Synthesis and Biodegradation of Poly(l-lactide-co-β-propiolactone)



Abstract
:
1. Introduction
2. Results
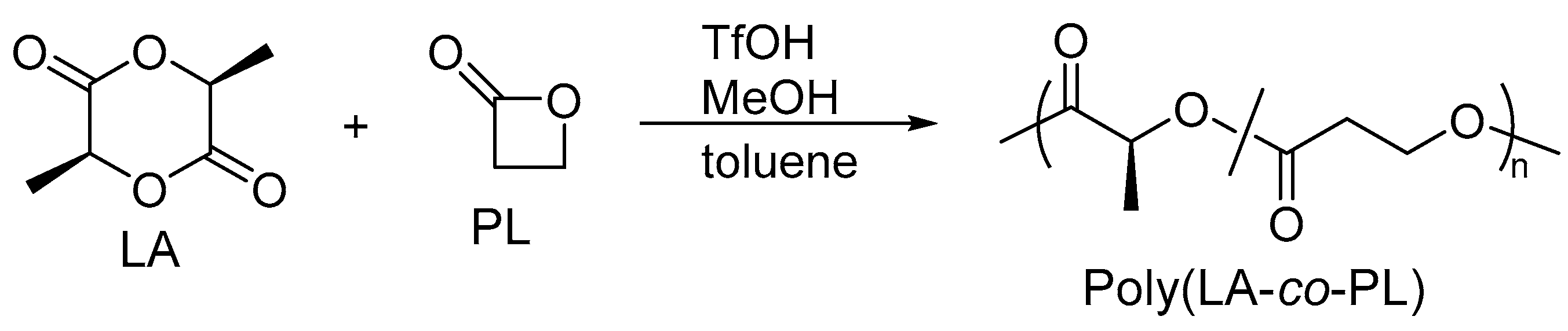
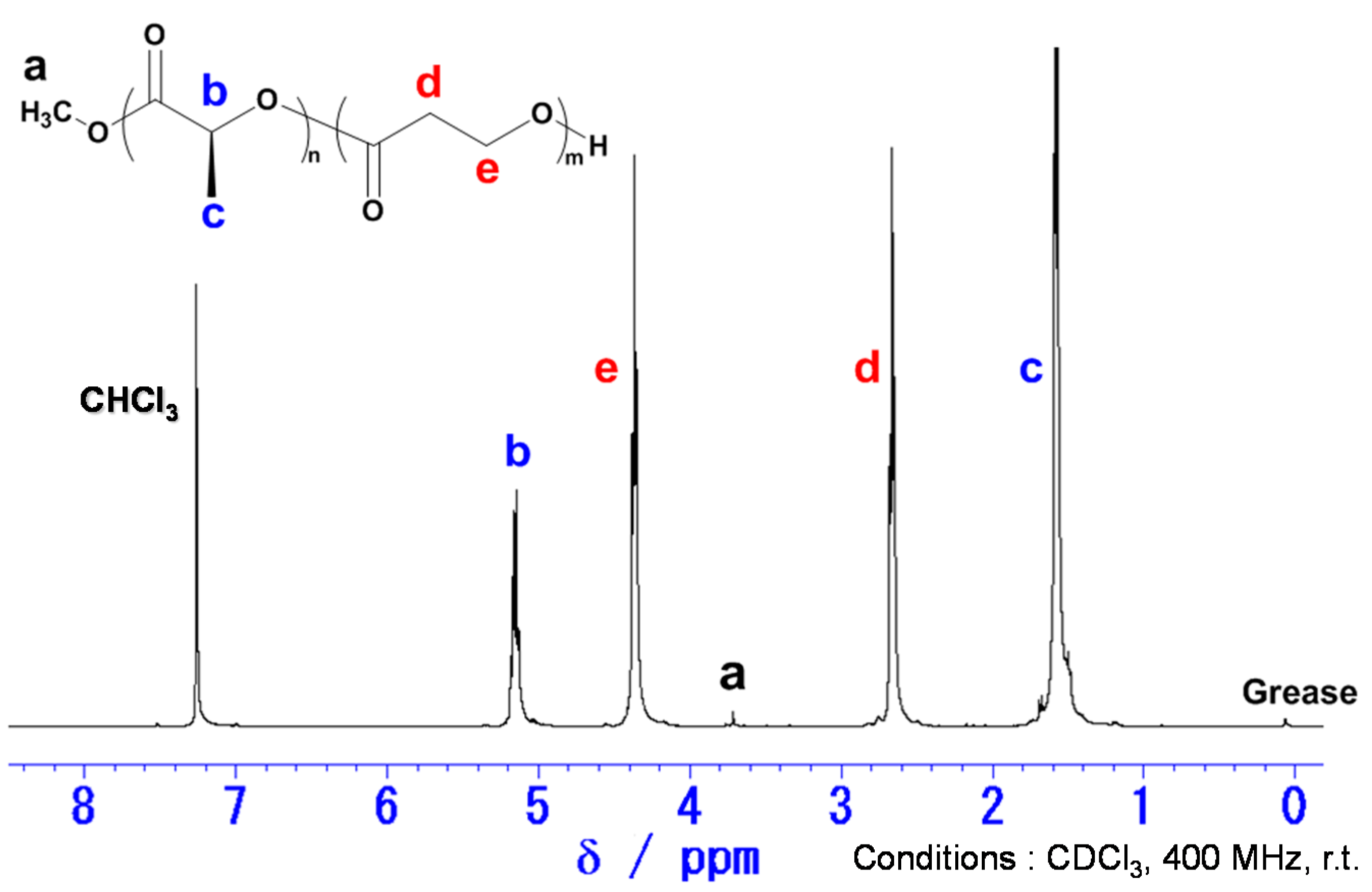
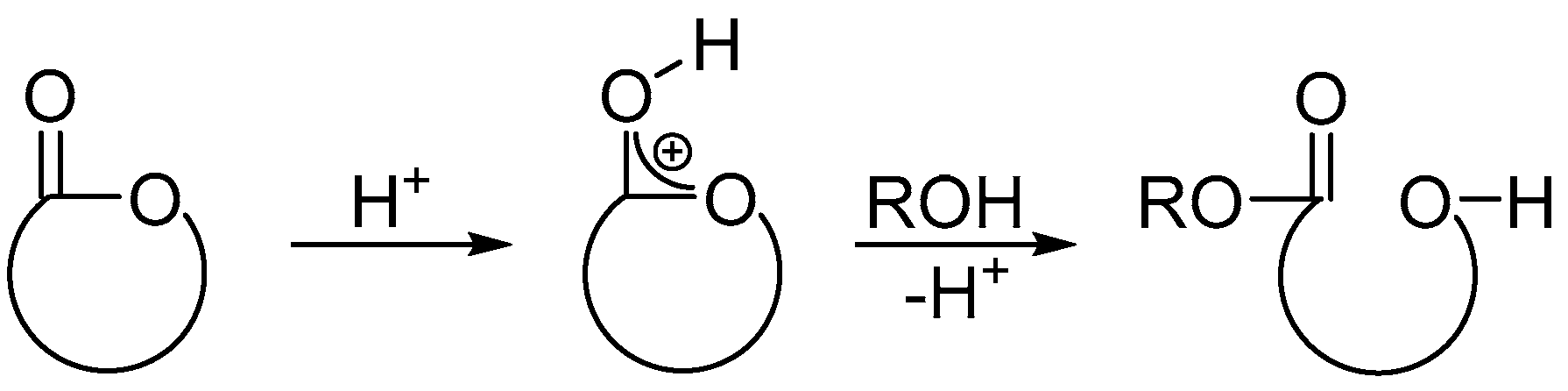
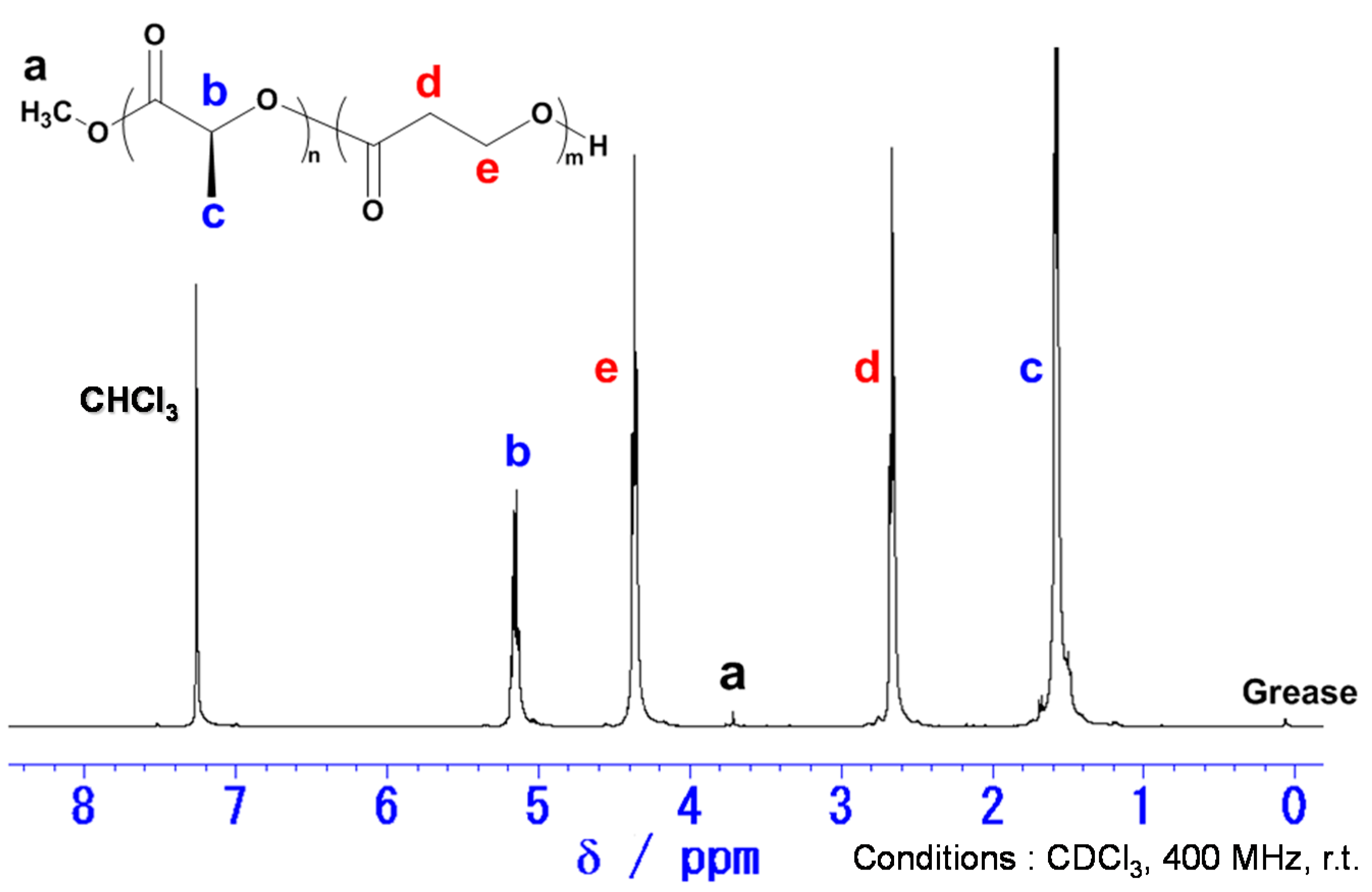
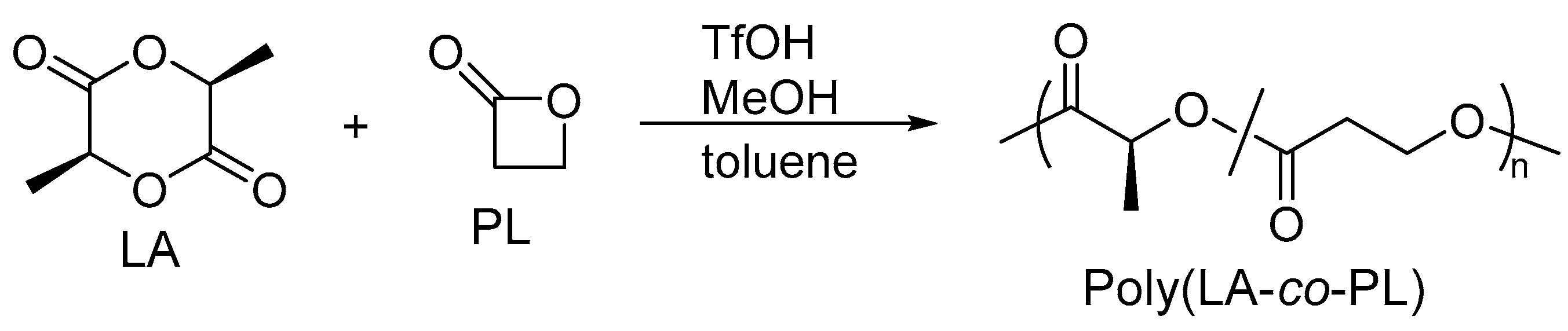
2.1. Copolymerization of LA with PL
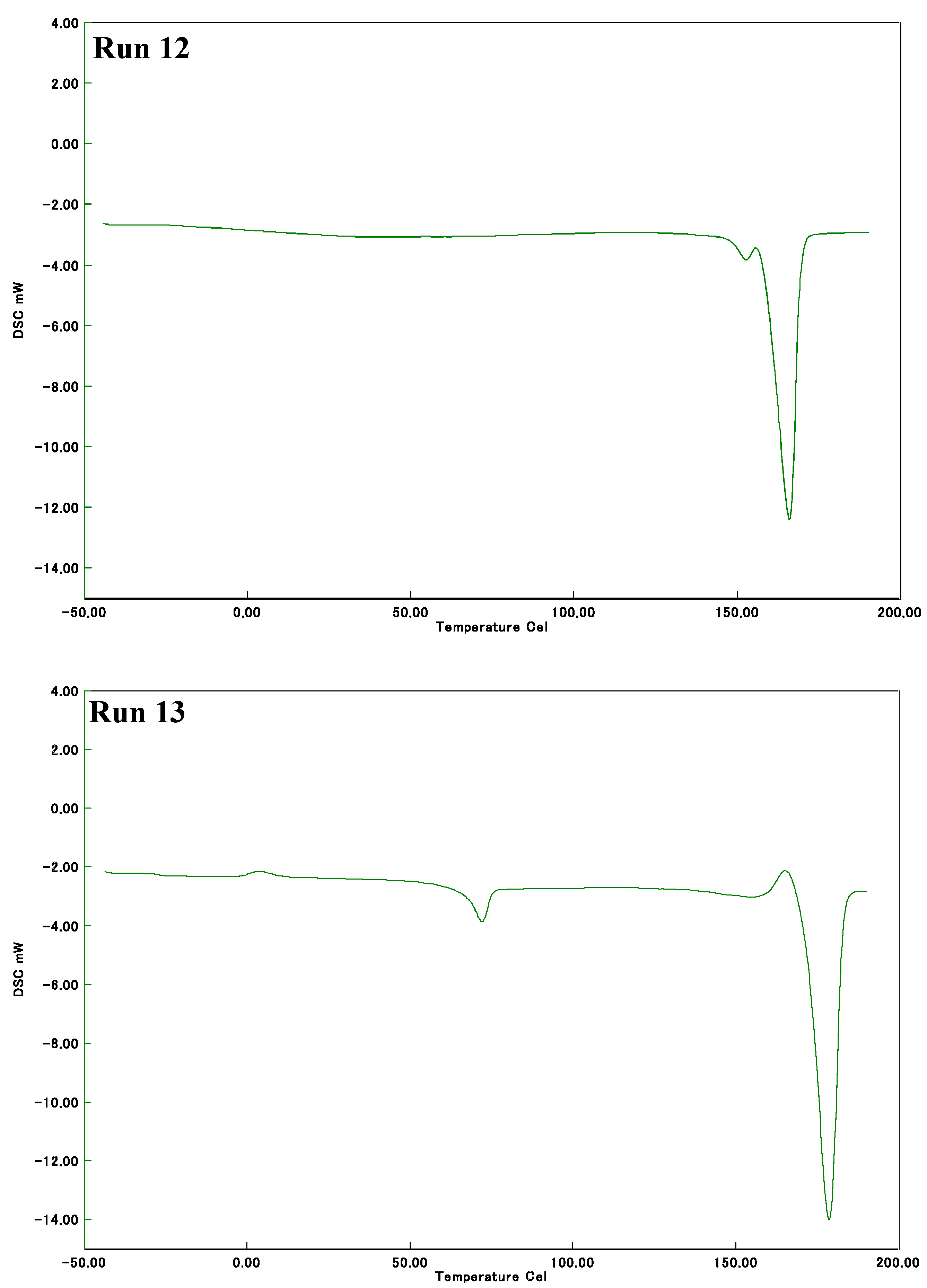
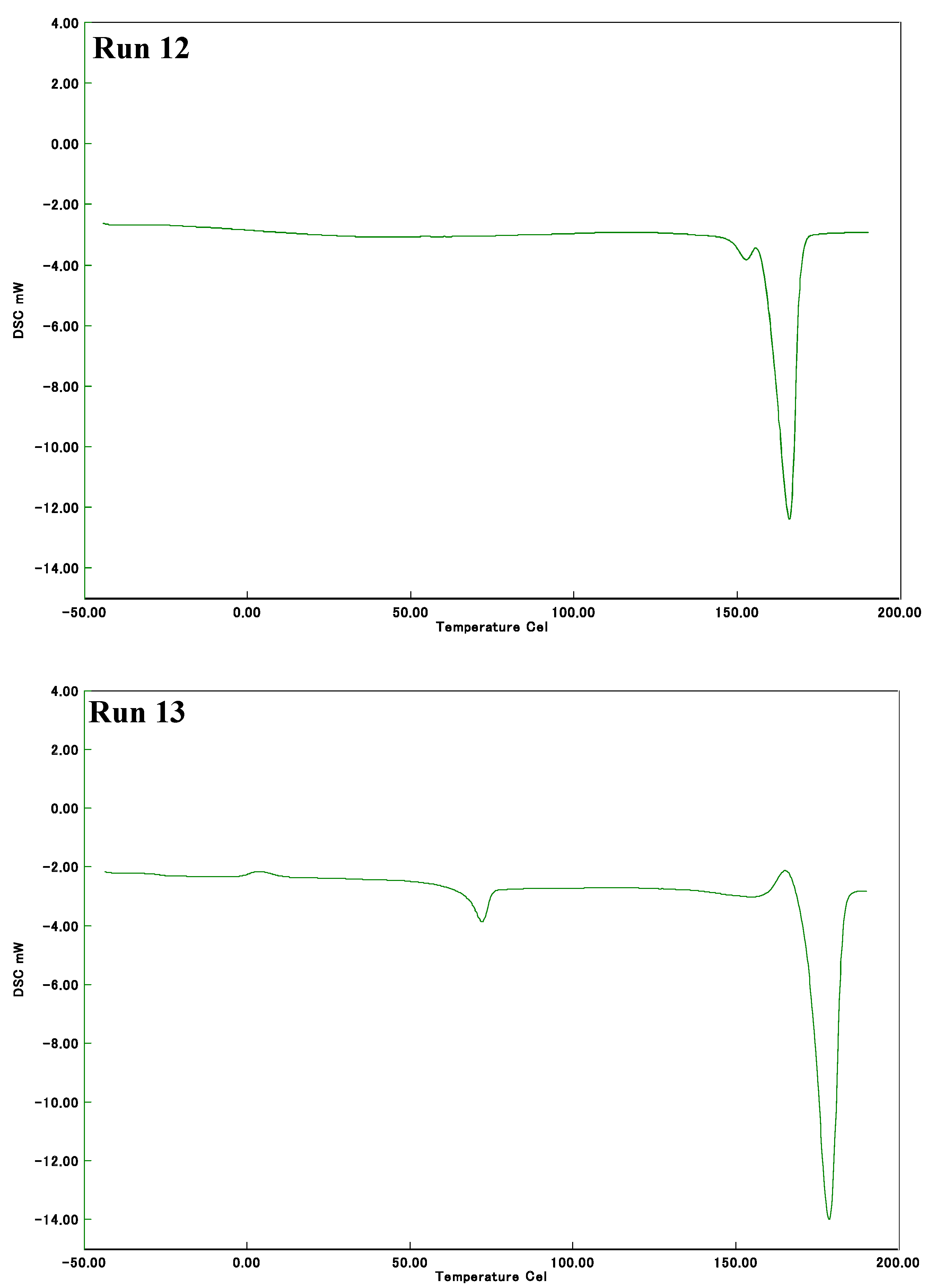
2.2. Thermal Properties of Poly(LA-co-PL)
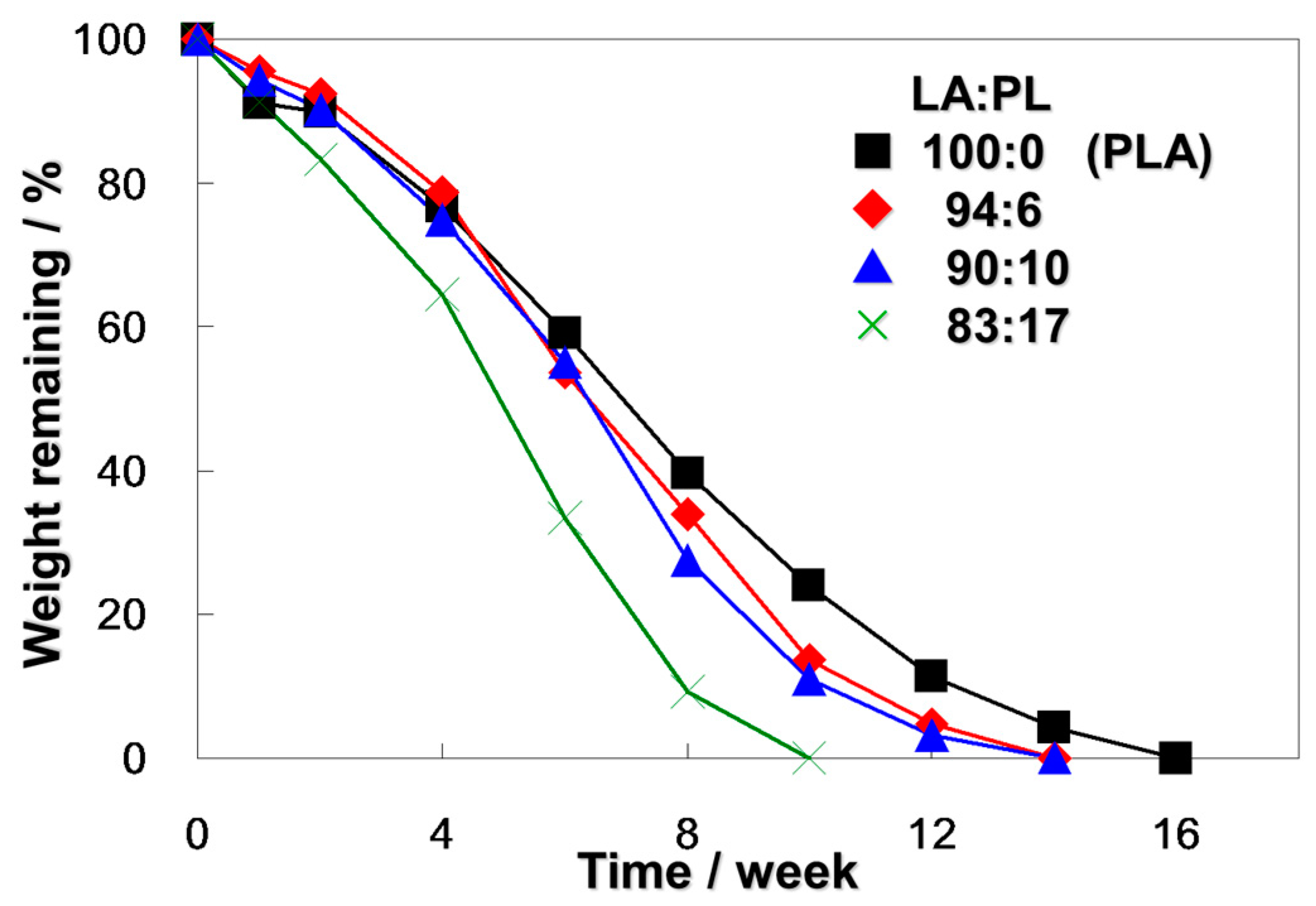
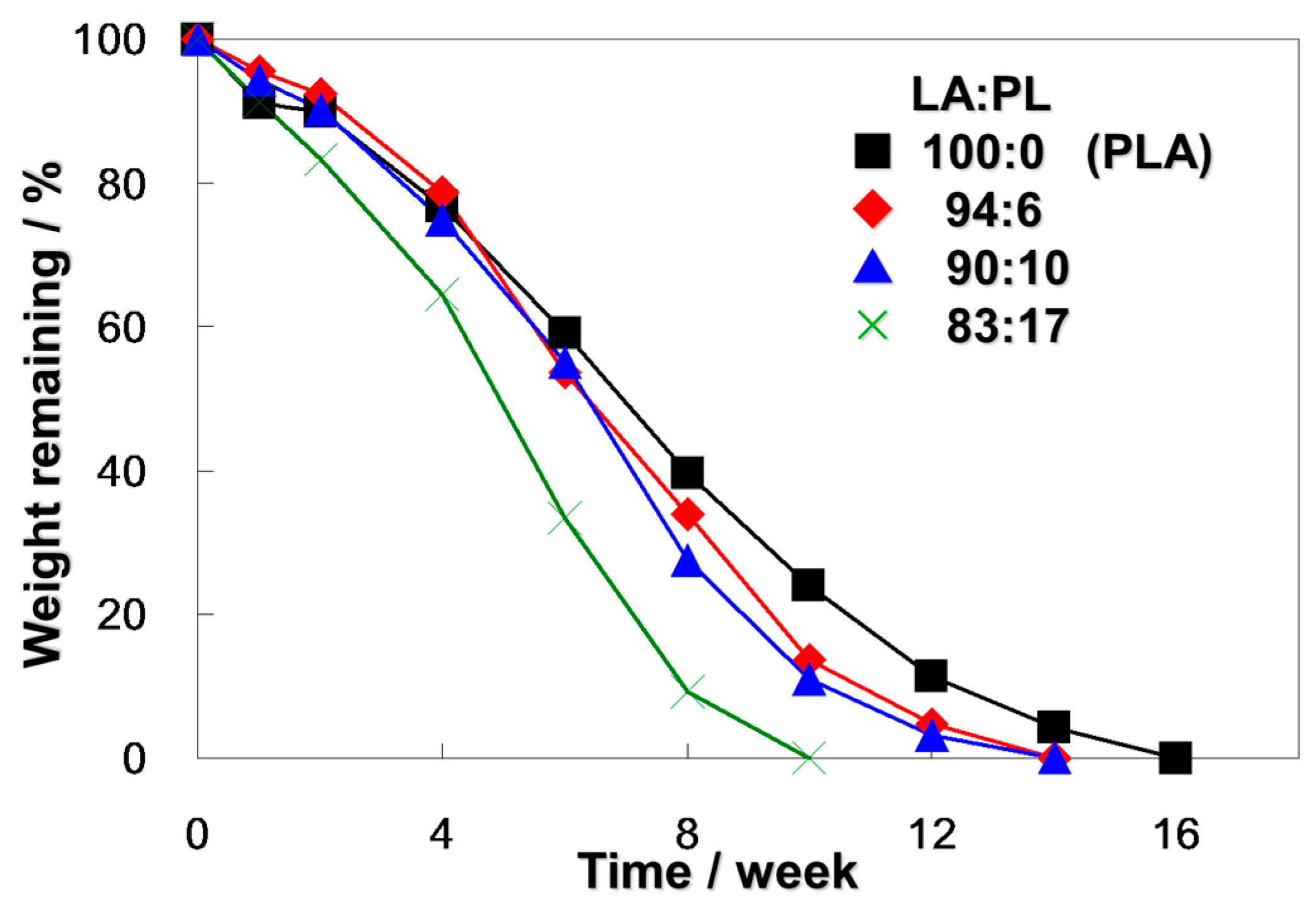
2.3. Biodegradation of Poly(LA-co-PL) in a Compost
3. Discussion
4. Materials and Methods
4.1. General
4.2. Materials
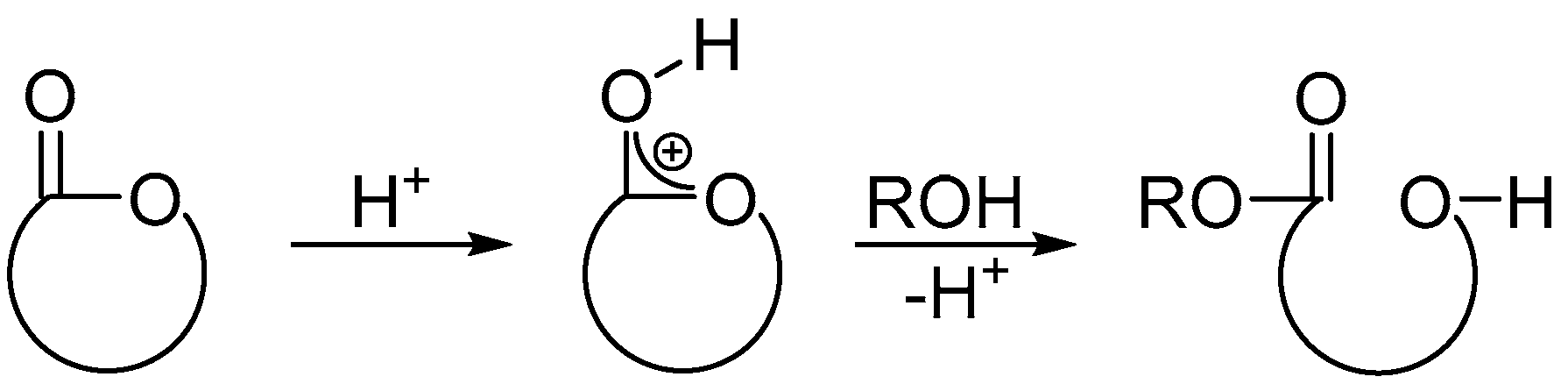
4.3. Copolymerization of LA and PL Catalyzed by TfOH
4.4. Copolymerization of LA and PL Catalyzed by Sm-1
4.5. Copolymerization of LA and PL Catalyzed by Sn(Oct)2
4.6. Degradation of the Polymers by a Compost
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Li, S. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J. Biomed. Mater. Res. 1999, 48, 342–353. [Google Scholar] [CrossRef]
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Gupta, A.P.; Kumar, V. New emerging trends in synthetic biodegradable polymers–Polylactide: A critique. Eur. Polym. J. 2007, 43, 4053–4074. [Google Scholar] [CrossRef]
- Williams, C.K.; Hillmyer, M.A. Polymers from renewable resources: A Perspective for a special issue of Polymer Reviews. Polym. Rev. 2008, 48, 1–10. [Google Scholar] [CrossRef]
- Sission, A.L.; Schroeter, M.; Lendlein, A. Polyesters. In Handbook of Biodegradable Polymers: Synthesis, Characterization and Applications; lndlein, A., Sission, A., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 1–21. [Google Scholar]
