascular Manifestations
5.4. Diagnosis of PXE
6. Therapeutic Intervention in PXE
7. The Role of ABCC6 in PXE
7.1. The Metabolic Hypothesis
7.2. The PXE Cell Hypothesis
7.3. The Extracellular ATP Release Hypothesis
8. Common Mutations and Variations in ABCC6 Associated with PXE
9. Next Generation Sequencing Approaches for Detecting Variants in ABCC6 and Modifier Genes
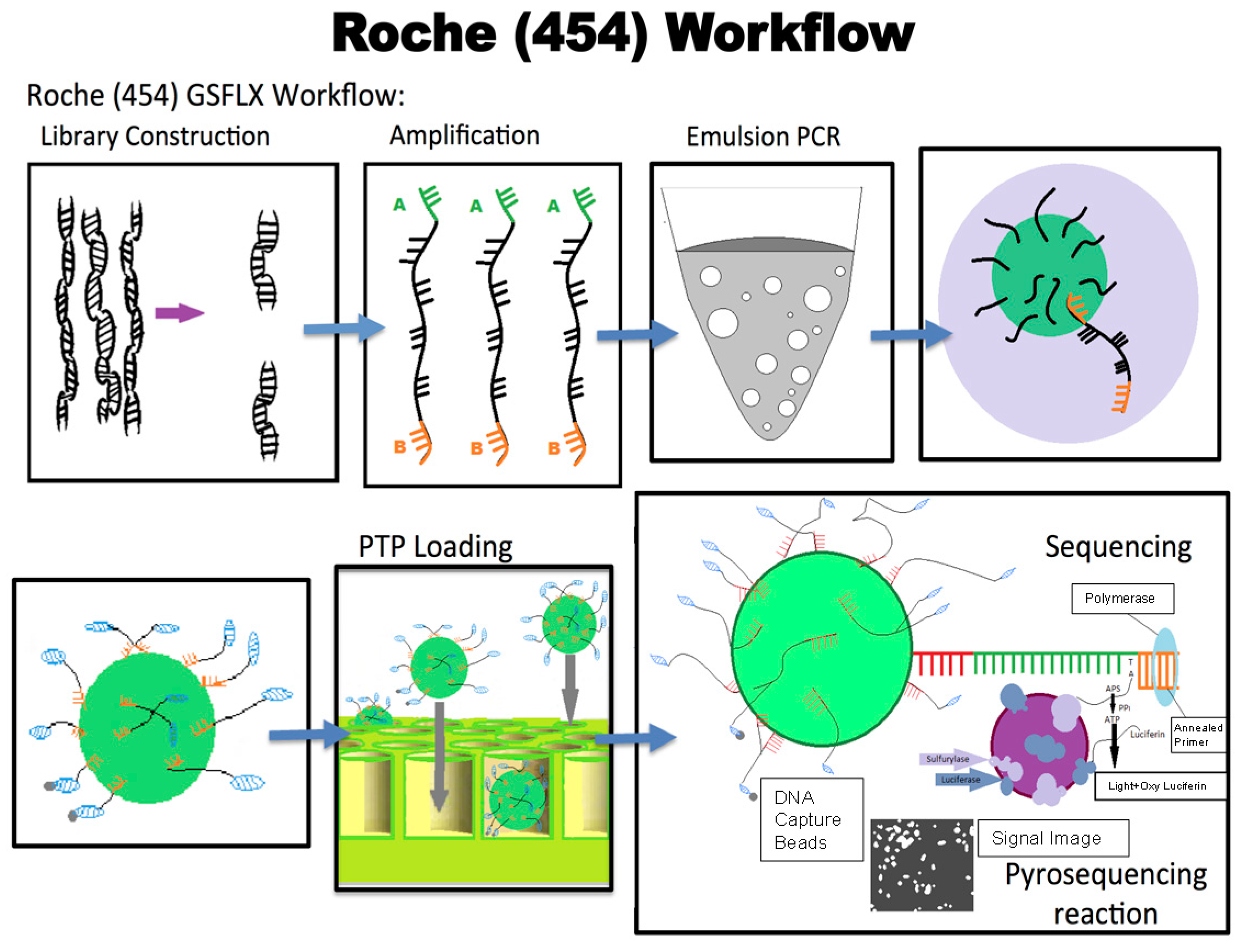
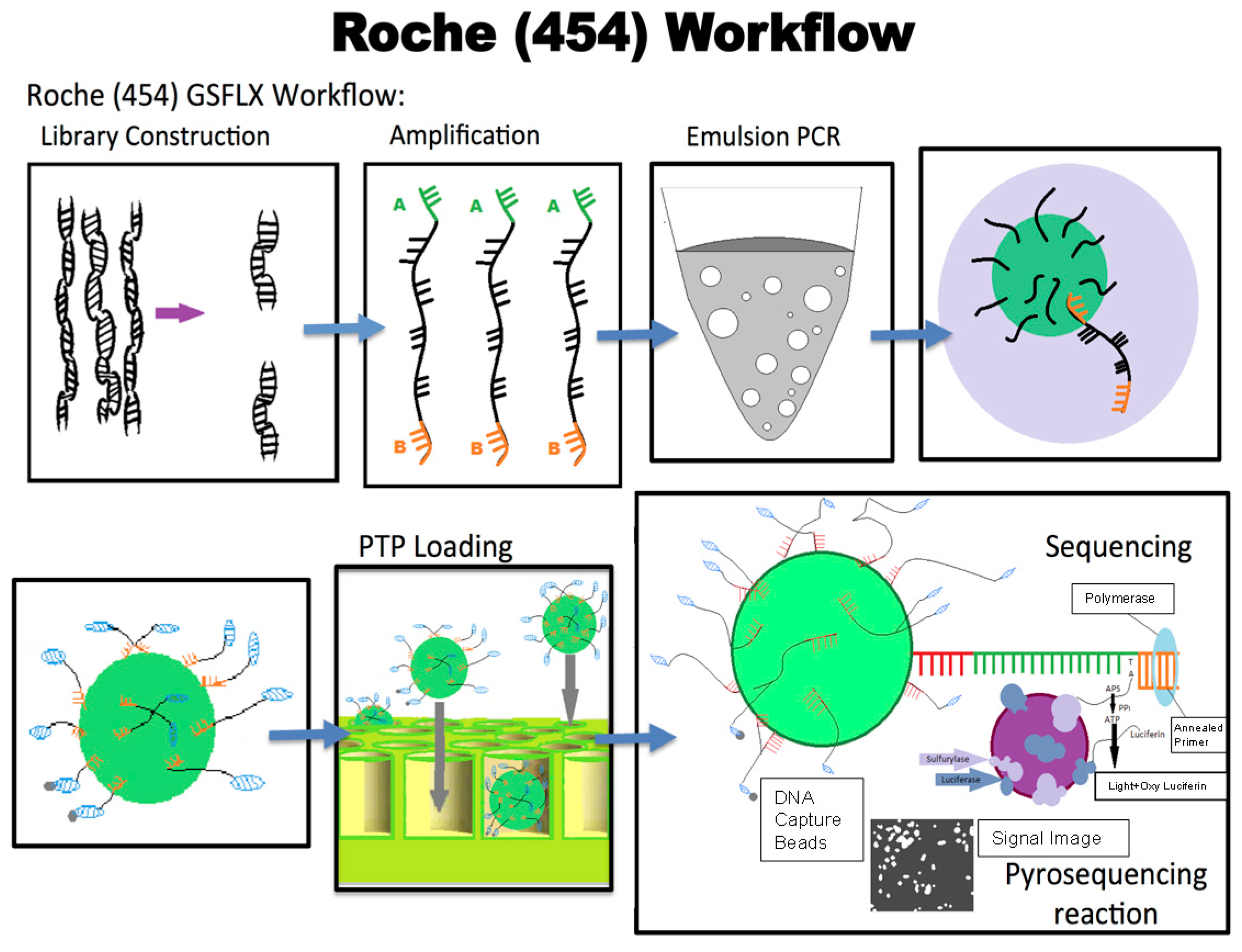
9.1. 454 Sequencing
9.2. Ion Torrent Sequencing
10. Modifier Genes and PXE
11. The Role of Disease Advocacy Groups in PXE and Other Rare Genetic Disorders
11.1. The PXE Story
11.2. The Birth of PXE International—For the People, of the People, by the People
11.3. The Role of Patient Advocates in the Discovery of the ABCC6 Gene
11.4. Other Genetic Disorders and the Role of Genetic Alliance
11.5. The Genetic Alliance Registry and BioBank (GARB)
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Qiaoli, L.; Qiujie, J.; Pfendner, E.; Váradi, A.; Uitto, J. Pseudoxanthoma elasticum: Clinical phenotypes, molecular genetics and putative pathomechanisms. Exp. Dermatol. 2009, 18, 1–11. [Google Scholar] [CrossRef]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaqlino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Pfendner, E.G.; Vanakker, O.M.; Terry, S.F.; Vourthis, S.; McAndrew, P.E.; McClain, M.R.; Fratta, S.; Marais, A.S.; Hariri, S.; Coucke, P.J.; et al. Mutation detection in the ABCC6 gene and genotype-phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J. Med. Genet. 2007, 44, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Ringpfeil, F.; Pulkkinen, L.; Uitto, J. Molecular genetics of pseudoxanthoma elasticum. Exp. Dermatol. 2001, 10, 221–228. [Google Scholar] [CrossRef] [PubMed]
- PXE International. Available online: WWW.PXE.org (accessed on 14 April 2017).
