A Tox21 Approach to Altered Epigenetic Landscapes: Assessing Epigenetic Toxicity Pathways Leading to Altered Gene Expression and Oncogenic Transformation In Vitro

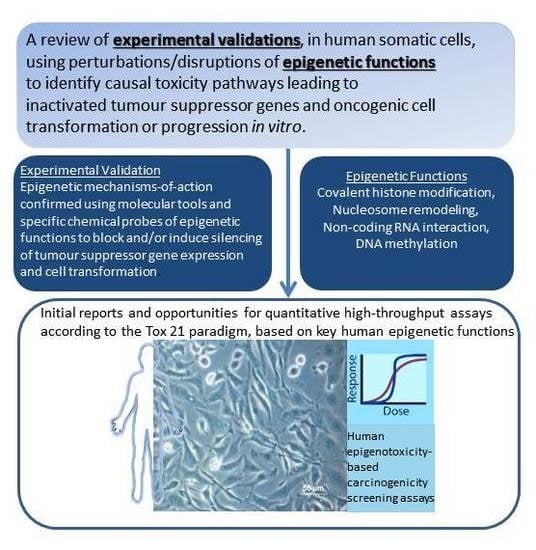
Abstract
:
1. Introduction
1.1. Considerations in Applying the Tox21 Strategy for Human Health Risk Assessment to the Epigenetic Mode of Action and Mechanistic Pathways Leading to Carcinogenesis
1.2. TSG Silencing Models in Cell Transformation
1.3. A Molecular Interpretation of the Waddington Epigenetic Landscape Model for the Development of Cellular Identity and as the Basis for Insight into the Epigenetic-Mode-of-Action of Carcinogens
2. Histone Post-Translational Modifications (HPTM), Histone Remodeling and Interaction with DNA Methylation Systems
2.1. Polycomb and Trithorax Group Proteins
2.1.1. Evidence for Causal Roles of Polycomb Complex H3K27 Methyltransferase Activities in Oncogenic Transformation of Human Cells
PcG Targets: Homeobox Genes, the “Bivalent State” and Epigenetic Switching during Carcinogenesis
Links between the pRB/p16 Tumour Suppressor Pathway and PRC2, PRC1 Proteins
Experimental Support for the Participation of PRC2 EZH2 H3K27 Methyltransferase in Driving the Progression of Oncogenic Phenotypes beyond the Extended Lifespan and Immortalization Stages
Stress- and Chemically-Induced Expression of PcG Proteins
2.1.2. Evidence for Causal Involvement of Trithorax H3K4me Histone Modifying Complexes (MLL/SET1 Complex) in Oncogenic Transformation
2.2. Evidence for Causal Participation of H3K9 Methyltransferases in Oncogenic Transformation of Human Cells
2.2.1. Enhanced Expressions of the H3K9 Writers Contribute to Human Cell Oncogenesis
2.2.2. H3K9 Methyltransferase Repression Reverts Oncogenic Phenotypes to More Controlled Cell Growth
2.3. Induction of “Histone Methylation Injuries” and Passage into Cellular Memory
3. ATP-Dependent Nucleosome Remodeling
4. DNA Methylation
4.1. DNA Methylation Enzymes
4.2. Active/Passive Demethylation Systems (Erasers/Editors)
4.3. Methyl Binding Proteins (Readers)
4.3.1. MeCP2
4.3.2. MBD1-6
4.3.3. SETDB and BAZ2
4.3.4. UHRF (Ubiquitin-Like with PHD and Ring Finger Domain Protein 1 and 2)
4.3.5. The KAISO Protein Family
4.4. Experimental Evidence for Perturbations to DNA Methylation Pathway Components as Contributors to Toxicity Pathways Leading to TSG Expression/Repression and Phenotypic Effects
4.4.1. DNMT Overexpression (Phenotypic Effects/TSG Suppression)
4.4.2. Experiments Creating Epigenetically Modified Promoters to Silence TSGs
4.4.3. TET1 as a Regulator of Gene Expression Networks and Oncogenic Cell Transformation
5. CTCF and Control of Epigenetic Modifications in TSGs
5.1. P16/INK4A
5.2. pRB
5.3. p53
5.4. Genome-Scale Epigenomic Changes
6. Non-Coding RNA: Roles in Both Oncogenic and Tumour Suppressive Pathways
6.1. H19
6.2. HOTAIR
6.3. ANRIL
6.4. MIR31HG
7. Experimental Evidence from In Vitro Cell Transformation Models Supporting the Roles of Chemically-Induced Epigenetic Perturbations as key Steps in Developing Cancer-Related Adverse Phenotypes
7.1. The “Epigenetic Progenitor” Hypothesis and Multi-Step Tumourigenesis
- Epigenetic alterations of stem or progenitor cells create disturbances to the regulated expression of tumour-progenitor genes that normally function to promote “stemness,” as characterized by pluripotency and replication capacity while repressing differentiation (examples include Igf2 up-regulation by hypomethylation/loss of imprinting [289]; Latexin down-regulation by hypermethylation [290]). The alterations in expression at this early stage probably frequently result from environmental perturbations in epigenetic processes (writers, erasers, editors or readers). Consequently, increases or persistence of the more primitive precursor cells within a tissue, lead to the disruption of the balance between undifferentiated progenitor cells and differentiated cells, as well as disrupting the capacity to differentiate along specific differentiation program paths. It should be noted that the epigenetic progenitor hypothesis includes the idea that, in the relatively undifferentiated progenitor or tissue stem cells, some properties characteristic of advanced tumours may already exist (e.g., self-renewal, invasiveness, or drug resistance).
- Tumour-suppressor gene inactivations and/or oncogene activations occur within the expanded or altered progenitor compartment, removing further constraints on cell replication and survival.
- Gains of constitutive genetic, chromosomal and epigenetic instability lead onward to an increased pace of tumour evolution.
7.2. The Syrian Hamster Embryo (SHE) Cell Transformation Assay
7.3. p16 in Syrian Hamster Dermal Fibroblast and Embryo Cell Immortalization
7.4. p16 in Human Mammary Epithelial Cell Escape from Culture “Stasis”
7.5. Roles for Epigenetic Changes during Chemical Transformation of Human Cell Lines
8. Current Developments in Phenotypic Screening Assays for Chemicals Causing Epigenetic Perturbations in Human Cells
- Capability to be performed in 96- or 1536-well plate formats
- 10 steps or less
- Fast assay times (24–48 h)
- Minimal assay steps, 4 or less
- Robust signals, greater than 3-fold
9. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, J.; Perlmann, T. Maintaining differentiated cellular identity. Nat. Rev. Genet. 2012, 13, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, X.; Wang, D.; Baccarelli, A. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 2012, 41, 79–105. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Lochhead, P.; Chan, A.T.; Nishihara, R.; Cho, E.; Wolpin, B.M.; Meyerhardt, J.A.; Meissner, A.; Schernhammer, E.S.; Fuchs, C.S.; et al. Molecular pathological epidemiology of epigenetics: Emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013, 26, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Rusyn, I. Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol. 2013, 754, 215–232. [Google Scholar] [PubMed]
- Herceg, Z.; Lambert, M.P.; van Veldhoven, K.; Demetriou, C.; Vineis, P.; Smith, M.T.; Straif, K.; Wild, C.P. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis 2013, 34, 1955–1967. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, E.L.; Jacobs, M.N.; Gant, T.W. Environmentally induced epigenetic toxicity: Potential public health concerns. Crit. Rev. Toxicol. 2016, 46, 676–700. [Google Scholar] [CrossRef] [PubMed]
- Huen, K.; Calafat, A.M.; Bradman, A.; Yousefi, P.; Eskenazi, B.; Holland, N. Maternal phthalate exposure during pregnancy is associated with DNA methylation of LINE-1 and Alu repetitive elements in Mexican-American children. Environ. Res. 2016, 148, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.I.; Valdés, A.; Fernández, A.F.; Torrecillas, R.; Fraga, M.F. The effect of exposure to nanoparticles and nanomaterials on the mammalian epigenome. Int. J. Nanomed. 2016, 11, 6297–6306. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, S.P.; Kaiser, J.P.; Wesselkamper, S.C. Epigenetic regulation in environmental chemical carcinogenesis and its applicability in human health risk assessment. Int. J. Toxicol. 2015, 34, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Yosim, A.; Fry, R.C. Incorporating epigenetic data into the risk assessment process for the toxic metals arsenic, cadmium, chromium, lead, and mercury: Strategies and challenges. Front. Genet. 2014, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Guyton, K.Z.; Gibbons, C.F.; Fritz, J.M.; Portier, C.J.; DeMarini, D.M.; Caldwell, J.C.; Kavlock, R.J.; Lambert, P.; Hecht, S.S.; et al. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.N.; Colacci, A.; Louekari, K.; Luijten, M.; Hakkert, B.C.; Paparella, M.; Vasseur, P. International regulatory needs for development of an IATA for non-genotoxic carcinogenic chemical substances. Altex 2016, 33, 359–392. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Asada, K. Aberrant DNA methylation in contrast with mutations. Cancer Sci. 2010, 101, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic alterations in colorectal cancer: Emerging biomarkers. Gastroenterology 2015, 149, 1204–1225. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Acosta, D.; Andersen, M.; Anderson, H.; Bailar, J.C.; Boekelheide, K.; Brent, R.; Charnley, G.; Cheung, V.G.; Green, S.; et al. Toxicity testing in the 21st century: A vision and a strategy. J. Toxicol. Environ. Health Part B 2010, 13, 51–138. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.E.; Krewski, D. Toxicity testing in the 21st century: Bringing the vision to life. Toxicol. Sci. 2009, 107, 324–330. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (NRC). Toxicity Testing in the 21st Century: A Vision and A Strategy; National Academy Press: Washington, DC, USA, 2007. [Google Scholar]
- Krewski, D.; Westphal, M.; Andersen, M.E.; Paoli, G.M.; Chiu, W.A.; Al-Zoughool, M.; Croteau, M.C.; Burgoon, L.D.; Cote, I. A framework for the next generation of risk science. Environ. Health Perspect. 2014, 122, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Bouhifd, M.; Hogberg, H.T.; Kleensang, A.; Maertens, A.; Zhao, L.; Hartung, T. Mapping the human toxome by systems toxicology. Basic Clin. Pharmacol. Toxicol. 2014, 115, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Kleensang, A.; Maertens, A.; Rosenberg, M.; Fitzpatrick, S.; Lamb, J.; Auerbach, S.; Brennan, R.; Crofton, K.M.; Gordon, B.; Fornace, A.J., Jr.; et al. t4 workshop report. Altex 2014, 31, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Bultman, S.J. Metaboloepigenetics: Interrelationships between energy metabolism and epigenetic control of gene expression. J. Cell. Physiol. 2012, 227, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Piunti, A.; Pasini, D. Epigenetic methylations and their connections with metabolism. Cell. Mol. Life Sci. 2013, 70, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Kolybaba, A.; Classen, A.K. Sensing cellular states—Signaling to chromatin pathways targeting Polycomb and Trithorax group function. Cell Tissue Res. 2014, 356, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Extended Advisory Group on Molecular Screening and Toxicogenomics. Users’ Handbook Supplement to the Guidance Document for Developing and Assessing Adverse Outcome Pathways. 2016. Available online: https://aopwiki.org/training/aops/story_content/external_files/OECD%20Users%20Handbook%20-2016.pdf (accessed on 30 May 2017).
- Hartung, T. Utility of the adverse outcome pathway concept in drug development. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.; Kavlock, R.; Martin, M.; Reif, D.; Houck, K.; Knudsen, T.; Richard, A.; Tice, R.R.; Whelan, M.; Xia, M.; et al. Perspectives on validation of high-throughput assays supporting 21st century toxicity testing. Altex 2013, 30, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Adeleye, Y.; Andersen, M.; Clewell, R.; Davies, M.; Dent, M.; Edwards, S.; Fowler, P.; Malcomber, S.; Nicol, B.; Scott, A.; et al. Implementing toxicity testing in the 21st century (TT21C): Making safety decisions using toxicity pathways, and progress in a prototype risk assessment. Toxicology 2015, 332, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, J. Functional mechanisms for human tumor suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, J.; Zhao, Z. TSGene: A web resource for tumor suppressor genes. Nucleic Acids Res. 2013, 41, D970–D976. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Ink4-Arf locus in cancer and aging. Wiley Interdiscip. Rev. 2012, 1, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- LaPak, K.M.; Burd, C.E. The molecular balancing act of p16ink4a in cancer and aging. Mol. Cancer Res. 2014, 12, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, E.A.; Soliman, K.F.A. Basic concepts of epigenetics impact of environmental signals on gene expression. Epigenetics 2012, 7, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R. Epigenetic alterations in oncogenesis. Preface. Adv. Exp. Med. Biol. 2013, 754, v–vii. [Google Scholar] [PubMed]
- Fischle, W. Molecular mechanisms of histone modification function. Biochim. Biophys. Acta 2014, 1839, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R. Methylation: A multifaceted modification—Looking at transcription and beyond. Biochim. Biophys. Acta 2014, 1839, 1351–1352. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.K.; Imhof, A. Fast signals and slow marks: The dynamics of histone modifications. Trends Biochem. Sci. 2010, 35, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.; Letunic, I.; Bork, P.; Lim, W.A. Evolution of the phospho-tyrosine signaling machinery in premetazoan lineages. Proc. Natl. Acad. Sci. USA 2008, 105, 9680–9684. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Turner, B.M. Altered histone modifications in cancer. Adv. Exp. Med. Biol. 2013, 754, 81–107. [Google Scholar] [PubMed]
- Dumont, N.; Wilson, M.B.; Crawford, Y.G.; Reynolds, P.A.; Sigaroudinia, M.; Tlsty, T.D. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 14867–14872. [Google Scholar] [CrossRef] [PubMed]
- Bedi, U.; Mishra, V.K.; Wasilewski, D.; Scheel, C.; Johnsen, S.A. Epigenetic plasticity: A central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 2014, 5, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B. Chromatin: Constructing the big picture. EMBO 2011, 30, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Margueron, R. Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Khare Satyajeet, P.; Habib, F.; Sharma, R.; Gadewal, N.; Gupta, S.; Galande, S. HIstome—A relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 2011. Available online: http://nar.oxfordjournals.org/content/early/2011/12/02/nar.gkr1125.abstract (accessed on 30 May 2017).