- Garrison, T.F.; Murawski, A.; Quirino, R.L. Bio-based polymers with potential for biodegradability. Polymers 2016, 8, 262. [Google Scholar] [CrossRef]
- Nampoothiri, K.M.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Tech. 2010, 101, 8493–8501. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K.; Feijoo, J.L.; Yang, M.-C. Comparison of abiotic and biotic degradation of PDLLA, PCL and partially miscible PDLLA/PCL blend. Eur. Polym. J. 2013, 49, 706–717. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Preziosi, R.; Robson, G.D. Abiotic and biotic environmental degradation of the bioplastic polymer poly(lactic acid): A review. Polym. Degrad. Stab. 2017, 137, 122–130. [Google Scholar] [CrossRef]
- Yasuda, H.; Yamamoto, K.; Nakayama, Y.; Tsutsumi, C.; Lecomte, P.; Jerome, R.; McCarthy, S.; Kaplan, D. Comparison of Sm complexes with Sn compounds for syntheses of copolymers composed of lactide and ε-caprolactone and their biodegradabilities. React. Funct. Polym. 2004, 61, 277–292. [Google Scholar] [CrossRef]
- Shirahama, H.; Ichimaru, A.; Tsutsumi, C.; Nakayama, Y.; Yasuda, H. Characteristics of the biodegradability and physical properties of stereocomplexes between poly(l-lactide) and poly(d-lactide) copolymers. J. Polym. Sci. A Polym. Chem. 2005, 43, 438–454. [Google Scholar] [CrossRef]
- Nakayama, Y.; Aihara, K.; Yamanishi, H.; Fukuoka, H.; Tanaka, R.; Cai, Z.G.; Shiono, T. Synthesis of biodegradable thermoplastic elastomers from epsilon-caprolactone and lactide. J. Polym. Sci. Pol. Chem. 2015, 53, 489–495. [Google Scholar] [CrossRef]
- Anderson, A.J.; Dawes, E.A. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 1990, 54, 450–472. [Google Scholar] [PubMed]