- Klement, J.F.; Matsuzaki, Y.; Jiang, Q.J.; Terlizzi, J.; Choi, H.Y.; Fujimoto, N.; Li, K.; Pulkkinen, L.; Birk, D.E.; Sundberg, J.P.; et al. Targeted ablation of the ABCC6 gene results in ectopic mineralization of connective tissues. Mol. Cell. Biol. 2005, 25, 8299–8310. [Google Scholar] [CrossRef] [PubMed]
- Borths, E.L.; Locher, K.P.; Lee, A.T.; Rees, D.C. The structure of Escherichia coli BtuF and binding to its cognate ATP binding cassette transporter. Proc. Natl. Acad. Sci. USA 2002, 99, 16642–16647. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: Physiology, structure and mechanism—An overview. Res. Microbiol. 2001, 3, 205–210. [Google Scholar] [CrossRef]
- Schneider, E.; Hunke, S. ATP-binding-cassette (ABC) transport systems: Functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 1998, 22, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Miksch, S.; Lumsden, A.; Guenther, U.P.; Foernzler, D.; Christen-Zäch, S.; Daugherty, C.; Ramesar, R.K.; Lebwohl, M.; Hohl, D.; Neldner, K.H.; et al. Molecular genetics of pseudoxanthoma elasticum: Type and frequency of mutations in ABCC6. Hum. Mutat. 2005, 26, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Torrington, M.; Viljoen, D.L. Founder effect in 20 Afrikaner kindreds with pseudoxanthoma elasticum. S. Afr. Med. J. 1991, 79, 7–11. [Google Scholar] [PubMed]
- Le Saux, O.; Beck, K.; Sachsinger, C.; Treiber, C.; Göring, H.H.; Curry, K.; Johnson, E.W.; Bercovitch, L.; Marais, A.S.; Terry, S.F.; et al. Evidence for a founder effect for pseudoxanthoma elasticum in the Afrikaner population of South Africa. Hum. Genet. 2002, 111, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, N.; Martin, L.; Calvas, P.; Bert, M.L.; Hovnanian, A. Pseudoxanthoma Elasticum: A Clinical, Pathophysiological And Genetic Update Including 11 Novel ABCC6 Mutations. J. Med. Genet. 2005, 42, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Laube, S.; Moss, C. Pseudoxanthoma elasticum. Arch. Dis. Child. 2005, 90, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Phelps, R.G.; Yannuzzi, L.; Chang, S.; Schwartz, I.; Fuchs, W. Diagnosis of pseudoxanthoma elasticum by scar biopsy in patients without characteristic skin lesions. N. Engl. J. Med. 1987, 317, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Bunda, S.; Van Wart, C.M.; Douet, V.; Got, L.; Martin, L.; Hinek, A. Serum factors from pseudoxanthoma elasticum patients alter elastic fiber formation in vitro. J. Investig. Dermatol. 2006, 126, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, D.; Sartor, L.; Garbisa, S.; Boraldi, F.; Croce, A.; Passi, A.; De Luca, G.; Tiozzo, R.; Pasquali-Ronchetti, I. Dermal fibroblasts from pseudoxanthoma elasticum patients have raised MMP-2 degradative potential. BBA Mol. Basis Dis. 2005, 1741, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.S.; Küçükosmanoğlu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.; Bergen, A.A.; Gorgels, T.G.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.; Bergen, A.A.; Elferink, R.P.; Borst, P.; et al. ATP-Binding Cassette Subfamily C Member 6-Mediated ATP Secretion by the Liver Is the Main Source of the Mineralization Inhibitor Inorganic Pyrophosphate in the Systemic Circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 1, 114. [Google Scholar]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Hendig, D.; Arndt, M.; Szliska, C.; Kleesiek, K.; Götting, C. SPP1 promoter polymorphisms: Identification of the first modifier gene for pseudoxanthoma elasticum. Clin. Chem. 2007, 53, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J. Rare heritable skin diseases: Targets for regenerative medicine. J. Investig. Dermatol. 2012, 132, 2485. [Google Scholar] [CrossRef] [PubMed]
- Pseudoxanthoma Elasticum. Pseudoxanthoma Elasticum. Available online: http://emedicine.medscape.com/article/1074713-overview (accessed on 14 April 2017).