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.; Schneider, R. Targeting histone modifications—Epigenetics in cancer. Curr. Opin. Cell Biol. 2013, 25, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fang, L.; Li, H.; Tang, M.S.; Jin, C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J. Biol. Chem. 2013, 288, 21678–21687. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.D.; Tepperman, K.; Huang, M.Y.; Sartor, M.A.; Puga, A. Chromium inhibits transcription from polycyclic aromatic hydrocarbon-inducible promoters by blocking the release of histone deacetylase and preventing the binding of p300 to chromatin. J. Biol. Chem. 2004, 279, 4110–4119. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ke, Q.; Kluz, T.; Yan, Y.; Costa, M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol. Cell. Biol. 2006, 26, 3728–3737. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kluz, T.; Zhang, R.; Costa, M. Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis 2010, 31, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Chervona, Y.; Costa, M. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic. Biol. Med. 2012, 53, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Hickok, J.R.; Vasudevan, D.; Antholine, W.E.; Thomas, D.D. Nitric oxide modifies global histone methylation by inhibiting jumonji C domain-containing demethylases. J. Biol. Chem. 2013, 288, 16004–16015. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Liu, Y.; Xie, C.; Tu, W.; Xia, Y.; Costa, M.; Zhou, X. Cadmium induces histone H3 lysine methylation by inhibiting histone demethylase activity. Toxicol. Sci. 2015, 145, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Sun, H.; Ellen, T.P.; Chen, H.; Costa, M. Arsenite alters global histone H3 methylation. Carcinogenesis 2008, 29, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Sahay, S.; Tiwari, P.; Upadhyay, D.S.; Sultana, S.; Gupta, K.P. Involvement of EZH2, SUV39H1, G9a and associated molecules in pathogenesis of urethane induced mouse lung tumors: Potential targets for cancer control. Toxicol. Appl. Pharmacol. 2014, 280, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A. Throwing the cancer switch: Reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Occupying Chromatin: Polycomb Mechanisms for Getting to Genomic Targets, Stopping Transcriptional Traffic, and Staying Put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.; Healy, E.; Bracken, A.P. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 2015, 37, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Francis, N.J.; Kingston, R.E.; Woodcock, C.L. Chromatin compaction by a polycomb group protein complex. Science 2004, 306, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Isono, K.; Sharif, J.; Ohara, O.; Toyoda, T.; Ito, T.; Eskeland, R.; Bickmore, W.A.; et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet. 2012, 8, e1002774. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury Iii, W.J.; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Ji, G.; Gao, Z.; Han, X.; Ye, M.; Yuan, Z.; Luo, H.; Huang, X.; Natarajan, K.; Wang, J.; et al. Direct ChIP-bisulfite sequencing reveals a role of H3K27me3 mediating aberrant hypermethylation of promoter CpG islands in cancer cells. Genomics 2014, 103, 204–210. [Google Scholar] [CrossRef] [PubMed]
- O'Hagan, H.; Wang, W.; Sen, S.; DeStefano-Shields, C.; Lee, S.; Zhang, Y.; Clements, E.; Cai, Y.; Van-áNeste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Bonham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O'Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol. 2016, 8, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; Ueshima, K.; Sakurai, T.; et al. Reactive Oxygen Species Induce Epigenetic Instability through the Formation of 8-Hydroxydeoxyguanosine in Human Hepatocarcinogenesis. Dig. Dis. 2013, 31, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Reddington, J.P.; Sproul, D.; Meehan, R.R. DNA methylation reprogramming in cancer: Does it act by re-configuring the binding landscape of Polycomb repressive complexes? BioEssays 2014, 36, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Reddington, J.P.; Perricone, S.M.; Nestor, C.E.; Reichmann, J.; Youngson, N.A.; Suzuki, M.; Reinhardt, D.; Dunican, D.S.; Prendegast, J.G.; Mjoseng, H.; et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb-target genes. Genome Biol. 2013, 14, R25. [Google Scholar] [CrossRef] [PubMed]
- Putiri, E.L.; Tiedemann, R.L.; Liu, C.; Choi, J.H.; Robertson, K.D. Impact of human MLL/COMPASS and polycomb complexes on the DNA methylome. Oncotarget 2014, 5, 6338–6352. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Cipriany, B.R.; Wallin, C.B.; Ju, C.Y.; Szeto, K.; Hagarman, J.A.; Benitez, J.J.; Craighead, H.G.; Soloway, P.D. Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7772–7777. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Wakabayashi, M.; Hattori, N.; Yamashita, S.; Ushijima, T. Identification of coexistence of DNA methylation and H3K27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis 2014, 36, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.A.; Sigaroudinia, M.; Zardo, G.; Wilson, M.B.; Benton, G.M.; Miller, C.J.; Hong, C.; Fridlyand, J.; Costello, J.F.; Tlsty, T.D. Tumor Suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J. Biol. Chem. 2006, 281, 24790–24802. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Dimri, M.; Dimri, G.P. A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J. Biol. Chem. 2013, 288, 3406–3418. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Murphy, C.G.; Johnson, R.; Lay, J.M.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; King, B.L.; Rosenstein, M.C.; Wiegers, T.C.; et al. The comparative toxicogenomics database: Update 2013. Nucleic Acids Res. 2013, 41, D1104–D1114. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, N.; Bracken, A.P.; Trinh, E.; Schjerling, C.K.; Koseki, H.; Rappsilber, J.; Helin, K.; Hansen, K.H. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO 2007, 26, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Bernard, D.; Martínez, D.; Beach, D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2004, 6, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Haga, K.; Ohno, S.I.; Yugawa, T.; Narisawa-Saito, M.; Fujita, M.; Sakamoto, M.; Galloway, D.A.; Kiyono, T. Efficient immortalization of primary human cells by p16INK4a-specific short hairpin RNA or Bmi-1, combined with introduction of hTERT. Cancer Sci. 2007, 98, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Cheng, A.S.; Ahmed, S.; Boumber, Y.; Charo, C.; Yamochi, T.; Urano, T.; Furukawa, K.; Kwabi-Addo, B.; et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008, 40, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Karanikolas, B.D.W.; Figueiredo, M.L.; Wu, L. Polycomb group protein enhancer of zeste 2 is an oncogene that promotes the neoplastic transformation of a benign prostatic epithelial cell line. Mol. Cancer Res. 2009, 7, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.L.; Lau, S.S.; Chen, Y.; Kondo, Y.; Li, M.S.; Feng, H.; Ching, A.K.; Cheung, K.F.; Wong, H.K.; Tong, J.H.; et al. EZH2-mediated concordant repression of Wnt antagonists promotes β -catenin-dependent hepatocarcinogenesis. Cancer Res. 2011, 71, 4028–4039. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Jene-Sanz, A.; Váraljai, R.; Vilkov, A.V.; Khramtsov, G.F.; Khramtsov, A.I.; Olopade, O.I.; Lopez-Bigas, N.; Benevolenskaya, E.V. Expression of polycomb targets predicts breast cancer prognosis. Mol. Cell. Biol. 2013, 33, 3951–3961. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Song, L.B.; Li, J.; Liao, W.T.; Feng, Y.; Yu, C.P.; Hu, L.J.; Kong, Q.L.; Xu, L.H.; Zhang, X.; Liu, W.L.; et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Reipas, K.M.; Pambid, M.R.; Berns, R.; Stratford, A.L.; Fotovati, A.; Firmino, N.; Astanehe, A.; Hu, K.; Maxwell, C.; et al. YB-1 transforms human mammary epithelial cells through chromatin remodeling leading to the development of basal-like breast cancer. Stem Cells 2014, 32, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, A.; Mourtzoukou, D.; Kosmidou, V.; Avlonitis, S.; Kontogeorgos, G.; Zografos, G.; Pintzas, A. EZH2 is regulated by ERK/AKT and targets integrin α2 gene to control epithelial-mesenchymal transition and anoikis in colon cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Carcagno, A.L.; Ogara, M.F.; Sonzogni, S.V.; Marazita, M.C.; Sirkin, P.F.; Ceruti, J.M.; Cánepa, E.T. E2F1 transcription is induced by genotoxic stress through ATM/ATR activation. IUBMB Life 2009, 61, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009, 69, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Kapoor, A.; Wang, X.; Aksoy, O.; Evertts, A.G.; Zhang, M.Q.; Garcia, B.A.; Bernstein, E.; Lowe, S.W. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 8971–8976. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Kasiri, S.; Mandal, S.S. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene 2013, 32, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Salz, T.; Li, G.; Kaye, F.; Zhou, L.; Qiu, Y.; Huang, S. HSETD1A regulates Wnt target genes and controls tumor growth of colorectal cancer cells. Cancer Res. 2014, 74, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Lüscher-Firzlaff, J.; Gawlista, I.; Vervoorts, J.; Kapelle, K.; Braunschweig, T.; Walsemann, G.; Rodgarkia-Schamberger, C.; Schuchlautz, H.; Dreschers, S.; Kremmer, E.; et al. The human trithorax protein hASH2 functions as an oncoprotein. Cancer Res. 2008, 68, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Volkel, P.; Angrand, P.O. The control of histone lysine methylation in epigenetic regulation. Biochimie 2007, 89, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F. DNA methylation and histone modifications: Teaming up to silence genes. Curr. Opin. Genet. Dev. 2005, 15, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Fuks, F. A methylation rendezvous: Reader meets writers. Dev. Cell 2007, 12, 843–844. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, A.; Estève, P.O.; Pradhan, S.; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007, 21, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M.; Sharif, T.; Mousli, M.; Etienne-Selloum, N.; Fuhrmann, G.; Schini-Kerth, V.; Bronner, C. Down-regulation of UHRF1, associated with re-expression of tumor suppressor genes, is a common feature of natural compounds exhibiting anti-cancer properties. J. Exp. Clin. Cancer Res. 2011, 30, 41. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.; Krifa, M.; Mousli, M. Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem. Pharmacol. 2013, 86, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Babbio, F.; Pistore, C.; Curti, L.; Castiglioni, I.; Kunderfranco, P.; Brino, L.; Oudet, P.; Seiler, R.; Thalman, G.N.; Roggero, E.; et al. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene 2012, 31, 4878–4887. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Estève, P.O.; Jacobsen, S.E.; Pradhan, S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009, 37, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Kokura, K.; Sun, L.; Bedford, M.T.; Fang, J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J. 2010, 29, 3673–3687. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Watanabe, S.; Ichimura, T.; Tsuruzoe, S.; Shinkai, Y.; Tachibana, M.; Chiba, T.; Nakao, M. Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 2003, 278, 24132–24138. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Stancheva, I. Methyl-CpG Binding Protein MBD1 Couples Histone H3 Methylation at Lysine 9 by SETDB1 to DNA Replication and Chromatin Assembly. Mol. Cell 2004, 15, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Li, D.; Wang, B.; Kovalchuk, I.; Kovalchuk, O. SUV39H1 downregulation induces deheterochromatinization of satellite regions and senescence after exposure to ionizing radiation. Ageing 2014, 5, 411. [Google Scholar] [CrossRef] [PubMed]