- Sudesh, K.; Abe, H.; Doi, Y. Synthesis, structure and properties of polyhydroxyalkanoates: Biological polyesters. Prog. Polym. Sci. 2000, 25, 1503–1555. [Google Scholar] [CrossRef]
- Coulembier, O.; Dubois, P. Polyesters from β-lactones. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulembier, O., Raquez, J.-M., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp. 227–254. [Google Scholar]
- Jaffredo, C.G.; Carpentier, J.-F.; Guillaume, S.M. Organocatalyzed controlled ROP of β-lactones towards poly(hydroxyalkanoate)s: From β-butyrolactone to benzyl β-malolactone polymers. Polym. Chem. 2013, 4, 3837–3850. [Google Scholar] [CrossRef]
- Asano, M.; Yoshida, M.; Fukuzaki, H.; Kumakura, M.; Mashimo, T.; Yuasa, H.; Imai, K.; Yamanaka, H. Effect of crystallinity on the in-vivo degradation of poly(β-propiolactone) prepared by radiation-induced solid polymerization in organic solvent system at low temperature. Eur. Polym. J. 1990, 26, 29–33. [Google Scholar] [CrossRef]
- Mathisen, T.; Lewis, M.; Albertsson, A.-C. Hydrolytic degradation of nonoriented poly(β-propiolactone). J. Appl. Polym. Sci. 1991, 42, 2365–2370. [Google Scholar] [CrossRef]
- Furuhashi, Y.; Iwata, T.; Kimura, Y.; Doi, Y. Structural characterization and enzymatic degradation of α-, β-, and γ-crystalline forms for poly(β-propiolactone). Macromol. Biosci. 2003, 3, 462–470. [Google Scholar] [CrossRef]
- Cortizo, M.S.; Molinuevo, M.S.; Cortizo, A.M. Biocompatibility and biodegradation of polyester and polyfumarate based-scaffolds for bone tissue engineering. J. Tissue Eng. Regen. Med. 2008, 2, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Togo, M.; Sanui, K.; Ogata, N.; Kobayashi, T.; Ohtaki, Z. Ionic conductivity of polymer complexes formed by poly(β-propiolactone) and lithium perchlorate. Macromolecules 1984, 17, 2908–2912. [Google Scholar] [CrossRef]