- Terry, S.F.; Terry, P.F.; Rauen, K.A.; Uitto, J.; Bercovitch, L.G. Advocacy groups as research organizations: The PXE International example. Nat. Rev. Genet. 2007, 8, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Landy, D.C.; Brinich, M.A.; Colten, M.E.; Horn, E.J.; Terry, S.F.; Sharp, R.R. How disease advocacy organizations participate in clinical research: A survey of genetic organizations. Genet. Med. 2012, 14, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Allen, A. Who Owns My Disease? Mother Jones. Available online: http://www.motherjones.com/politics/2001/11/who-owns-my-disease (accessed on 14 April 2017).
- Genetic Alliance. Available online: www.geneticalliance.org (accessed on 14 April 2017).
- Levenson, D. Genetic alliance marks 25 years. Am. J. Med. Genet. Part A 2011, 155. [Google Scholar] [CrossRef]
- Moitra, K. ABC transporters in human disease. In Colloquium Series on The Genetic Basis of Human Disease; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2012; Volume 1, pp. 1–84. [Google Scholar]
- Uitto, J.; Váradi, A.; Bercovitch, L.; Terry, P.F.; Terry, S.F. Pseudoxanthoma elasticum: Progress in research toward treatment: Summary of the 2012 PXE International Research Meeting. J. Investig. Dermatol. 2013, 133, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J.; Li, Q.; van de Wetering, K.; Váradi, A.; Terry, S.F. Insights into Pathomechanisms and Treatment Development in Heritable Ectopic Mineralization Disorders: Summary of the PXE International Biennial Research Symposium-2016. J. Investig. Dermatol. 2017, 137, 790–795. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moitra, K.; Garcia, S.; Jaldin, M.; Etoundi, C.; Cooper, D.; Roland, A.; Dixon, P.; Reyes, S.; Turan, S.; Terry, S.; et al. ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy. Int. J. Mol. Sci. 2017, 18, 1488. https://doi.org/10.3390/ijms18071488
Moitra K, Garcia S, Jaldin M, Etoundi C, Cooper D, Roland A, Dixon P, Reyes S, Turan S, Terry S, et al. ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy. International Journal of Molecular Sciences. 2017; 18(7):1488. https://doi.org/10.3390/ijms18071488
Chicago/Turabian StyleMoitra, Karobi, Sonia Garcia, Michelle Jaldin, Clementine Etoundi, Donna Cooper, Anna Roland, Patrice Dixon, Sandra Reyes, Sevilay Turan, Sharon Terry, and et al. 2017. "ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy" International Journal of Molecular Sciences 18, no. 7: 1488. https://doi.org/10.3390/ijms18071488
APA StyleMoitra, K., Garcia, S., Jaldin, M., Etoundi, C., Cooper, D., Roland, A., Dixon, P., Reyes, S., Turan, S., Terry, S., & Dean, M. (2017). ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy. International Journal of Molecular Sciences, 18(7), 1488. https://doi.org/10.3390/ijms18071488