- Cherrier, T.; Suzanne, S.; Redel, L.; Calao, M.; Marban, C.; Samah, B.; Mukerjee, R.; Schwartz, C.; Gras, G.; Sawaya, B.E.; et al. p21WAF1 gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 2009, 28, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 Histone Methyltransferase G9a Promotes Lung Cancer Invasion and Metastasis by Silencing the Cell Adhesion Molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Soejima, K.; Yasuda, H.; Kawada, I.; Nakachi, I.; Yoda, S.; Naoki, K.; Ishizaka, A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.T.; Wang, M.Y.; Chen, M.W.; Wei, L.H.; Chen, C.K.; Ko, C.H.; Jeng, Y.M.; Sung, P.L.; Jan, Y.H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [PubMed]
- Bachman, K.E.; Park, B.H.; Rhee, I.; Rajagopalan, H.; Herman, J.G.; Baylin, S.B.; Kinzler, K.W.; Vogelstein, B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell 2003, 3, 89–95. [Google Scholar] [CrossRef]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A.E. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, C.; Guilbaud, G.; Schiavone, D.; Sale, J.E. Nucleotide Pool Depletion Induces G-Quadruplex-Dependent Perturbation of Gene Expression. Cell Rep. 2015, 13, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Jasencakova, Z.; Groth, A. Replication stress, a source of epigenetic aberrations in cancer? BioEssays 2010, 32, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Jasencakova, Z.; Scharf, A.N.D.; Ask, K.; Corpet, A.; Imhof, A.; Almouzni, G.; Groth, A. Replication Stress Interferes with Histone Recycling and Predeposition Marking of New Histones. Mol. Cell 2010, 37, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Vieyra, C.V.; Liang, G. Nucleosome occupancy and gene regulation during tumorigenesis. Adv. Exp. Med. Biol. 2013, 754, 109–134. [Google Scholar] [PubMed]
- Swygert, S.G.; Peterson, C.L. Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 2014, 1839, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Wade, P.A. Cancer biology and NuRD: A multifaceted chromatin remodelling complex. Nat. Rev. Cancer 2011, 11, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Wu, K.J. Epigenetic regulation of hypoxia-responsive gene expression: Focusing on chromatin and DNA modifications. Int. J. Cancer 2014, 134, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, E.C.; Carmody, L.C.; Tolliday, N.; Crabtree, G.R.; Palmer, M.A.J. Screening for inhibitors of an essential chromatin remodeler in mouse embryonic stem cells by monitoring transcriptional regulation. J. Biomol. Screen. 2012, 17, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.E.; Cho, Y.J.; Koellhoffer, E.C.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W.M. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Farias, N.; Ho, N.; Butler, S.; Delaney, L.; Morrison, J.; Shahrzad, S.; Coomber, B.L. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 2015, 26, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, B.; Cheng, Y.; Yang, Y.; Huang, C.; Meng, X.; Wu, B.; Zhang, L.; Lv, X.; Li, J. Melittin induces PTCH1 expression by down-regulating MeCP2 in human hepatocellular carcinoma SMMC-7721 cells. Toxicol. Appl. Pharmacol. 2015, 288, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Gil, J.; Dumont, P.; Rizzo, S.; Monté, D.; Quatannens, B.; Hudson, D.; Visakorpi, T.; Fuks, F.; De Launoit, Y. The methyl-CpG-binding protein MECP2 is required for prostate cancer cell growth. Oncogene 2006, 25, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, B.F.; Chan, W.Y. An epigenetic regulator: Methyl-CpG-binding domain protein 1 (MBD1). Int. J. Mol. Sci. 2015, 16, 5125–5140. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.K.; Mei, S.C.; Brenner, C. RFTS-deleted DNMT1 enhances tumorigenicity with focal hypermethylation and global hypomethylation. Cell Cycle 2014, 13, 3222–3231. [Google Scholar] [CrossRef] [PubMed]
- Pacaud, R.; Brocard, E.; Lalier, L.; Hervouet, E.; Vallette, F.M.; Cartron, P.F. The DNMT1/PCNA/UHRF1 disruption induces tumorigenesis characterized by similar genetic and epigenetic signatures. Sci. Rep. 2014, 4, 4230. [Google Scholar] [CrossRef] [PubMed]
- Teneng, I.; Tellez, C.S.; Picchi, M.A.; Klinge, D.M.; Yingling, C.M.; Snider, A.M.; Liu, Y.; Belinsky, S.A. Global identification of genes targeted by DNMT3b for epigenetic silencing in lung cancer. Oncogene 2015, 34, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Teng, I.W.; Hou, P.C.; Lee, K.D.; Chu, P.Y.; Yeh, K.T.; Jin, V.X.; Tseng, M.J.; Tsai, S.J.; Chang, Y.S.; Wu, C.S.; et al. Targeted methylation of two tumor suppressor genes is sufficient to transform mesenchymal stem cells into cancer stem/initiating cells. Cancer Res. 2011, 71, 4653–4663. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Shang, Y.; Jin, Z.; Zhang, W.; Lv, C.; Zhao, X.; Liu, Y.; Li, N.; Liang, J. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Biol. Ther. 2015, 16, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Boukhari, A.; Alhosin, M.; Bronner, C.; Sagini, K.; Truchot, C.; Sick, E.; Schini-Kerth, V.B.; Andre, P.; Mely, Y.; Mousli, M.; et al. CD47 activation-induced UHRF1 over-expression is associated with silencing of tumor suppressor gene p16INK4A in glioblastoma cells. Anticancer Res. 2015, 35, 149–157. [Google Scholar] [PubMed]
- Zhang, Y.; Huang, Z.; Zhu, Z.; Zheng, X.; Liu, J.; Han, Z.; Ma, X.; Zhang, Y. Upregulated UHRF1 promotes bladder cancer cell invasion by epigenetic silencing of KiSS1. PLoS ONE 2014, 9, e104252. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ou, H.; Xiang, L.; Li, X.; Huang, Y.; Yang, D. Elevated UHRF1 expression contributes to poor prognosis by promoting cell proliferation and metastasis in hepatocellular carcinoma 3. Oncotarget 2017, 8, 10510–10522. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Song, C.X.; Huang, H.; Frankenberger, C.A.; Sankarasharma, D.; Gomes, S.; Chen, P.; Chen, J.; Chada, K.K.; He, C.; et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 9920–9925. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.K.; Brenner, C. Suppression of TET1-Dependent DNA Demethylation Is Essential for KRAS-Mediated Transformation. Cell Rep. 2014, 9, 1827–1841. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Poliseno, L.; Song, M.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.; Yuan, X.; Cantley, L.; et al. MicroRNA-Antagonism Regulates Breast Cancer Stemness and Metastasis via TET-Family-Dependent Chromatin Remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Dettori, D.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Maldotti, M.; Parlato, C.; Paliogiannis, P.; Oliviero, S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 2015, 34, 4168–4176. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Wei, L.; Law, C.T.; Ho, D.W.H.; Tsang, F.H.C.; Au, S.L.K.; Sze, K.M.F.; Lee, J.M.F.; Wong, C.C.L.; Ng, I.O.L. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology 2016, 63, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Babbio, F.; Castiglioni, I.; Cassina, C.; Gariboldi, M.B.; Pistore, C.; Magnani, E.; Badaracco, G.; Monti, E.; Bonapace, I.M. Knock-down of methyl CpG-binding protein 2 (MeCP2) causes alterations in cell proliferation and nuclear lamins expression in mammalian cells. BMC Cell Biol. 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Nishikawaji, T.; Akiyama, Y.; Shimada, S.; Kojima, K.; Kawano, T.; Eishi, Y.; Yuasa, Y.; Tanaka, S. Oncogenic roles of the SETDB2 histone methyltransferase in gastric cancer. Oncotarget 2016, 7, 67251–67265. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, O.; Li, R.; Hung, J.H.; Chen, P.B.; Dong, X.; Ee, L.S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Frommel, S.C.; Oakes, C.C.; Simon, R.; Grupp, K.; Gerig, C.Y.; Bär, D.; Robinson, M.D.; Baer, C.; Weiss, M.; et al. BAZ2A (TIP5) is involved in epigenetic alterations in Prostate cancer and its overexpression predicts disease recurrence. Nat. Genet. 2015, 47, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Kinde, B.; Gabel, H.W.; Gilbert, C.S.; Griffith, E.C.; Greenberg, M.E. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. USA 2015, 112, 6800–6806. [Google Scholar] [CrossRef] [PubMed]
- Shorter, K.R.; Felder, M.R.; Vrana, P.B. Consequences of dietary methyl donor supplements: Is more always better? Prog. Biophys. Mol. Biol. 2015, 118, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Kiani, J.; Grandjean, V.; Liebers, R.; Tuorto, F.; Ghanbarian, H.; Lyko, F.; Cuzin, F.; Rassoulzadegan, M. RNA-Mediated Epigenetic Heredity Requires the Cytosine Methyltransferase Dnmt2. PLoS Genet. 2013, 9, e1003498. [Google Scholar] [CrossRef] [PubMed]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Xiang, B.; Ghosh, S.; Ren, T.; Lewis, P.W.; Cochrane, J.C.; Allis, C.D.; Picketts, D.J.; Patel, D.J.; Li, H.; et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 2011, 18, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; De Carvalho, D.D.; Jeong, S.; Jones, P.A.; Liang, G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS. Genet. 2011, 7, e1001286. [Google Scholar] [CrossRef] [PubMed]
- Locke, W.J.; Zotenko, E.; Stirzaker, C.; Robinson, M.D.; Hinshelwood, R.A.; Stone, A.; Reddel, R.R.; Huschtscha, L.I.; Clark, S.J. Coordinated epigenetic remodelling of transcriptional networks occurs during early breast carcinogenesis. Clin. Epigenet. 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, Y.Y.; Dai, Y.J.; Zhang, W.; Zhang, W.N.; Xiong, S.M.; Gu, Z.H.; Wang, K.K.; Zeng, R.; Chen, Z.; et al. DNMT3A Arg882 mutation drives chronic myelomonocytic leukemia through disturbing gene expression/DNA methylation in hematopoietic cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2620–2625. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. Myelodysplastic Syndromes: Diagnosis and Treatment. Mayo Clin. Proc. 2015, 90, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. In Epigenetics; Allis, C.D., Jenuwein, T., Reinberg, D., Caparros, M.-L., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2007; pp. 457–476. [Google Scholar]
- Ko, M.; An, J.; Pastor, W.A.; Koralov, S.B.; Rajewsky, K.; Rao, A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol. Rev. 2015, 263, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Kinney, S.R.; Pradhan, S. Ten eleven translocation enzymes and 5-hydroxymethylation in mammalian development and cancer. Adv. Exp. Med. Biol. 2013, 754, 57–79. [Google Scholar] [PubMed]
- Kroeze, L.I.; van der Reijden, B.A.; Jansen, J.H. 5-Hydroxymethylcytosine: An epigenetic mark frequently deregulated in cancer. Biochim. Biophys. Acta 2015, 1855, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Brocato, J.; Costa, M. 10th NTES Conference: Nickel and arsenic compounds alter the epigenome of peripheral blood mononuclear cells. J. Trace Elem. Med. Biol. 2014, 31, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. TET1: An epigenetic guardian of lymphomagenesis. Nat. Immunol. 2015, 16, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Peng, K.L.; Kang, M.L.; Chen, Y.R.; Yang, Y.C.; Tsai, C.H.; Chu, C.S.; Jeng, Y.M.; Chen, Y.T.; Lin, F.M.; et al. TET1 Suppresses Cancer Invasion by Activating the Tissue Inhibitors of Metalloproteinases. Cell Rep. 2012, 2, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of Melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Masuda, K.; Sato, T.; Sakaguchi, Y.; Suzuki, T.; Suzuki, T.; Koyama-Nasu, R.; Nasu-Nishimura, Y.; Katou, Y.; Ogawa, H.; et al. 5-Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP-methylosome complex. Cell Rep. 2014, 9, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.H.; Barton, M.C.; Issa, J.P.J. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, Y.; Xu, L.; Xiao, R.; Lu, X.; Chen, L.; Chong, J.; Li, H.; He, C.; Fu, X.D.; et al. Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex. Nature 2015, 523, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Buck-Koehntop, B.A.; Defossez, P.A. On how mammalian transcription factors recognize methylated DNA. Epigenetics 2013, 8, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Pohodich, A.E.; Zoghbi, H.Y. Rett syndrome: Disruption of epigenetic control of postnatal neurological functions. Hum. Mol. Genet. 2015, 24, R10–R16. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Loh, D.H.; Kudo, T.; Truong, D.; Derakhshesh, M.; Kaswan, Z.M.; Ghiani, C.A.; Tsoa, R.; Cheng, Y.; Sun, Y.E.; et al. Circadian rhythm disruption in a mouse model of Rett syndrome circadian disruption in RTT. Neurobiol. Dis. 2015, 77, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Hite, K.C.; Adams, V.H.; Hansen, J.C. Recent advances in MeCP2 structure and function. Biochem. Cell Biol. 2009, 87, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Bergo, A.; Strollo, M.; Gai, M.; Barbiero, I.; Stefanelli, G.; Sertic, S.; Gigli, C.C.; Di Cunto, F.; Kilstrup-Nielsen, C.; Landsberger, N. Methyl-CpG binding protein 2 (MeCP2) localizes at the centrosome and is required for proper mitotic spindle organization. J. Biol. Chem. 2015, 290, 3223–3237. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Song, J.Z.; Ng, W.; Du, Q.; Armstrong, N.J.; Locke, W.J.; Statham, A.L.; French, H.; Pidsley, R.; Valdes-Mora, F.; et al. Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene 2017, 36, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Schmitz, K.M.; Mayer, C.; Yuan, X.; Akhtar, A.; Grummt, I. Reversible acetylation of the chromatin remodelling complex NoRC is required for non-coding RNA-dependent silencing. Nat. Cell Biol. 2009, 11, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Tallant, C.; Valentini, E.; Fedorov, O.; Overvoorde, L.; Ferguson, F.M.; Filippakopoulos, P.; Svergun, D.I.; Knapp, S.; Ciulli, A. Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure 2015, 23, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Amato, A.; Lucas, X.; Blank, M.; Ciulli, A. Structural Basis of Molecular Recognition of Helical Histone H3 Tail by PHD Finger Domains. Biochem. J. 2017, 474, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- Anosova, I.; Melnik, S.; Tripsianes, K.; Kateb, F.; Grummt, I.; Sattler, M. A novel RNA binding surface of the TAM domain of TIP5/BAZ2A mediates epigenetic regulation of rRNA genes. Nucleic Acids Res. 2015, 43, 5208–5220. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Lacey, M. DNA hypomethylation and hemimethylation in cancer. Adv. Exp. Med. Biol. 2013, 754, 31–56. [Google Scholar] [PubMed]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forne, I.; Pichler, G.; Horl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, Q.; Li, P.; Liu, X.; Jia, Y.; Wu, W.; Li, J.; Dong, S.; Koseki, H.; Wong, J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011, 21, 1723–1739. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xiong, J.; Wang, M.; Yang, N.; Wong, J.; Zhu, B.; Xu, R.M. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol. Cell 2014, 54, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Cui, S.; Bian, C.; Yu, X. Uhrf2 is important for DNA damage response in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2013, 441, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, A.; Hendrich, B.; Jorgensen, H.; Ruzov, A.; Wilm, M.; Georgiev, G.; Bird, A.; Prokhortchouk, E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001, 15, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Zhang, B.; Tian, W.; Gu, L.; Lu, Z.; Deng, D. Kaiso mainly locates in the nucleus in vivo and binds to methylated, but not hydroxymethylated DNA. Chin J. Cancer Res. 2015, 27, 148–155. [Google Scholar] [PubMed]
- Uysal-Onganer, P.; Kypta, R.M. Wnt11 in 2—The regulation and function of a non-canonical Wnt. Acta Physiol. 2012, 204, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Schackmann, R.C.; Tenhagen, M.; van de Ven, R.A.; Derksen, P.W. p120-catenin in cancer—Mechanisms, models and opportunities for intervention. J. Cell Sci. 2013, 126, 3515–3525. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, A.; Sansom, O.; Selfridge, J.; Caballero, I.M.; Salozhin, S.; Aithozhina, D.; Cerchietti, L.; Meng, F.G.; Augenlicht, L.H.; Mariadason, J.M.; et al. Kaiso-deficient mice show resistance to intestinal cancer. Mol. Cell Biol. 2006, 26, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Pierre, C.C.; Longo, J.; Mavor, M.; Milosavljevic, S.B.; Chaudhary, R.; Gilbreath, E.; Yates, C.; Daniel, J.M. Kaiso overexpression promotes intestinal inflammation and potentiates intestinal tumorigenesis in Apc mice. Biochim. Biophys. Acta 2015, 1852, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2010, 101, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.K.; Wang, Y.C. Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 2014, 4, 46. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.V.; Bisht, K.S.; Sun, L.; Muldoon-Jacobs, K.; Awwad, R.; Kaushal, A.; Nguyen, P.; Huang, L.; Pennington, J.D.; Markovina, S.; et al. DNMT1 as a molecular target in a multimodality-resistant phenotype in tumor cells. Mol. Cancer Res. 2008, 6, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Vertino, P.M.; Yen, R.W.C.; Gao, J.; Baylin, S.B. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol. Cell. Biol. 1996, 16, 4555–4565. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.H.; Lee, K.D.; Hsu, C.C.; Tseng, M.J.; Jin, V.X.; Sun, W.S.; Hung, Y.C.; Yeh, K.T.; Yan, P.S.; Lai, Y.Y.; et al. DNA methylation of the Trip10 promoter accelerates mesenchymal stem cell lineage determination. Biochem. Biophys. Res. Commun. 2010, 400, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.R.; Hu, J.F. Directing DNA methylation to inhibit gene expression. Cell. Mol. Neurobiol. 2006, 26, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, D.; Xiao, G.-H.; Cummings-Lorbetskie, C.; Stubbert, L.; Parfett, C. DNA methylation of repeated elements and cancer-related genes in normal human liver, and in cancer and non-cancer liver cell lines, treated with 5adC or PCB126. Curr. Top. Toxicol. 2016, 12, 47–74. [Google Scholar]
- Yu, D.H.; Waterland, R.A.; Zhang, P.; Schady, D.; Chen, M.H.; Guan, Y.; Gadkari, M.; Shen, L. Targeted p16Ink4a epimutation causes tumorigenesis and reduces survival in mice. J. Clin. Investig. 2014, 124, 3708–3712. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Gan, Y.; Gu, L.; Wilson, J.; Liu, Z.; Zhang, B.; Deng, D. P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biol. 2015, 16, 252. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Valentini, E.; Bacalini, M.G.; Zampieri, M.; Calabrese, R.; Guastafierro, T.; Mariano, G.; Reale, A.; Franceschi, C.; Caiafa, P. Poly(ADP-ribosyl)ation is involved in the epigenetic control of TET1 gene transcription. Oncotarget 2014, 5, 10356–10367. [Google Scholar] [CrossRef] [PubMed]
- Nestor, C.E.; Ottaviano, R.; Reinhardt, D.; Cruickshanks, H.A.; Mjoseng, H.K.; McPherson, R.C.; Lentini, A.; Thomson, J.P.; Dunican, D.S.; Pennings, S.; et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease 1. Cell. Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Sajadian, S.O.; Tripura, C.; Samani, F.S.; Ruoss, M.; Dooley, S.; Baharvand, H.; Nussler, A.K. Vitamin C enhances epigenetic modifications induced by 5-azacytidine and cell cycle arrest in the hepatocellular carcinoma cell lines HLE and Huh7 1. Clin. Epigenet. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment 1. Clin. Epigenet. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Huntley, M.; Durand, N.; Stamenova, E.; Bochkov, I.; Robinson, J.; Sanborn, A.; Machol, I.; Omer, A.; Lander, E.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Bailey, C.G.; Rasko, J.E. CTCF and BORIS in genome regulation and cancer. Curr. Opin. Genet. Dev. 2014, 24, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Tang, G.; Wang, H.; Hao, L.; He, T.; Sun, X.; Ting, A.H.; Deng, A.; Sun, S. DNA methylation reactivates GAD1 expression in cancer by preventing CTCF-mediated polycomb repressive complex 2 recruitment. Oncogene 2016, 35, 3995–4008. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.P.; Giordano, A. The tumor suppressor role of CTCF. J. Cell. Physiol. 2012, 227, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell. Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic Silencing of the p16INK4a Tumor Suppressor Is Associated with Loss of CTCF Binding and a Chromatin Boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Leick, M.B.; Schoff, C.J.; Wang, E.C.; Congress, J.L.; Galloway, D.A. Loss of imprinting of IGF2 and the epigenetic progenitor model of cancer. Am. J. Stem Cells 2012, 1, 59–74. [Google Scholar] [PubMed]
- Severson, P.L.; Tokar, E.J.; Vrba, L.; Waalkes, M.P.; Futscher, B.W. Coordinate H3K9 and DNA methylation silencing of ZNFs in toxicant-induced malignant transformation. Epigenetics 2013, 8, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wagner, J.; Damaschke, N.; Yao, T.; Wuerzberger-Davis, S.M.; Lee, M.H.; Svaren, J.; Miyamoto, S.; Jarrard, D.F. A novel pathway links oxidative stress to loss of Insulin Growth Factor-2 (IGF2) imprinting through NF-kB activation. PLoS ONE 2014, 9, e88052. [Google Scholar]
- Gao, A.; Song, S.; Zuo, X.; Guo, W.; Niu, P.; Tian, L. Epigenetic mediated transcriptional activation of PARP-1 participates in silica-associated malignant transformation of human bronchial epithelial cells. Toxicol. Lett. 2010, 193, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Zuo, X.; Liu, Q.; Lu, X.; Guo, W.; Tian, L. Methylation of PARP-1 promoter involved in the regulation of benzene-induced decrease of PARP-1 mRNA expression. Toxicol. Lett. 2010, 195, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Wnek, S.M.; Kuhlman, C.L.; Camarillo, J.M.; Medeiros, M.K.; Liu, K.J.; Lau, S.S.; Gandolfi, A.J. Interdependent genotoxic mechanisms of monomethylarsonous acid: Role of ROS-induced DNA damage and poly(ADP-ribose) polymerase-1 inhibition in the malignant transformation of urothelial cells. Toxicol. Appl. Pharmacol. 2011, 257, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Amati, M.; Nocchi, L.; Saccucci, F.; Strafella, E.; Staffolani, S.; Tarquini, L.M.; Carbonari, D.; Alleva, R.; Borghi, B.; et al. Asbestos exposure affects poly(ADP-ribose) polymerase-1 activity: Role in asbestos-induced carcinogenesis. Mutagenesis 2011, 26, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Guastafierro, T.; Calabrese, R.; Ciccarone, F.; Bacalini, M.G.; Reale, A.; Perilli, M.; Passananti, C.; Caiafa, P. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem. J. 2012, 441, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Guastafierro, T.; Catizone, A.; Calabrese, R.; Zampieri, M.; Martella, O.; Bacalini, M.G.; Reale, A.; Di Girolamo, M.; Miccheli, M.; Farrar, D.; et al. ADP-ribose polymer depletion leads to nuclear Ctcf re-localization and chromatin rearrangement. Biochem. J. 2013, 449, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Livide, G.; Epistolato, M.C.; Amenduni, M.; Disciglio, V.; Marozza, A.; Mencarelli, M.A.; Toti, P.; Lazzi, S.; Hadjistilianou, T.; De Francesco, S.; et al. Epigenetic and copy number variation analysis in retinoblastoma by MS-MLPA. Pathol. Oncol. Res. 2012, 18, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab. Investig. 2001, 81, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Hibberts, N.A.; McNicol, A.M.; Clayton, R.N.; Farrell, W.E. Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res. 2000, 60, 1211–1216. [Google Scholar] [PubMed]
- De La Rosa-Velázquez, I.; Rincón-Arano, H.; Benitez-Bribiesca, L.; Recillas-Targa, F. Epigenetic Regulation of the Human Retinoblastoma Tumor Suppressor Gene Promoter by CTCF. Cancer Res. 2007, 67, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Dávalos-Salas, M.; Furlan-Magaril, M.; Gonzalez-Buendia, E.; Valdes-Quezada, C.; Ayala-Ortega, E.; Recillas-Targa, F. Gain of DNA methylation is enhanced in the absence of CTCF at the human retinoblastoma gene promoter. BMC Cancer 2011, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Saldaña-Meyer, R.; Recillas-Targa, F. Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 2011, 6, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Rasko, J.E.J.; Klenova, E.M.; Leon, J.; Filippova, G.N.; Loukinov, D.I.; Vatolin, S.; Robinson, A.F.; Hu, Y.J.; Ulmer, J.; Ward, M.D.; et al. Cell Growth Inhibition by the Multifunctional Multivalent Zinc-Finger Factor CTCF. Cancer Res. 2001, 61, 6002–6007. [Google Scholar] [PubMed]