- Kumagai, Y.; Doi, Y. Enzymatic degradation of binary blends of microbial poly(3-hydroxybutyrate) with enzymatically active polymers. Polym. Degrad. Stab. 1992, 37, 253–256. [Google Scholar] [CrossRef]
- Shimamura, E.; Scandola, M.; Doi, Y. Microbial Synthesis and characterization of poly(3-hydroxybutyrate-co-3-hydroxypropionate). Macromolecules 1994, 27, 4429–4435. [Google Scholar] [CrossRef]
- Crescenzi, V.; Manzini, G.; Calzolari, G.; Borri, C. Thermodynamics of fusion of poly-β-propiolactone and poly-ε-caprolactone. comparative analysis of the melting of aliphatic polylactone and polyester chains. Eur. Polym. J. 1972, 8, 449–463. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, T.; Yamaguchi, A.; Nishishita, T. Ring-opening copolymerization of optically active β-butyrolactone with several lactones catalyzed by distannoxane complexes: Synthesis of new biodegradable polyesters. Macromolecules 1993, 26, 4388–4390. [Google Scholar] [CrossRef]
- Hori, Y.; Suzuki, M.; Yamaguchi, A.; Nishishita, T. Ring-opening polymerization of optically active .beta.-butyrolactone using distannoxane catalysts: Synthesis of high-molecular-weight poly(3-hydroxybutyrate). Macromolecules 1993, 26, 5533–5534. [Google Scholar] [CrossRef]
- Cross, E.D.; Allan, L.E.N.; Decken, A.; Shaver, M.P. Aluminum salen and salan complexes in the ring-opening polymerization of cyclic esters: Controlled immortal and copolymerization of rac-β-butyrolactone and rac-lactide. J. Polym. Sci. A Polym. Chem. 2013, 51, 1137–1146. [Google Scholar] [CrossRef]
- MacDonald, J.P.; Parker, M.P.; Greenland, B.W.; Hermida-Merino, D.; Hamley, I.W.; Shaver, M.P. Tuning thermal properties and microphase separation in aliphatic polyester ABA copolymers. Polym. Chem. 2015, 9, 1445–1453. [Google Scholar] [CrossRef]
- Fagerland, J.; Finne-Wistrand, A.; Pappalardo, D. Modulating the thermal properties of poly(hydroxybutyrate) by the copolymerization of rac-β-butyrolactone with lactide. New J. Chem. 2016, 40, 7671–7679. [Google Scholar] [CrossRef]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Polyesters from Dilactones. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulembier, O., Raquez, J.-M., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp. 255–286. [Google Scholar]
- Santos, M.; Gangoiti, J.; Llama, M.J.; Serra, J.L.; Keul, H.; Möller, M. Poly(3-hydroxyoctanoate) depolymerase from Pseudomonas fluorescens GK13: Catalysis of ester-forming reactions in non-aqueous media. J. Mol. Catal. B Enzym. 2012, 77, 81–86. [Google Scholar] [CrossRef]
- Jaipuri, F.A.; Bower, B.D.; Pohl, N.L. Protic acid-catalyzed polymerization of -lactones for the synthesis of chiral polyesters. Tetrahedron Asymmetry 2003, 14, 3249–3252. [Google Scholar] [CrossRef]
- Yamashita, Y.; Takemoto, Y.; Ihara, E.; Yasuda, H. Organolanthanide-initiated living polymerizatin of ε-caprolactone, δ-valerolactone, and β-propiolactone. Macromolecules 1996, 29, 1798–1806. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Dunsing, R. Polylactones, 8. Mechanism of the cationic polymerization of L,L-dilactide. Macromol. Chem. Phys. 1986, 187, 1611–1625. [Google Scholar] [CrossRef]