- Kemp, C.; Moore, J.; Moser, R.; Bernard, B.; Teater, M.; Smith, L.; Rabaia, N.; Gurley, K.; Guinney, J.; Busch, S.; et al. CTCF Haploinsufficiency Destabilizes DNA Methylation and Predisposes to Cancer. Cell Rep. 2014, 7, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Kugel, J.F.; Goodrich, J.A. Non-coding RNAs: Key regulators of mammalian transcription. Trends Biochem. Sci. 2012, 37, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Constancia, M.; Dean, W.; Lopes, S.; Moore, T.; Kelsey, G.; Reik, W. Deletion of a silencer element in Igf2 results in loss of imprinting independent of H19. Nat. Genet. 2000, 26, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Fauque, P.; Jouannet, P.; Lesaffre, C.; Ripoche, M.A.; Dandolo, L.; Vaiman, D.; Jammes, H. Assisted Reproductive Technology affects developmental kinetics, H19 Imprinting Control Region methylation and H19 gene expression in individual mouse embryos. BMC Dev. Biol. 2007, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Gabory, A.; Ripoche, M.A.; Le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forné, T.; Jammes, H.; Ainscough, J.F.X.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Monnier, P.; Martinet, C.; Pontis, J.; Stancheva, I.; Ait-Si-Ali, S.; Dandolo, L. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc. Natl. Acad. Sci. USA 2013, 110, 20693–20698. [Google Scholar] [CrossRef] [PubMed]
- Matouk, I.; Raveh, E.; Ohana, P.; Lail, R.A.; Gershtain, E.; Gilon, M.; De Groot, N.; Czerniak, A.; Hochberg, A. The increasing complexity of the oncofetal H19 gene locus: Functional dissection and therapeutic intervention. Int. J. Mol. Sci. 2013, 14, 4298–4316. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhou, L.; Wu, L.M.; Lai, M.C.; Xie, H.Y.; Zhang, F.; Zheng, S.S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Niinuma, T.; Suzuki, H.; Nojima, M.; Nosho, K.; Yamamoto, H.; Takamaru, H.; Yamamoto, E.; Maruyama, R.; Nobuoka, T.; Miyazaki, Y.; et al. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 2012, 72, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jutooru, I.; Chadalapaka, G.; Johnson, G.; Frank, J.; Burghardt, R.; Kim, S.; Safe, S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 2013, 32, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Z.; Mei, Q.; Li, X.; Guo, M.; Fu, X.; Han, W. Long non-coding RNA HOTAIR, a driver of malignancy, predicts negative prognosis and exhibits oncogenic activity in oesophageal squamous cell carcinoma. Br. J. Cancer 2013, 109, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Kasiri, S.; Bashyal, A.; Mandal, S.S. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J. Mol. Biol. 2013, 425, 3707–3722. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15 INK4B tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Montes, M.; Nielsen, M.M.; Maglieri, G.; Jacobsen, A.; Højfeldt, J.; Agrawal-Singh, S.; Hansen, K.; Helin, K.; Van De Werken, H.J.G.; Pedersen, J.S.; Lund, A.H. The lncRNA MIR31HG regulates p16 INK4A expression to modulate senescence. Nat. Commun. 2015, 6, 6967. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Vaissière, T. Epigenetic mechanisms and cancer an interface between the environment and the genome. Epigenetics 2011, 6, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T. Oncogenic transformation of human cells: Shortcomings of rodent model systems. Trends Mol. Med. 2004, 10, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Weinberg, R.A. Modelling the molecular circuitry of cancer. Nat. Rev. Cancer 2002, 2, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Combes, R.; Balls, M.; Curren, R.; Fischbach, M.; Fusenig, N.; Kirkland, D.; Lasne, C.; Landolph, J.; LeBoeuf, R.; Marquardt, H.; et al. Cell transformation assays as predictors of human carcinogenicity: The report and recommendations of ECVAM Workshop 39. ATLA Altern. Lab. Anim. 1999, 27, 745–767. [Google Scholar] [PubMed]
- Karpinets, T.V.; Foy, B.D. Tumorigenesis: The adaptation of mammalian cells to sustained stress environment by epigenetic alterations and succeeding matched mutations. Carcinogenesis 2005, 26, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Cui, H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis. Markers 2007, 23, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.; Premi, S.; Soper, C.; Platt, J.; Bosenberg, M. The Hematopoietic Stem Cell Regulatory Gene Latexin Has Tumor-Suppressive Properties in Malignant Melanoma. J. Investig. Dermatol. 2013, 133, 1827–1833. [Google Scholar] [CrossRef] [PubMed]
- Isfort, R.J.; Cody, D.B.; Kerckaert, G.A.; Tycko, B.; LeBoeuf, R.A. Role of the H19 gene in Syrian hamster embryo cell tumorigenicity. Mol. Carcinog. 1997, 20, 189–193. [Google Scholar] [CrossRef]
- Vanparys, P.; Corvi, R.; Aardema, M.J.; Gribaldo, L.; Hayashi, M.; Hoffmann, S.; Schechtman, L. Application of in vitro cell transformation assays in regulatory toxicology for pharmaceuticals, chemicals, food products and cosmetics. Mutat. Res. 2012, 744, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.W.; Barrilleaux, B.L.; Varlakhanova, N.; Bush, K.M.; Chan, V.; Knoepfler, P.S. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013, 22, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Yasaei, H.; Gilham, E.; Pickles, J.C.; Roberts, T.P.; O'Donovan, M.; Newbold, R.F. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumour-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 2013, 32, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Pickles, J.C.; Pant, K.; Mcginty, L.A.; Yasaei, H.; Roberts, T.; Scott, A.D.; Newbold, R.F. A mechanistic evaluation of the Syrian hamster embryo cell transformation assay (pH 6.7) and molecular events leading to senescence bypass in SHE cells. Mutat. Res. 2016, 802, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Maslov, A.Y.; Lee, M.; Gundry, M.; Gravina, S.; Strogonova, N.; Tazearslan, C.; Bendebury, A.; Suh, Y.; Vijg, J. 5-Aza-2′-deoxycytidine-induced genome rearrangements are mediated by DNMT1. Oncogene 2012, 31, 5172–5179. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Grusby, L.; Laird, P.W.; Magge, S.N.; Moeller, B.J.; Jaenisch, R. Mutagenicity of 5-aza-2′-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci. USA 1997, 94, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Jensen, T.J.; Garbe, J.C.; Stampfer, M.R.; Futscher, B.W. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res. 2009, 69, 5251–5258. [Google Scholar] [CrossRef] [PubMed]
- McDermott, K.M.; Zhang, J.; Holst, C.R.; Kozakiewicz, B.K.; Singla, V.; Tlsty, T.D. p16INK4a Prevents Centrosome Dysfunction and Genomic Instability in Primary Cells. PLoS Biol. 2006, 4, e51. [Google Scholar] [CrossRef] [PubMed]
- Bistulfi, G.; Pozzi, S.; Ren, M.; Rossetti, S.; Sacchi, N. A repressive epigenetic domino effect confers susceptibility to breast epithelial cell transformation: Implications for predicting breast cancer risk. Cancer Res. 2006, 66, 10308–10314. [Google Scholar] [CrossRef] [PubMed]
- Corlazzoli, F.; Rossetti, S.; Bistulfi, G.; Ren, M.; Sacchi, N. Derangement of a factor upstream of RARα triggers the repression of a pleiotropic epigenetic network. PLoS ONE 2009, 4, e4305. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Xu, X.C. Benzo[a]pyrene diol epoxide suppresses retinoic acid receptor-β2 expression by recruiting DNA (cytosine-5-)-methyltransferase 3A. Mol. Cancer 2010, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Severson, P.L.; Tokar, E.J.; Vrba, L.; Waalkes, M.P.; Futscher, B.W. Agglomerates of aberrant DNA methylation are associated with toxicant-induced malignant transformation. Epigenetics 2012, 7, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Treas, J.; Tyagi, T.; Singh, K.P. Chronic exposure to arsenic, estrogen, and their combination causes increased growth and transformation in human prostate epithelial cells potentially by hypermethylation-mediated silencing of MLH1. Prostate 2013, 73, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Benbrahim-Tallaa, L.; Waterland, R.A.; Dill, A.L.; Webber, M.M.; Waalkes, M.P. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de Novo DNA methyltransferase. Environ. Health Perspect. 2007, 115, 1454–1459. [Google Scholar] [PubMed]
- Ji, W.; Yang, L.; Yu, L.; Yuan, J.; Hu, D.; Zhang, W.; Yang, J.; Pang, Y.; Li, W.; Lu, J.; et al. Epigenetic silencing of O6-methylguanine DNA methyltransferase gene in NiS-transformed cells. Carcinogenesis 2008, 29, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Damiani, L.A.; Yingling, C.M.; Leng, S.; Romo, P.E.; Nakamura, J.; Belinsky, S.A. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 2008, 68, 9005–9014. [Google Scholar] [CrossRef] [PubMed]
- Tellez, C.S.; Juri, D.E.; Do, K.; Bernauer, A.M.; Thomas, C.L.; Damiani, L.A.; Tessema, M.; Leng, S.; Belinsky, S.A. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011, 71, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Rao, M.; Humphries, A.E.; Hong, J.A.; Liu, F.; Yang, M.; Caragacianu, D.; Sehrump, D.S. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009, 69, 3570–3578. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, Y.; Boggs, S.E.; Belinsky, S.A.; Liu, J. Cigarette smoke induces demethylation of prometastatic oncogene synuclein-gamma in lung cancer cells by downregulation of DNMT3B. Oncogene 2007, 26, 5900–5910. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Xu, H.; Shan, J.; Tao, Y.; Hong, J.A.; Inchauste, S.; Zhang, M.; Kunst, T.F.; Mercedes, L.; Schrump, D.S. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J. Clin. Investig. 2013, 123, 1241–1261. [Google Scholar] [CrossRef] [PubMed]
- Brodie, S.A.; Li, G.; El-Kommos, A.; Kang, H.; Ramalingam, S.S.; Behera, M.; Gandhi, K.; Kowalski, J.; Sica, G.L.; Khuri, F.R.; et al. Class I HDACs are mediators of smoke carcinogen-induced stabilization of dnmt1 and serve as promising targets for chemoprevention of lung cancer. Cancer Prev. Res. 2014, 7, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Cartularo, L.; Kluz, T.; Cohen, L.; Shen, S.S.; Costa, M. Molecular mechanisms of malignant transformation by low dose cadmium in normal human bronchial epithelial cells. PLoS ONE 2016, 11, e0155002. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.S.; Swanson, H.I. Dioxin-induced immortalization of normal human keratinocytes and silencing of p53 and p16INK4a. J. Biol. Chem. 2004, 279, 27187–27193. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Li, Y.; Zhang, A.; Wang, B.; Xu, Y.; Xu, W.; Zhao, Y.; Luo, F.; Liu, Q. The acquisition of cancer stem cell-like properties and neoplastic transformation of human keratinocytes induced by arsenite involves epigenetic silencing of let-7c via Ras/NF-kB. Toxicol. Lett. 2014, 227, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Wu, M.M.; Chan, M.W.Y.; Pu, Y.S.; Chen, C.J.; Lee, T.C. Long-term low-dose exposure of human urothelial cells to sodium arsenite activates lipocalin-2 via promoter hypomethylation. Arch. Toxicol. 2014, 88, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Wozniak, R.J.; Eblin, K.E.; Wnek, S.M.; Gandolfi, A.J.; Futscher, B.W. Epigenetic mediated transcriptional activation of WNT5A participates in arsenical-associated malignant transformation. Toxicol. Appl. Pharmacol. 2009, 235, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Ye, S.; Pan, Y.; Bao, Y.; Chen, H.; Shao, C. Long-term cadmium exposure leads to the enhancement of lymphocyte proliferation via down-regulating p16 by DNA hypermethylation. Mutat. Res. 2013, 757, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Kleinstreuer, N.C.; Yang, J.; Berg, E.L.; Knudsen, T.B.; Richard, A.M.; Martin, M.T.; Reif, D.M.; Judson, R.S.; Polokoff, M.; Dix, D.J.; et al. Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat. Biotechnol. 2014, 32, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Tice, R.R.; Austin, C.P.; Kavlock, R.J.; Bucher, J.R. Improving the human hazard characterization of chemicals: A Tox21 update. Environ. Health Perspect. 2013, 121, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. Phenotypic vs. Target-based drug discovery for first-in-class medicines. Clin. Pharmacol. Ther. 2013, 93, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Thorne, N.; McKew, J.C. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today 2013, 18, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Björkman, M.; Östling, P.; Härmä, V.; Virtanen, J.; Mpindi, J.P.; Rantala, J.; Mirtti, T.; Vesterinen, T.; Lundin, M.; Sankila, A.; et al. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene 2012, 31, 3444–3456. [Google Scholar] [CrossRef] [PubMed]