- Bourissou, D.; Martin-Vaca, B.; Dumitrescu, A.; Graullier, M.; Lacombe, F. Controlled cationic polymerization of lactide. Macromolecules 2005, 38, 9993–9998. [Google Scholar] [CrossRef]
- Nakayama, Y.; Yasuda, H.; Yamamoto, K.; Tsutsumi, C.; Jerome, R.; Lecomte, P. Comparison of Sm complexes with Sn compounds for syntheses of copolymers composed of lactide and cyclic carbonates and their biodegradabilities. React. Funct. Polym. 2005, 63, 95–105. [Google Scholar] [CrossRef]
- Degée, P.; Dubois, P.; Jérǒme, R.; Jacobsen, S.; Fritz, H.-G. New catalysis for fast bulk ring-opening polymerization of lactide monomers. Macromol. Symp. 1999, 144, 289–302. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Kreiser-Saunders, I.; Stricker, A. Polylactones 48. SnOct2-Initiated polymerizations of lactide: A mechanistic study. Macromolecules 2000, 33, 702–709. [Google Scholar] [CrossRef]
- Schumann, H.; Albrecht, I.; Gallagher, M.; Hahn, E.; Muchmore, C.; Pickardt, J. Organometallic compounds of the lanthanides XLIV. Bis(cyclopentadienyl)lutetium alkoxides, thiolates and selenolates. Crystal structures of (C5Me5)2Lu(μ-StC4H9)2Li(THF)2 and (C5H5)2Lu(μ-SeC6H5)2Li(THF)2. J. Organomet. Chem. 1988, 349, 103–115. [Google Scholar] [CrossRef]
- Chen, C.F.; Lin, C.T.Y.; Chu, I.M. Study of novel biodegradable thermo-sensitive hydrogels of methoxy-poly(ethylene glycol)-block-polyester diblock copolymers. Polym. Int. 2010, 59, 1428–1435. [Google Scholar] [CrossRef]
- Save, M.; Soum, A. Controlled ring-opening polymerization of lactones and lactide initiated by lanthanum isopropoxide, 2. Mechanistic studies. Macromol. Chem. Phys. 2002, 203, 2591–2603. [Google Scholar] [CrossRef]
- Gazeau-Bureau, S.; Delcroix, D.; Martín-Vaca, B.; Bourissou, D.; Navarro, C.; Magnet, S. Organo-catalyzed ROP of ε-caprolactone: Methanesulfonic acid competes with trifluoromethanesulfonic acid. Macromolecules 2008, 41, 3782–3784. [Google Scholar] [CrossRef]
- Save, M.; Schappacher, M.; Soum, A. Controlled ring-opening polymerization of lactones and lactides initiated by lanthanum isopropoxide, 1 General aspects and kinetics. Macromol. Chem. Phys. 2002, 203, 889–899. [Google Scholar] [CrossRef]
- Kale, G.; Auras, R.; Singh, S.P.; Narayan, R. Biodegradability of polylactide bottles in real and simulated composting conditions. Polym. Test. 2007, 26, 1049–1061. [Google Scholar] [CrossRef]
- Evans, W.J.; Chamberlain, L.R.; Ulibarri, T.A.; Ziller, J.W. Reactivity of trimethylaluminum with (C5Me5)2Sm(THF)2: Synthesis, structure, and reactivity of the samarium methyl complexes (C5Me5)2[(μ-Me)AlMe2(µ-Me)]2Sm(C5Me5)2 and (C5Me5)2SmMe(THF). J. Am. Chem. Soc. 1988, 110, 6423–6432. [Google Scholar] [CrossRef]
- Nakayama, Y.; Inaba, T.; Toda, Y.; Tanaka, R.; Cai, Z.; Shiono, T.; Shirahama, H.; Tsutsumi, C. Synthesis and properties of cationic ionomers from poly(ester-urethane)s based on polylactide. J. Polym. Sci. A Polym. Chem. 2013, 51, 4423–4428. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Catalyst | [LA]0/[PL]0/[init.]/[cat.] | Temp. (°C) | LA-Conv. 2 (%) | PL-Conv. 3 (%) | Mn 4 (103) | Mw/Mn 4 | PL-Cont. 5 (mol%) |
|---|---|---|---|---|---|---|---|---|
| 1 6 | TfOH | 125/125/1/1 | 50 | 48 | 98 | 11 | 1.2 | 67 |
| 2 6,7 | Sm-1 | 250/250/1/1 | 0 | 0 | 0 | - | - | - |
| 3 8 | Sn(Oct)2 | 500/500/4/1 | 100 | 0 | 0 | - | - | - |
| Run | [LA]0/[PL]0 | [init.]/[cat.] | Time (h) | LA-Conv. 2 (%) | PL-Conv. 3 (%) | Mn 4 (103) | Mw/Mn 4 | PL-Cont. 5 (mol %) |
|---|---|---|---|---|---|---|---|---|