- Engle, S.J.; Vincent, F. Small Molecule Screening in Human Induced Pluripotent Stem Cell-derived Terminal Cell Types. J. Biol. Chem. 2014, 289, 4562–4570. [Google Scholar] [CrossRef] [PubMed]
- National Center for Advancing Translational Sciences (NCATS). Assay Development & Screening; Assay Guidance Criteria. 2017. Available online: https://ncats.nih.gov/preclinical/drugdev/assay#criteria (accessed on 30 May 2017).
- Larman, H.B.; Scott, E.R.; Wogan, M.; Oliveira, G.; Torkamani, A.; Schultz, P.G. Sensitive, multiplex and direct quantification of RNA sequences using a modified RASL assay. Nucleic Acids Res. 2014, 42, 9146–9157. [Google Scholar] [CrossRef] [PubMed]
- Grimm, F.A.; Iwata, Y.; Sirenko, O.; Chappell, G.A.; Wright, F.A.; Reif, D.M.; Braisted, J.; Gerhold, D.L.; Yeakley, J.M.; Shepard, P.; et al. A chemical-biological similarity-based grouping of complex substances as a prototype approach for evaluating chemical alternatives. Green Chem. 2016, 18, 4407–4419. [Google Scholar] [CrossRef] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.J.; Barrier, M.; Jeffay, S.; Nichols, H.P.; Kleinstreuer, N.C.; Singh, A.V.; Reif, D.M.; Sipes, N.S.; Judson, R.S.; Dix, D.J.; et al. Evaluation of 309 environmental chemicals using a mouse embryonic stem cell adherent cell differentiation and cytotoxicity assay. PLoS ONE 2011, 6, e18540. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Shou, D.; Huang, R.; Khuc, T.; Dai, S.; Zheng, W.; Klumpp-Thomas, C.; Xia, M. Identification of HDAC Inhibitors Using a Cell-Based HDAC I/II Assay. J. Biomol. Screen. 2016, 21, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.A.; Wichterman-Kouznetsova, J.; Varghese, D.; Huang, R.; Huang, W.; Becker, M.; Austin, C.P.; Inglese, J.; Johnson, R.L.; Martinez, E.D. Identification of small molecule modulators of gene transcription with anticancer activity. ACS Chem. Biol. 2014, 9, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Shaw, A.Y.; Wang, S.G.; Hsu, C.C.; Teng, I.W.; Tseng, M.J.; Huang, T.H.; Chen, C.S.; Leu, Y.W.; Hsiao, S.H. Identification of novel DNA methylation inhibitors via a two-component reporter gene system. J. Biomed. Sci. 2011, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Gertych, A.; Oh, J.H.; Wawrowsky, K.A.; Weisenberger, D.J.; Tajbakhsh, J. 3-D DNA methylation phenotypes correlate with cytotoxicity levels in prostate and liver cancer cell models. BMC Pharmacol. Toxicol. 2013, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Platchek, M.; Kement, B.; Bee, W.T.; Truong, M.; Zeng, X.; Hung, S.; Lin, H.; Morrow, D.; Kallal, L.A.; et al. A novel approach applying a chemical biology strategy in phenotypic screening reveals pathway-selective regulators of histone 3 K27 tri-methylation. Mol. Biosyst. 2014, 10, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Luense, S.; Denner, P.; Fernández-Montalván, A.; Hartung, I.; Husemann, M.; Stresemann, C.; Prechtl, S. Quantification of histone H3 lys27 trimethylation (H3K27me3) by high-throughput microscopy enables cellular large-scale screening for small-molecule EZH2 inhibitors. J. Biol. Screen. 2015, 20, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Mulji, A.; Haslam, C.; Brown, F.; Randle, R.; Karamshi, B.; Smith, J.; Eagle, R.; Munoz-Muriedas, J.; Taylor, J.; Sheikh, A.; et al. Configuration of a High-Content Imaging Platform for Hit Identification and Pharmacological Assessment of JMJD3 Demethylase Enzyme Inhibitors. J. Biomol. Screen. 2012, 17, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Greally, J.M.; Jacobs, M.N. In vitro and in vivo testing methods of epigenomic endpoints for evaluating endocrine disruptors. Altex 2013, 30, 445–471. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Beland, F.A.; Pogribny, I.P. Role of epigenetic events in chemical carcinogenesis-a justification for incorporating epigenetic evaluations in cancer risk assessment. Toxicol. Mech. Methods 2011, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
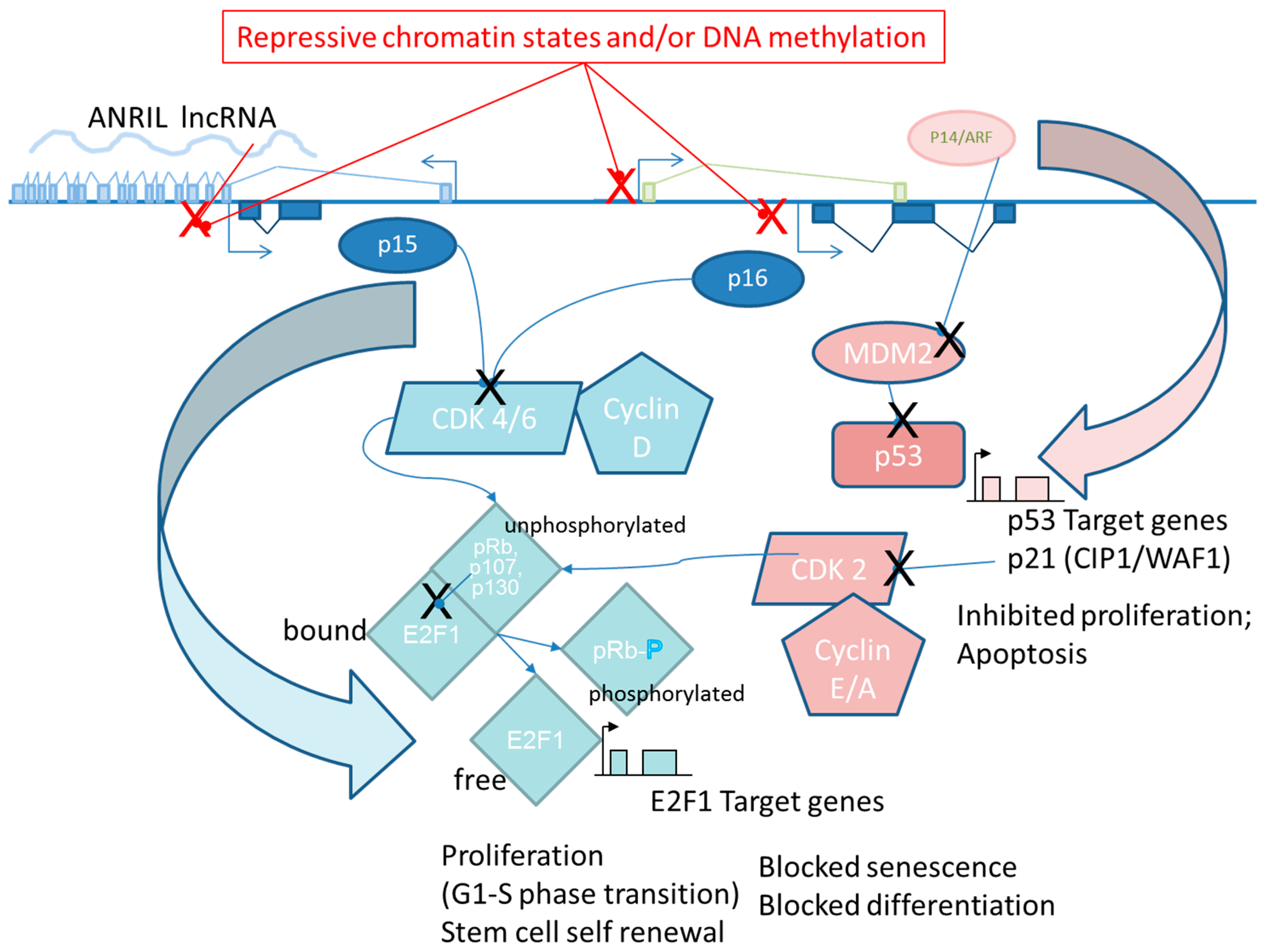
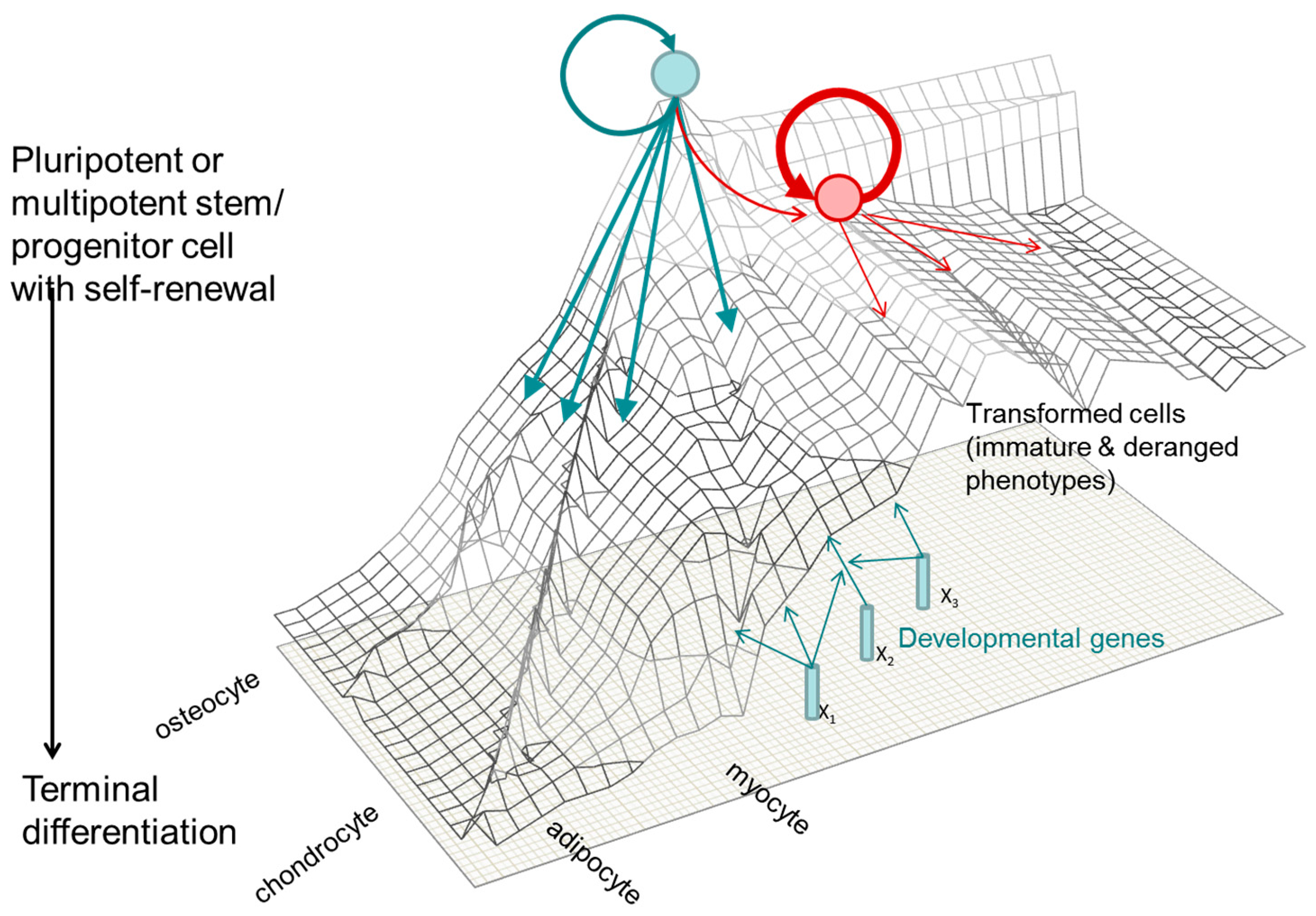
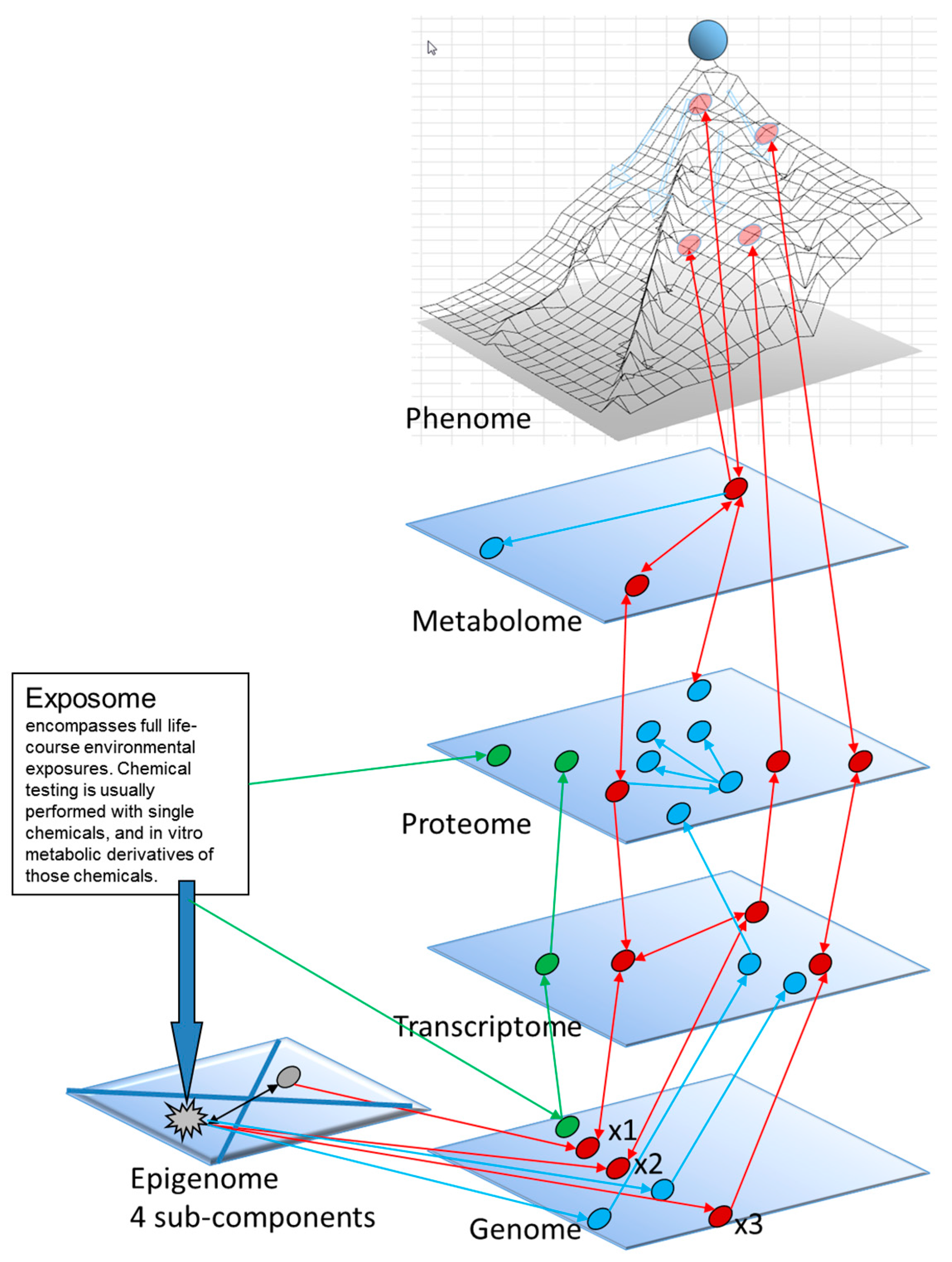
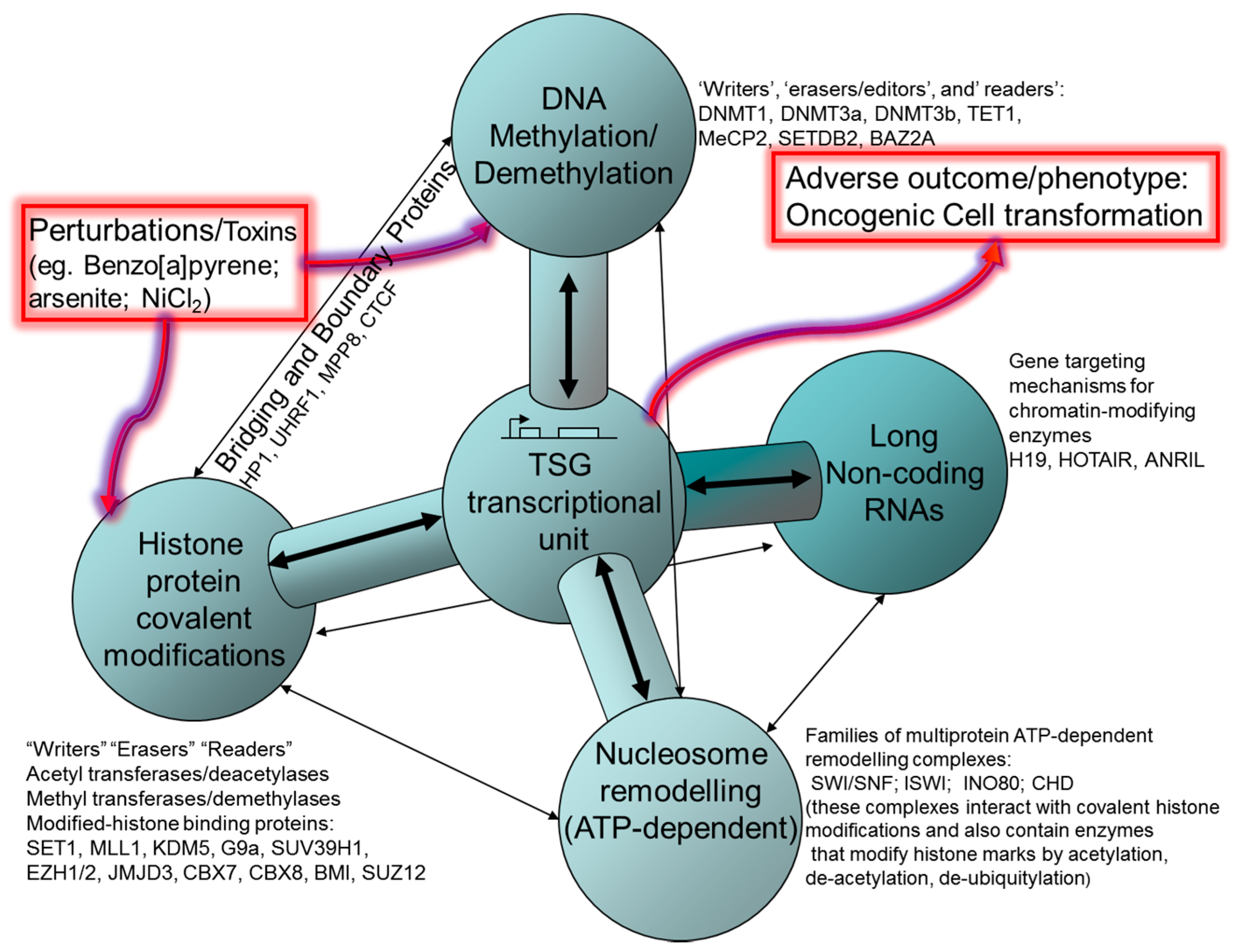
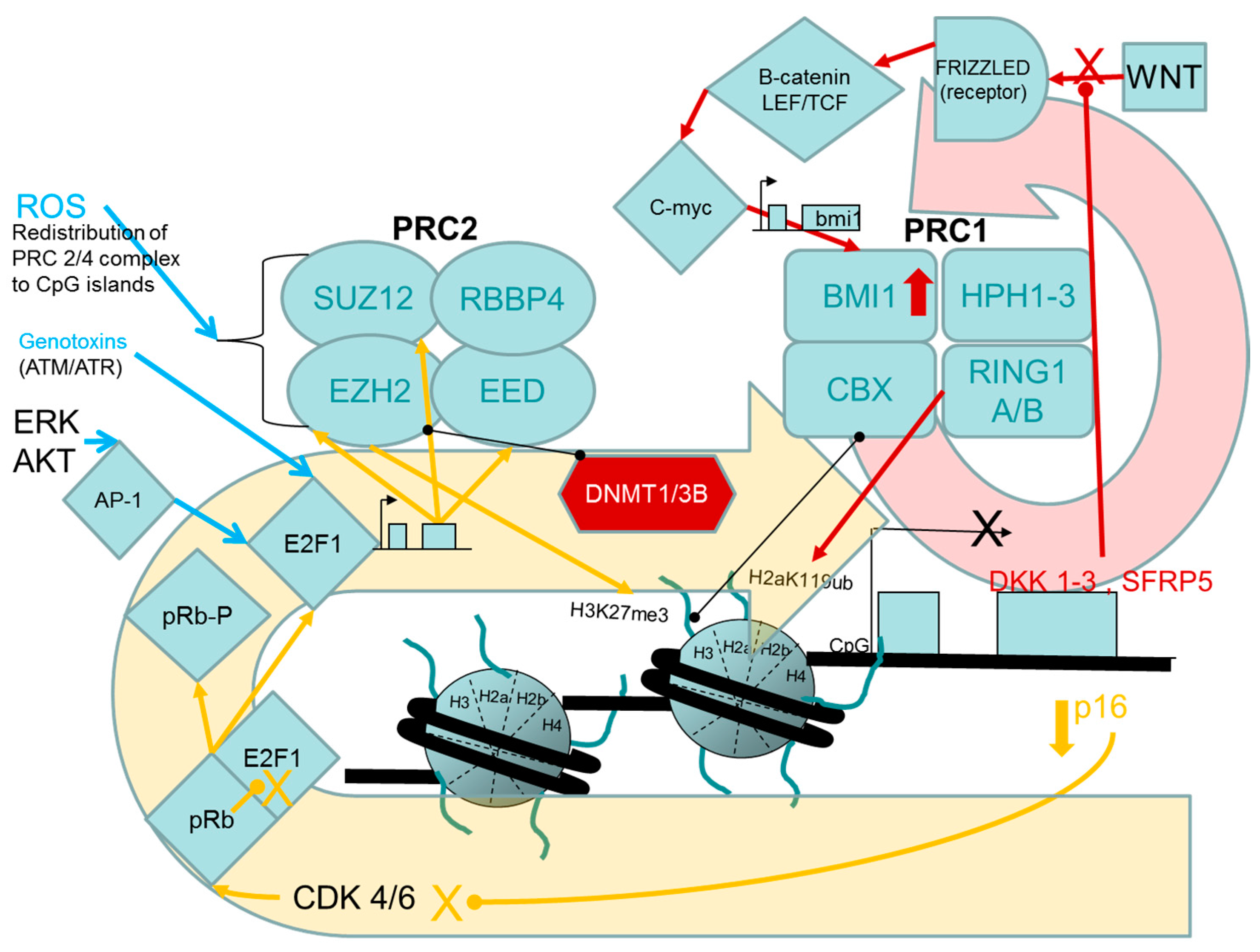
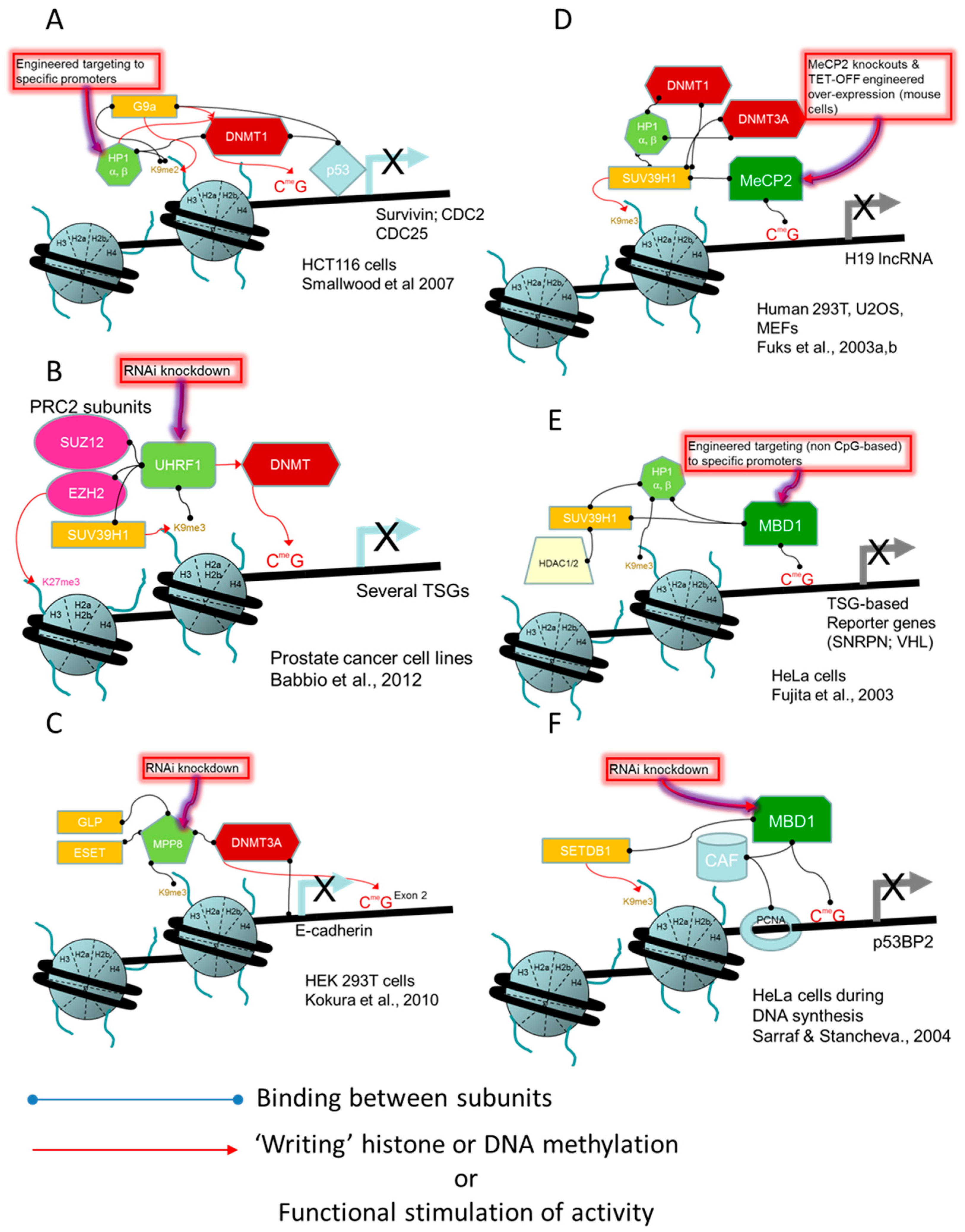
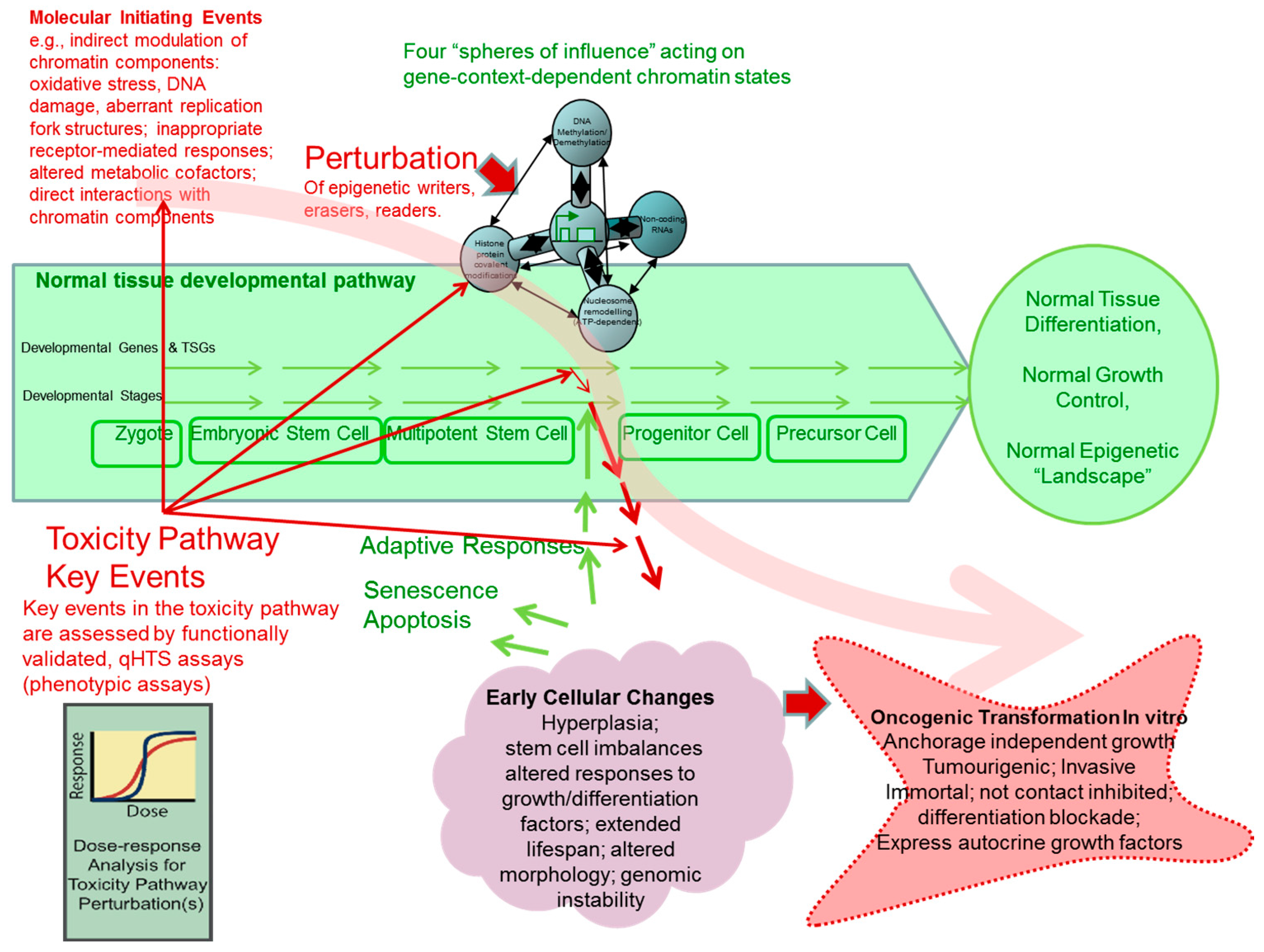

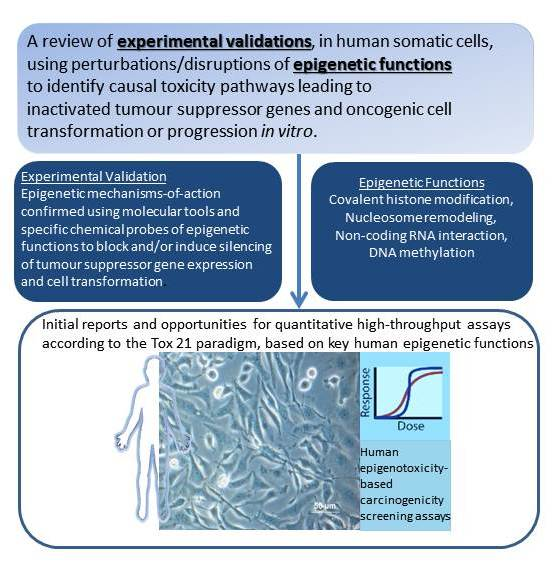{kind=link}
{kind=link}
{kind=link}
{kind=link}
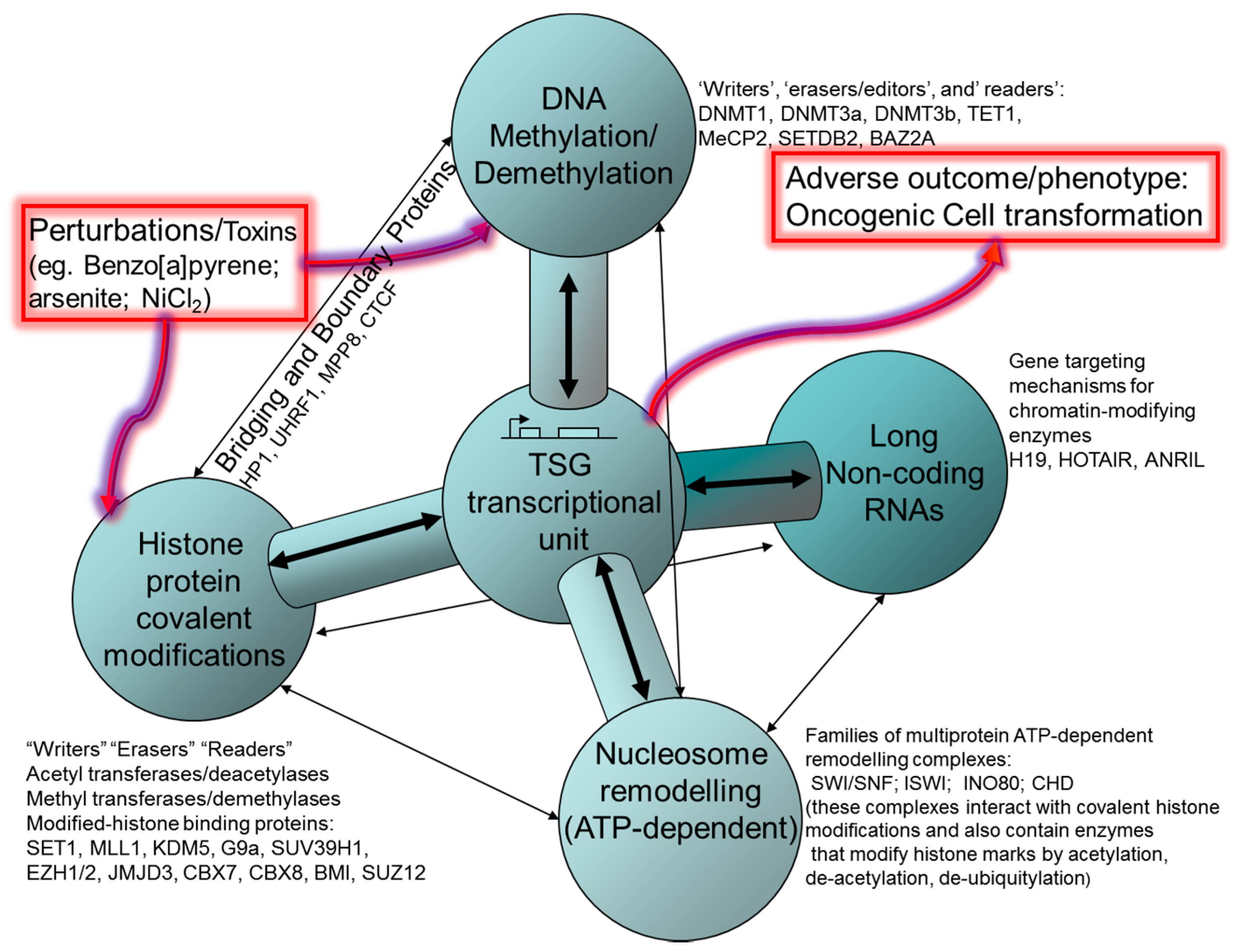{kind=link}
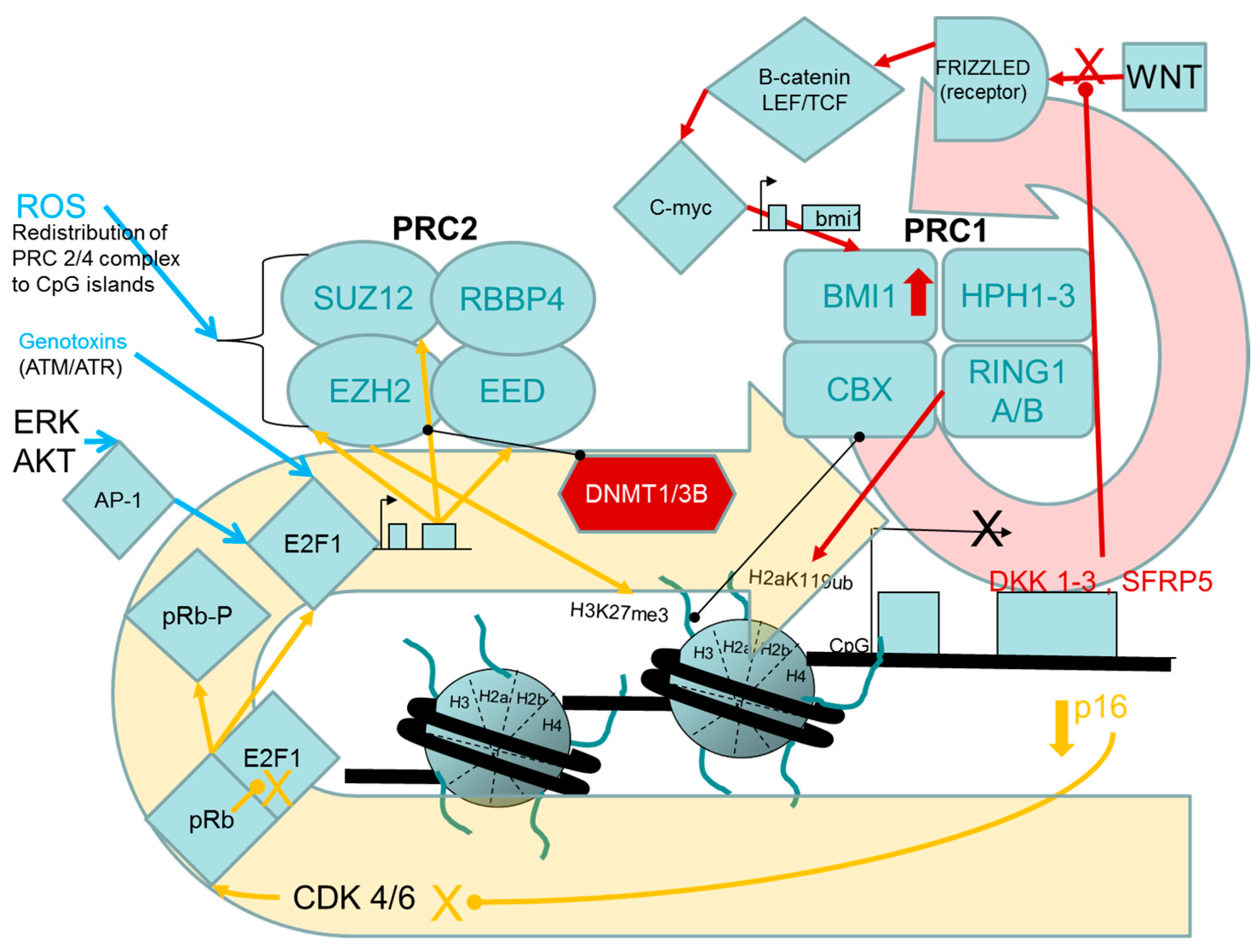{kind=link}
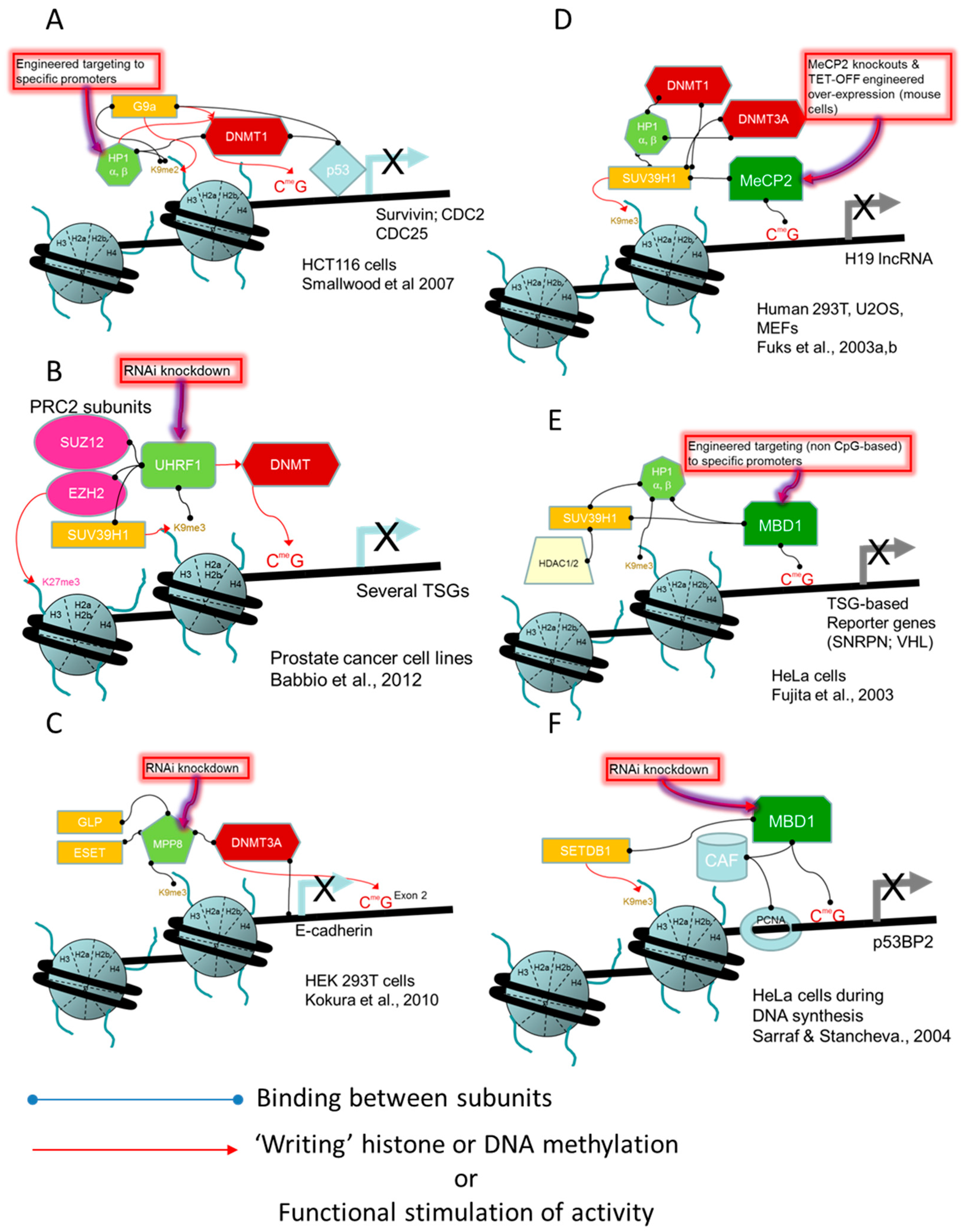{kind=link}
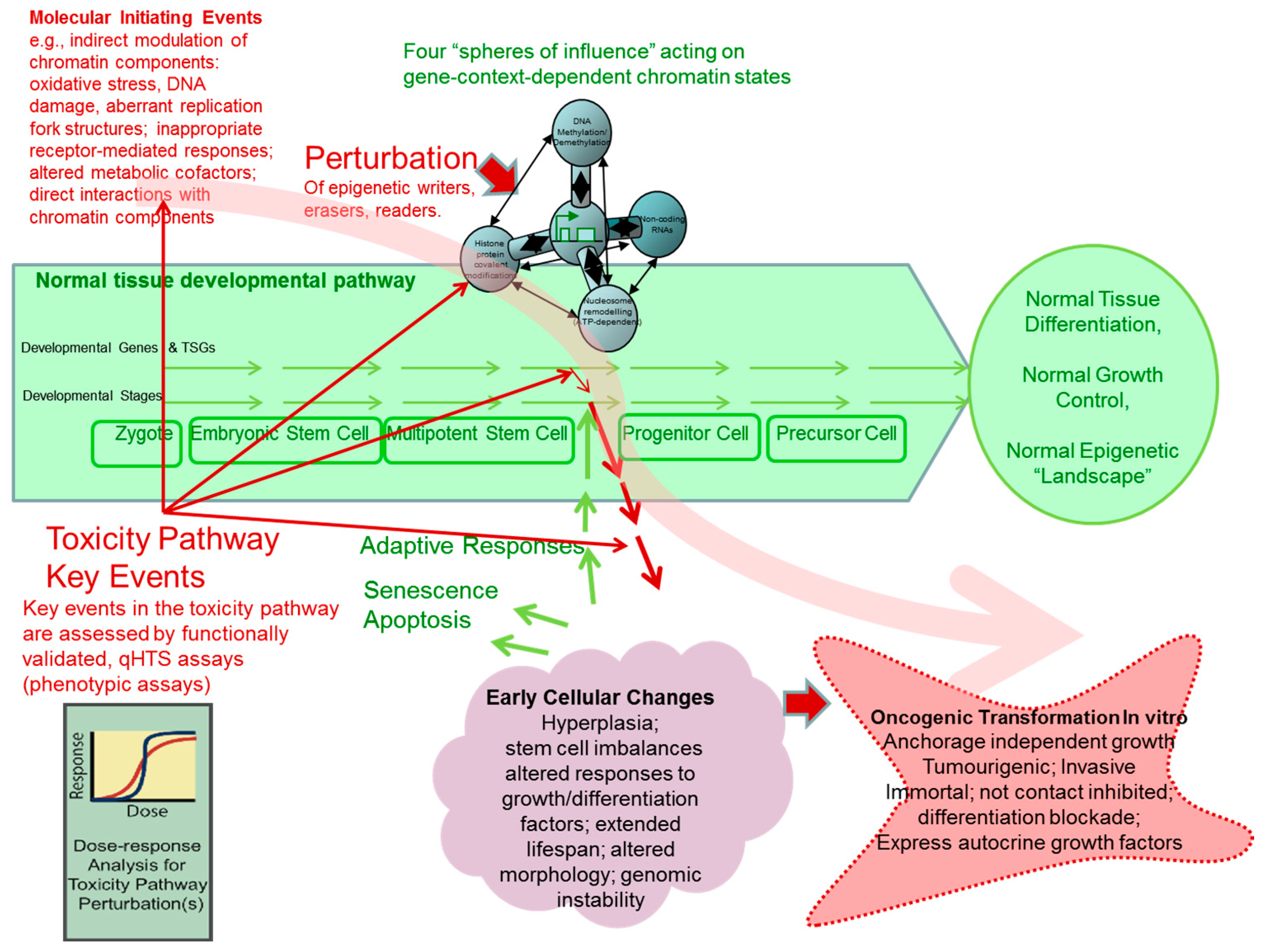{kind=link}
| Histone Modification/Marks (Associations with Activated or Repressed Transcription) | “Writer” Enzymes (Transferases) | “Eraser” Enzymes | “Reader” Proteins and Protein Domains that Transduce Epigenetic Information |
|---|---|---|---|
| 1. Lysine Acetylation (active transcriptional start sites, enhancers) | Lysine acetyl transferases (KAT) | Histone deacetylases (HDACs) | Tandem bromodomains; (e.g., BRG1 ATPase of SWI/SNF remodelling complex) |
| H3K9ac | KAT2A | SIRTUIN1,6 (class III HDAC; NAD+ dependent) | Tandem PHD fingers |
| H3K27ac | p300, CBP | ||
| H4K16ac | MOZ/MYST3/KAT6A | SIRTUIN 1,2 | |
| 2. Lysine Methylation | lysine methyltransferases (KMT) | Lysine demethylases (KDM) | Bromodomains |
| H3K4me3 (active transcriptional start sites/TSS) | SETD1A,B; MLL1/KMT2A; MLL2; MLL3; MLL4 | KDM4/JMJD2; KDM5/JARID1A,B | PHD fingers (inhibitor of growth/ING); Tudor domains (53BP1); |
| H3K4me1 (activated enhancers) | SETD7 | KDM1/LSD1 (H3K4me2, H3K4me1) | ZF-CW proteins; WD40 |
| H3K36me3 (activated gene bodies) | SET2 (H3K36me3) | JHDM1B/KDM2B (H3K36me3) | PWWP (e.g., DNMT3 A/B, bind H3K36me3) |
| H3K9me3 (repressed; in constitutive heterochromatin, e.g., pericentomeric, inactive X) | EHMT1/GLP, EHMT2/G9A (H3K9me1,2); SUV39H1, SUV39H2, SETDB1 and SETDB2 (H3K9me2, me3) | KDM3/JMJD1 (H3K9me2, H3K9me1); KDM4A/JUMD2A (H3K9me3) | UHRF1, HP1 (an heterochomatin adaptor protein); MPP8 |
| H3K27me3 (Polycomb- repressed TSS) | EZH1,2 (H3K27me1–me3) | KDM6/UTX, JMJD3 (H3K27me3) | Chromodomains (PRC1/CBX7,CBX8, HP1 bind H3K27me3); |
| 3. Serine/Threonine and Tyrosine Phosphorylation; H3S10, H3S28 | Protein kinases (ATM/ATR, PKC, AURORA B, JAK2, etc.) | Protein tyrosine and serine/threonine phosphatases DUSP1 | Chromoshadow domains (phosphotyrosine); 14-3-3, BRCA1 C Terminus (BRCT) domain (phosphoserine or phosphothreonine) |
| 4. Lysine Ubiquitination H2AK119ub1 (repressed gene transcription) | Ubiquitin E2 conjugases; E3 ligases (e.g., polycomb repressive complex 1 RING1A/B protein; UHRF1) | Ubiquitin-specific proteases MYSM1 | JARID2 from the PRC2 complex [48]. |
| 5. Arginine Methylation | Protein Arginine methyltransferases (PRMTs) | Histone demethylases | Tudor domains (for asymmetrically dimethylated arginine); PHD domain (for symmetrically dimethylated arginine) |
| TSG Target | “Reader/Writer/Erasers” Experimentally Perturbed | Experimental Perturbation | PRC or Histone Mark Verification | Cell Model | Adverse Phenotype Modified by Perturbation: ← Reversal of Oncogenic Phenotype; → Enhancement of Oncogenic Phenotype | Reference |
|---|---|---|---|---|---|---|
| p16/INK4A | SUZ12, CBX8, BMI1; mRNA down-regulation; loss of binding to p16 locus | Knockdown (stable shRNA expression from retroviral vector) | Knockdowns of CBX8 or BMI1 reduced the recruitment of both proteins and the p16/INK4A locus | TIG3-T telomerase-immortalized human fibroblasts | ← p16 up-regulation with decrease growth of TIG3-T cells and reduction of colony formation in U2OS cells | [91] |
| p16/INK4A | JMJD3 (H3K27me3 demethylase) down-regulation or up-regulation | Knockdown (stable shRNA expression from retroviral vector); up-regulation by ectopic expression | Exogenous JMJD3 was recruited to the p16/INK4A locus, caused reduction in H3K27me3 | 1° human fibroblasts | → p16 down-regulation and partial bypass of RAS oncogene-induced senescence response ← Induced senescence | [92] |
| p16/INK4A and ARF | CBX7 up-regulation/down-regulation | Over expression and knockdown (stable shRNA expression from retroviral vector) | No measures of changes to CBX7 abundance at p16 or ARF loci | Variety of human 1° cells | → CBX7 overexpression causes p16 down-regulation and extended replicative capacity CBX7 repression caused severely impaired growth | [93] |
| p16/INK4A | BMI1 up-regulation | Overexpression by expression vector transfection | No measures of histone ubiquitination on p16/INK4A chromatin | 1° human mammary epithelial cells | → BMI1 overexpression suppressed p16/INK4A expression and weakly induced hTERT activity; p16/INK4A was shown to be required replicative lifetime extension | [94] |
| Writers * | Editors/Erasers ** | Readers | ||
|---|---|---|---|---|
| Share methyl-CpG- binding domain (MBD): | Bind hemimethylated DNA through their SET and RING finger associated domain | Share zinc finger DNA binding domain | ||
| DNMT1 (Maintenance) | TEt1 (acute myeloid leukemia) | MeCP2 (Rett syndrome; prostate cancer) | UHRF1 (Binds hemimethylated DNA, H3K9me3, and H3R2. E3 ubiquitin ligase. Essential to maintenance DNA methylation) | KAISO (interfere with WNT signaling pathway) |
| DNMT2 (no catalytic site) | TEt2 | MBD1 | UHRF2 (prefers fully hydroxymethylated over hemihydroxymethylated DNA) | ZBTB4 (KAISO paralogs) |
| DNMT3A (de novo) | TEt3 | MBD2 | ZBTB38 (paralogs) | |
| DNMT3B (de novo) | MBD3 | ZFP57 (imprints) | ||
| DNMT3L (cooperates with 3A, 3B) | MBD4 (glycosylase) | |||
| MBD5 | ||||
| MBD6 | ||||
| SETDB1 (H3K9MT) | ||||
| SETDB2 (H3K9MT) | ||||
| BAZ2A (NoRC complex; rDNA, centromeres, telomeres) | ||||
| BAZ2B | ||||
| Human Cells | Epigenetic Target | Transforming Event | Outcomes and Evidence of Cell Transformation | Reference |
|---|---|---|---|---|
| Colorectal cancer cell lines (p53 wild-type HCT116 and LS174T, p53 mutant SW480) | DNMTs | Folic acid supplementation. | Inverse dose-response effects on global genomic methylation but positive dose-response effects on colonosphere formation in vitro in HCT116 and LS174T cells, but not in the SW480 cell line. | [144] |
| Hepatocarcinoma tissues and SMMC-7721cell line | MeCP2 | Down- and up-regulation by siRNA and plasmid, respectively. | MeCP2 over-expressed in HCC tissues compared to adjacent noncancerous tissues. Up- and down-regulation increase or decrease proliferation, respectively, through modulation of the Sonic hedgehog signaling pathway. | [145,146] |
| Normal prostate epithelial cells and cancer prostate cell lines (LNCaP, PC3, DU-145) | MeCP2 | Down- and up-regulation by shRNA and plasmid, respectively. | Based on growth curves and colony formation assays, down-regulation reduces growth of the normal cells and prostate cell lines, while up-regulation tested in LNCaP cells induce androgen-independent growth and soft-agar colony formation associated with c-MYC protein stabilization. | [143] |
| Pancreatic (PANC-1, BxPC-3) and prostate cancer (PC3) cell lines | MBD1 | Down-regulation by siRNA. | Down-regulation inhibits cell growth and invasion, and induces apoptosis in pancreatic but not in the prostate cell line. MBD1 would be oncogenic in pancreatic, but tumour suppressive in the prostate cell line. | [147] |
| HBEC3 non-malignant bronchial epithelial cells, and H358 lung cancer cell line | DNMT1 | DNMT1 overexpression and DNMT1 mutant (deletion of the replication foci targeting sequence). | Overexpression of normal and of mutant DNMT1 increased anchorage-independent soft-agar colony formation. Pericentromeric DNA repeated sequence (SAT2) hypomethylation, but tumour suppressor gene promoter hypermethylation. | [148] |