| 4 | 90/10 | 1/1 | 24 | 32 | >99 | 10 | 1.11 | 27 |
| 5 | 90/10 | 1/2 | 24 | 67 | >99 | 26 | 1.16 | 18 |
| 6 | 90/10 | 1/3 | 24 | 64 | >99 | 30 | 1.17 | 15 |
| 7 | 90/10 | 1/2 | 48 | 86 | 96 | 37 | 1.11 | 11 |
| 8 | 90/10 | 1/2 | 96 | 98 | 98 | 55 | 1.53 | 10 |
| 9 | 100/0 | 1/2 | 96 | 95 | - | 41 | 1.59 | 0 |
| 10 | 95/5 | 1/2 | 96 | 89 | >99 | 35 | 1.73 | 6 |
| 11 | 85/15 | 1/2 | 96 | 80 | 93 | 32 | 1.51 | 17 |
| 12 | 80/20 | 1/2 | 96 | 68 | >99 | 25 | 1.25 | 27 |
| Run | PL-Content | Tg (°C) | Tm (°C) | ΔHm (J/g) |
|---|---|---|---|---|
| 9 | 0 | 62 | 172 | 55 |
| 10 | 6 | 45 | 170 | 53 |
| 8 | 10 | 41 | 167 | 49 |
| 11 | 17 | 24 | 165 | 45 |
| 12 | 27 | 11 | 166 | 42 |
| 13 2 | 20 | - | 72,178 | 4,46 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, Y.; Aihara, K.; Cai, Z.; Shiono, T.; Tsutsumi, C. Synthesis and Biodegradation of Poly(l-lactide-co-β-propiolactone). Int. J. Mol. Sci. 2017, 18, 1312. https://doi.org/10.3390/ijms18061312
Nakayama Y, Aihara K, Cai Z, Shiono T, Tsutsumi C. Synthesis and Biodegradation of Poly(l-lactide-co-β-propiolactone). International Journal of Molecular Sciences. 2017; 18(6):1312. https://doi.org/10.3390/ijms18061312
Chicago/Turabian StyleNakayama, Yuushou, Kazuki Aihara, Zhengguo Cai, Takeshi Shiono, and Chikara Tsutsumi. 2017. "Synthesis and Biodegradation of Poly(l-lactide-co-β-propiolactone)" International Journal of Molecular Sciences 18, no. 6: 1312. https://doi.org/10.3390/ijms18061312
APA StyleNakayama, Y., Aihara, K., Cai, Z., Shiono, T., & Tsutsumi, C. (2017). Synthesis and Biodegradation of Poly(l-lactide-co-β-propiolactone). International Journal of Molecular Sciences, 18(6), 1312. https://doi.org/10.3390/ijms18061312