| Non-tumorigenic astrocytes (Astro#40), mammary epithelial cells (MCF10A), lung fibroblasts (Wi-38), and mesothelial cells (Met5A) | DNMT1 | Disruption of the DNMT1/PNCA/UHRF1 complex by interfering DNMT1 plasmid. | Induction of cancer phenotype including increased cell proliferation, resistance to irradiation-induced ceall death, injections of cells caused tumours in mice. Global and promoter hypomethylation preceeding chromosomal aberrations. | [148,149] |
| Human bronchial epithelial cells (HBEC2 immortalized with hTERT and CDK4), and 22 non-small-cell lung cancer cell lines | DNMT3B | Overexpressed (5-20 fold over control) to human tumor levels. | Accelerate carcinogen-induced oncogenic transformation (colony formation in soft agar and acquired mesenchymal-like morphology). Three-fold increase in the number of genes epigenetically silenced. | [150] |
| Mesenchymal stem cell lineages | DNMT1 | (i) Engineered methylated ssDNA targetted to promoter attracting DNMT1. (ii) 5-aza-dC induced promoter hypomethylation. | Gene down-regulation (HIC1, RASSF1A). Enhanced growth in soft agar. Increased cell migratory transwell invasion. Drug resistence. Soft tissue sarcoma in nude mice. | [151] |
| Gastric (AGS, SGC7901, MKN45), colorectal (HT29, HT116, RKO, DLD1, SW620) | UHRF1 | cDNA up-regulation, or short-hairpin RNA down-regulation. | In both gastric and colorectal cancer cell lines, down-regulation of UFRF1 reduces cell proliferation, migration and invasion properties, and tumour growth in athymic mice. | [48,152,153] |
| Bladder invasive (253J, T24, and KU7) and non-invasive (RT4, RT112, DSH1) cancer cell lines | UHRF1 | Engineered up-regulation, or siRNA down-regulation. | Increase and decrease transwell invasion. UHRF1 up-regulation hypermethylates and silences the metastasis suppressor KiSS1. | [154] |
| Hepatocarcinoma cell lines (HepG2, HCCLM3) | UHRF1 | Down-regulation by siRNA | Down-regulation decrease cell proliferation, transwell migration and invasion, and tumour growth in nude mice. Up-regulated in patients with hepatocarcinomas. Patients with elevated expression had shorter relapse-free survival time. | [155] |
| Breast cancer cell line (MDA-MB-231-1833) | TET1 and its target HOXA gene cluster | Up-regulation of TET1 leading to its auto-up-regulation. | Suppress cell invasion in vitro, and xenograft tumour growth and invasiveness. | [156] |
| Non-malignant bronchial epithelial cells (HBEC3) and metastatic non-small cell lung cancer cell ine (H1299) | TET1 | Transfection of RAS oncogene | Suppressed TET1, TSG and H19 expression, and induce soft-agar colony formation as oncogenic cell transformation. | [157] |
| MCF7 and MCF10 breast cancer and epithelial cell lines, respectively | TET1, 2, and 3. | Engineered expression of miR-22 | Down-regulation of TETs. Epithelial-mesenchymal transition driven by miR-200 in MCF10, non-metastatic to metastatic properties in MCF7 cells. | [158] |
| Primary epithelial colon cells and colon cancer cell lines (Caco-2, SW48) | TET1 | Down- and up-regulation by shRNA and Doxycycline-inducible TET1-promoter. | Down- or up-regulation promote or decrease cell proliferation, respectively. Up-regulation decreases colonosphere formation in vitro and tumor size from injected cells in nude mice. Hypermethylation and down-regulation of WNT pathway inhibitors (DKK3, DKK4). | [159] |
| Primary cells and hepatocellular carcinoma cell lines (Hep3B, MHCC97L) | SETDB1 | Down-regulation by shRNA | Reduce cell proliferation in vitro, suppressed orthotopic tumorigenicity and metastasis in vivo. Associations with clinical prognosis. | [160,161] |
| Primary cells and gastric cancer cell lines (MKN74, MKN45, AGS, NUGC3) | SETDB2 | Down-regulation by siRNA, up-regulation by vector transfection. | Decreased and increased cell proliferation, migration, and invasion, respectively. Associations with clinical prognosis. | [162,163] |
| Prostate normal epithelial (RWPE1) and metastatic cancer cell lines (PC3, LNCaP, DU145) | BAZ2A | Down-regulation by siRNA | Reduce metastatic cancer cell proliferation, invasion and migration based on in vitro assays. Associations with clinical prognosis. | [164] |
| Cell Type | Carcinogen(s) | Epigenetic Perturbations Associated with Treatment | Experiments Blocking (Reversal) or Inducing a Molecular Perturbation Associated with Chemical Treatment | Phenotypic Effects of Molecular Perturbations | References |
|---|---|---|---|---|---|
| Syrian hamster embryonic (primary) | Benzo[a]pyrene, 3-MCA (5–10 µg/mL) | H19 long non-coding RNA expression suppressed in transformed colonies; loss of HpaII DNA restriction enzyme cutting; hypermethylation | Transfection/plasmid -based re-expression (reversal) | H19 re-expression caused faster growth in vitro; slower tumour growth in vivo | [291] |
| Syrian hamster dermal (primary) | Benzo[a]pyrene (1–2 µg/mL), NiCl2 (250 µM) | p16 promoter hypermethylation; silencing of p16 expression | Reversal of p16 repression with 5-azaC treatment (demethylating) | Associated with immortalization (post-stasis/SIPS); induced re-expression associated with return to senescence | [294] |
| Human breast epithelial cells (primary) | Benzo[a]pyrene (1 µg/mL) | 10 s–100 s–1000 s stepwise DNA hyper/hypomethylation events by CHIP-microarray analysis; p16 expression suppressed | Inducing low p16 expression (shRNA knockdown) permitted more frequent bypass of stasis | Associated with post-stasis (SIPS); post- replicative senescence bypass caused by reduced p16 expression. | [299] |
| RWPE-1, UROtsa immortal human prostate epithelial and urothelial cells | Arsenite (1 µM) | 100 s–1000 s hyper/hypomethylation events; link between histone3-lysine 9-trimethylation domains of stem cells and DNA hypermethylation | – | Associated events with oncogenic transformation | [304] |
| RWPE-1 human prostate epithelial cells (immortal) | Arsenic (100 ng/mL) and Estrogen (100 pg/mL), in combination | Hypermethylation-mediated silencing of MLH1 | – | Associated with oncogenic transformation | [305] |
| RWPE-1 human prostate epithelial cells (immortal) | Cadmium chloride (10 µM) | Global hypermethylation; p16, RASSF1A promoters hypermethylation; reduced expression of p16, RASSF1A tumour suppressor mRNAs; increased DNMT activity | Reversal of p16 and RASSF1A repression with 5-azaC treatment (demethylating); partial reversal of repression with procainamide (specific DNMT1 inhibitor) | Global and gene-specific hypermethylation associated with tumourigenicity in nude mice | [306] |
| 16HBE human bronchial epithelial cells (immortal) | NiS (1–2 µg/cm2) | DNMT1 up-regulation; MGMT gene silencing; promoter hypermethylation; reduced histone 4 acetylation; reduced histone 3 lysine 9 acetylation/methylation ratio | Block DNMT1 expression with shRNA in transformed cells | Blocked DNMT1 reduced growth of the transformed cells; caused re-expression of MGMT by reversion of epigenetic changes | [307] |
| Human bronchial epithelial cells (immortal) | MNU (500 µM) and benzo[a]pyrene diol epoxide (BPDE, 50 nM) | Growth in soft agar; DNMT up-regulated; several genes with hypermethylated promoters | Block DNMT1 expression with stable shRNA;reversal of repressed miR-200 family epithelial mediators with 5-azaC treatment (demethylating) | Blocked DNMT1 expression prevented chemical transformation; reversed transformed phenotype and gene silencing | [308,309] |
| Human bronchial epithelial cells (immortal) | MNU (1 mM) and benzo[a]pyrene diol epoxide (BPDE, 100 nM) | DNMT3B overexpressing clones were transformed more frequently by chemicals; many polycomb-targeted genes (MAL, OLIGO2) were hypermethylated and stably marked with H3K27Me3 and H3K9Me2 repressive marks in transformed cells (PRC2 thought to be recruited by the hypermethylation) | Re-expression of silenced genes was achieved by treatment with 5-Aza-dC (DNA methyltransferase inhibitor) and with Trichostatin A (histone de-acetyltransferase inhibitor) | Cells transformed to anchorage-independent growth; plasmid-based re-expression of MAL and OLIGO2 suppressed anchorage-independent growth in vitro and tumour growth in nude mice | [150] |
| Human lung tumour cell lines (A549, Calu-6) | Tobacco smoke condensate (0.0005–0.003 puffs/mL) | Tumours in nude mice enhanced; recruitment of Polycomb group proteins (BMI1, SUZ12, Ezh2, SirT1) to suppressed DKK1 gene (an inhibitor of WNT pathway) | Induced WNT achieved by blocking (using stable shRNA knockdown) of DKK1 (a WNT pathway inhibitor); knockdown of Polycomb proteins relieved DKK1 suppression | Experimental WNT up-regulation enhanced tumourigenicity in nude mice: DKK1 remained silenced in tumours | [310] |
| Human lung tumour cell lines (A549; H292) | Tobacco smoke extract (0.6–2.5%) | Demethylation of synuclein-gamma (SNCG) CpG island; increased expression of SNCG in A549 SNCG-nonexpressing cells | siRNA knockdown of SNCG mRNA; exogenous expression of DNMT3B decreased SNCG expression in H292 SNCG-expressing cells with an unmethylated CpG island | SNCG knockdown during tobacco smoke extract exposure suppressed the invasive phenotype induced by the extract | [311] |
| Primary airway epithelial cells; immortal bronchial epithelial cells; lung cancer cell lines | Cigarette smoke condensate (25 µg/mL) | Repression of microRNA miR-487b expression by CSC ; up-regulation mRNA for miR-487b targets that are promoters of malignant growth; increased miR-487 genomic methylation and nucleosome occupancy | miR-487b repression reversed with 5-aza-dC methylation inhibitor | Engineered expression of miR-487b restored proliferation and invasion of lung cancer cells in vitro and in vivo | [312] |
| Immortalized human bronchial epithelial line | MNU (500 µM) and benzo[a]pyrene (50 nM) | Increased histone deacetylase expression; increased DNMT1 expression; hypermethylation of oncogenesis-related gene promoters | HDAC overexpression causes DNMT1 expression throughout cell cycle; HDAC knockdown or inhibitor (valproic acid) decreases DNMT1 levels; the inhibitor decreased methylation of silenced genes, reactivating expression | Carcinogen-induced anchorage-independent colony formation; HDAC inhibitor decreased this | [313] |
| Adenovirus 12/SV40 hybrid virus-immortalized human bronchial epithelial cell line BEAS-2B | NiCl2 (100 µM), or 1% O2 (hypoxia) | H3K9me2 repressive mark increased at Spry2 gene promoter; JMJD1A histone demethylase activity decreased; SPRY2 gene expression (mRNA and protein) decreased | SPRY2 over-expression from stable transformation of an expression vector | Increased SPRY2 expression decreased NiCl2-induced anchorage independent growth | [57] |
| Adenovirus 12/ SV40 hybrid virus-immortalized human bronchial epithelial cell line BEAS-2B | CdCl2 (10-100 nM) | Transformants displayed increased anchorage-independent growth; increased migration; MGMT down-regulated by epigenetic mechanisms, with diminished capacity to repair alkylated DNA damage | 5-aza-dC inhibitor of DNA methylation; sodium butyrate inhibitor of histone deacetylation | MGMT expression was increased in the transformed cells | [314] |
| Neonatal human epithelial keratinocytes | 2,3,7,8-TCDD (1 nM) | TCDD repressed transcription of p16/INK4A, p14/ARF, p53, Rb genes; p16 promoter DNA was methylated in response to TCDD | 5-aza-dC treatment reversed p16/INK4A and p53 repression by TCDD | -Association with extended lifespan in culture | [315] |
| Immortal adult skin keratinocytes (HaCaT) | Arsenite (1 µM) | Arsenite repressed expression of Let-7c miRNA | 5-aza-dC inhibitor of DNA methyltransferases prevented arsenite-induced repression of Let-7c miRNA; overexpression of Let-7c by transfection blocked activation of the RAS/NF-kB signalling pathway | -Overexpression of Let-7c miRNA reduced spheroid formation in non-adherent dishes and colony formation in soft agar by arsenite-transfomed cell; -overexpression of Let-7c RNA abolished tumour growth in nude mice | [316] |
| Human urothelial cells (immortal) | Sodium arsenite 0.5 µM | Overexpression due to hypomethylation of lipocalin-2 gene promoter; anchorage-independent growth in vitro | Blocked expression: stable lipocalin-silenced transformed cells | Lipocalin-silenced transformed cells showed significantly less anchorage-independent growth | [317] |
| Immortal human urothelial (UROtsa) | As(III); monomethyl arsenous acid (MMA, 50 nM); NaAsO2 (1 µM) | WNT5A gene expression greatly increased in malignantly transformed variants of UROtsa cells even in absence of the agents; oncogenic transformation was associated with decreased repressive histone marks (H3K27me3; H3K9me2) and increased activating histone marks (H3K9 and14Ac; H3K4me2) in WNT5A promoter region | Histone deacetylase inhibitors trichostatin A and sodium butyrate activated WNT5A gene expression in the hypoacetylated parental UROtsa cells; siRNA knockdown of WNT5A expression | Knockdown of WNT5A expression inhibited anchorage independent growth | [318] |
| Human B lymphoblast HMy2.CIR line | CdCl2 (5–100 nM) | Increased cell proliferation; increased DNMT1, DNMT3B mRNA; increased global DNA methylation; decreased p16 mRNA, protein; increased p16 CpG island methylation | 5-aza-dC inhibition of DNA methylatransferase reversed the repression of p16 expression | Cd-stimulated cell proliferation was totally eliminated by 5-aza-dC treatment | [319] |
| Assay Name/Epigenetic Target | Brief Methodology | Application/Throughput | Confirming Assays | Comments/Reference |
|---|---|---|---|---|
| qRT-PCR BMI1 (PRC1 component, see Section 2.1.1)—a gene negatively regulated by the BAF trithorax SWI/SNF chromatin remodelling complex | Standard RT/PCR method; 18 h cell exposure; lysis; 384 well plates; up-regulated BMI1 mRNA indicated chemical inhibition of the BAF complex. | Library of ~30,000 small molecules (novel and pharmacologically active); one concentration: 10 μM | ▪ BMI1 luciferase knock-in reporter vector; ▪ qRT-PCR ▪ esBAF-regulated gene battery with other polycomb complex genes | 20 compounds identified that mimic a KO of the Brg1 subunit of the BAF complex [141] |
| RASL—Seq (modified) RNA species encoding epigenetic writers, erasers/editors/readers | ▪ Cell lysates, 384-well plates ▪ Ligation of modified oligonucleotide probes on mRNA targets ▪ Barcoding PCR amplification ▪ Pooled samples, Next Gen Seq | ▪ Probe sets for 77 RNAs in a small molecule screening (4 doses ea.) for effects on B-cell phenotypes; ▪ Can analyse up to 36,000 samples pooled per sequencing reaction | – | Up to 100 RNAs essential to epigenetic pathways could simultaneously be monitored across 1000+ treatments at multiple doses. With fewer RNA species targeted, more detailed dose-response curves could be generated. A focus would be warranted on the 23 epigenetic pathway components covered in this review with evidence for causal participation in repressing TSG expression and increasing cell transformation [327]. A related targeted high-throughput transcriptomics analysis, for 1767 genes, has recently been applied to induced pluripotent stem cell-derived cardiomyocytes and hepatocytes exposed to DMSO-soluble extracts of 21 petroleum substances [328] |
| MethyLight high throughput RT-PCR assay methylated or unmethylated genomic sequences | ▪ Isolate genomic DNA and bisulfite convert ▪ Fluorescence-based PCR with primers and a probe that overlap potential CpG methylation sites. | ▪ Mlh1 promoter (target); ▪ Methylation assessed in a panel of 25 human tumour/normal match-paired tissue samples | Bisulfite sequencing analysis | ▪ Sensitive to 1 methylated allele among 10,000 unmethylated equivalents. ▪ Authors suggest usage for 1000 s of samples, but MethyLight results have not yet been reported in a chemical library screening. Methylated promoters controlling TSG expression that influence cell transformation would be informative targets in treated cell populations [329] |
| Immunofluorescence detection Specific cell proteins | ▪ Proteins in fixed, cultured cells are detected directly in microplates ▪ Primary antibody, secondary antibody, and DNA/cell stains; In Cell Western™ | Detection of effects on stem cell differentiated protein (myoglobin), across 4 concentrations, for 30/309 environmental chemicals | – | No reported use to detect chemical effects on essential epigenetic pathway components [330] |
| Luminogenic HDAC (class I and II) assay (HDAC-Glo I/II) | ▪ Cell-based qHTS approach ▪ Cell lines (5) cultured in 1536-well plates ▪ Cells incubated with a cell-permeable HDAC I/II substrate which is converted to a luciferase substrate by deacetylation ▪ Optimized assay conditions | ▪ NCATS Pharmaceutical Collection screened for inhibitor activity (2527 compounds, 8 concentrations each) ▪ 43 active compounds identified from diverse structural classes | Selected compounds retested at 11 concentrations in the cell-based assay and in fluorogenic HDAC isoform-specific biochemical assays | Increased HDAC I/II activities were detected in cancer-derived cell lines, as compared to an embryonic kidney cell line, indicating that there is potential to detect activation of HDAC activity by chemicals, as well as inhibition of activity [331] |
| Single-Cell Assay Name/Epigenetic Target | Brief Methodology | Application/Throughput | Confirming Assays | Comments/Reference |
|---|---|---|---|---|
| Locus Derepression Assays Epigenetically silenced reporter gene(s) promoters | ▪ Based on a GFP reporter gene under the control of the cytomegalovirus (CMV) promoter stably integrated and epigenetically silenced in the genome of C127 mouse mammary adenocarcinoma cells ▪ 1536-well format; ▪ 30 h incubation ▪ Expression detected with a laser scanning microplate cytometer | Screening of a chemical library of over 280,000 compounds, each in a 6-point concentration series; identified 550 compounds with good quality response curves | ▪ GFP mRNA expression (q/RT-PCR) was increased by a tested subset of positive compounds ▪ Selected compounds activated other silenced genes, including TSG p21/Cdkn1a, in human cancer cells. ▪ Increased activating and repressive histone methylation marks at the activated promoter | Cancer selective activity of some of the compounds was explored by global gene expression profiling [332] |
| ▪Uses a two-component reporter-gene system: (1) Trip10 gene promoter positively controls Tet repressor expression, but is silenced by targeted methylation; (2) CMV promoter with two Tet repressor binding sites permits EGFP expression when Tet repressor expression is silenced. ▪ 5 day chemical treatment of human MCF7 cells; ▪ EGFP expression/repression monitored by fluorescence microscopy and high-content image analysis. | ▪ 169 compounds with presumptive DNA methyl transferase inhibitory activity (procainamide derivatives) were screened at a single concentration ▪ 46 compounds significantly repressed EGFP expression by reactivating the silenced Tet repressor. | ▪ ELISA and Western blotting confirmed suppression of EGFP expression ▪ Demethylation of silenced, endogenous gene, Gstp1, was detected ▪ Global demethylation measured by differential microarray hybridization. | The feasibility of the two-component reporter system for chemical screening was established. This system could be adapted to report on methylation of selected TSG promoters [333] | |
| Locus Repression Assay Actively expressing reporter gene promoter | ▪ Uses a two-component reporter gene system, similar to the derepression assay, above, except that reporter component 1 is an unmethylated TSG promoter (e.g., HIC1, or RASSF1) which allows expression of the Tet repressor; expressed Tet repressor inhibits EGFP expression of reporter component 2; ▪ Human bone marrow mesenchymal stem cells were transfected and used for targeted promoter methylation experiments | Targeted methylation and silencing of either HIC1 or RASSF1 promoters permitted expression of EGFP | ▪ Targeted promoter methylation confirmed by methylation-specific PCR ▪ Targeted methylation of TSGs induced genome-wide DNA methylation changes ▪ Concurrent targeted methylation of HIC1 and RASSF1A promoters oncogenically transformed MSCs and transformation was reversed by 5-aza-dC. | High throughput chemical screening for TSG locus repression may be feasible in this two component system, although none was attempted in this report. This approach may be useful to monitor a range of TSG promoter reporter constructs, including the p16/INK4A promoter, for silencing among cells during chemical treatments. [151] |
| MeC phenotyping (3D-qDNA methylation imaging) Genomic DNA in situ | ▪ Fixed DU145 prostate cancer and Huh7 hepatocarcinoma cells in 12-well plates; ▪ Processed for anti-5-methylcytosine antibody immunofluorescence; counterstained with DAPI; ▪ Image analysis of nuclear MeC and DAPI intensity distributions and mean intensity signals | Dose-response analysis of two DNA methyltransferase inhibitors was characterized in detail. | MethyLight assay, targeting DNA repetitive elements | The authors suggest that nuclear MeC/DAPI co-distribution is a more robust measure than just MeC intensity [334] |
| Immunofluorescence detection/high content imaging assays Global histone marks in situ | ▪ 384 well plates; fixed cells, human breast epithelial cancer; ▪ Anti H3K27me3 primary antibody; Alexa Fluor secondary antibody; nuclear stain; ▪ High content imaging | ▪ Screening of a Biologically Diverse Compound Set (5853 compounds), each at 5 micromolar ▪ 467 H3K27me3 enhancers; 28 H3K27me3 suppressors identified. | “Hits” were confirmed by an 11-point dose response curve and in further biochemical assays with peptide or nucleosome targets for EZH2 methyltransferase activity. | ▪ No cellular effects were measured for the compounds that affect H3K27me3 levels; ▪ Many active compounds may have “upstream” pathway activities controlling EZH2 enzyme activity. Other repressive HPTM targets controlling TSG silencing and cell transformation (H3K9me2/3) could be assayed in a similar manner [335] |
| ▪ 384-well plates; fixed cells; ▪ MDA-MB231, MCF7 breast adenocarcinoma and Hela cervical epithelium cell lines ▪ H3K27me3-, H3K27ac-specific primary antibody (and others); fluorescent-conjugated secondary antibody; ▪ Confocal image acquisition | Screening of 9 pyridone EZH2 inhibitors as proof-of-concept. | – | ▪ The authors suggest that miniaturized assay (1536 wells) would permit large-scale screening of several million compounds [336] | |
| ▪ Exogenous expression of JMJD3 H3K27me3 demethylase (Table 1, eraser) in human embryonic kidney HEK293 cells; 384-well plates; fixed cells ▪ H3K27me3-specific primary antibodies (and others) and species-specific fluorescent secondary antibodies ▪ Quantitative imaging analysis | Screening of an 87,500 compound library at 10 micromolar; 3524 “hits” inhibiting demethylase activity were identified | “Hits” were confirmed by an 11-point dose response curve, using the same assay and in a biochemical assay for JMJD3 demethylase activity | ▪ The single concentration screening design was shown to be reproducible in two independent screenings [337] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parfett, C.L.; Desaulniers, D. A Tox21 Approach to Altered Epigenetic Landscapes: Assessing Epigenetic Toxicity Pathways Leading to Altered Gene Expression and Oncogenic Transformation In Vitro. Int. J. Mol. Sci. 2017, 18, 1179. https://doi.org/10.3390/ijms18061179
Parfett CL, Desaulniers D. A Tox21 Approach to Altered Epigenetic Landscapes: Assessing Epigenetic Toxicity Pathways Leading to Altered Gene Expression and Oncogenic Transformation In Vitro. International Journal of Molecular Sciences. 2017; 18(6):1179. https://doi.org/10.3390/ijms18061179
Chicago/Turabian StyleParfett, Craig L., and Daniel Desaulniers. 2017. "A Tox21 Approach to Altered Epigenetic Landscapes: Assessing Epigenetic Toxicity Pathways Leading to Altered Gene Expression and Oncogenic Transformation In Vitro" International Journal of Molecular Sciences 18, no. 6: 1179. https://doi.org/10.3390/ijms18061179
APA StyleParfett, C. L., & Desaulniers, D. (2017). A Tox21 Approach to Altered Epigenetic Landscapes: Assessing Epigenetic Toxicity Pathways Leading to Altered Gene Expression and Oncogenic Transformation In Vitro. International Journal of Molecular Sciences, 18(6), 1179. https://doi.org/10.3390/ijms18061179