
Nanosecond-Pulsed DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress



 ,
,
Abstract
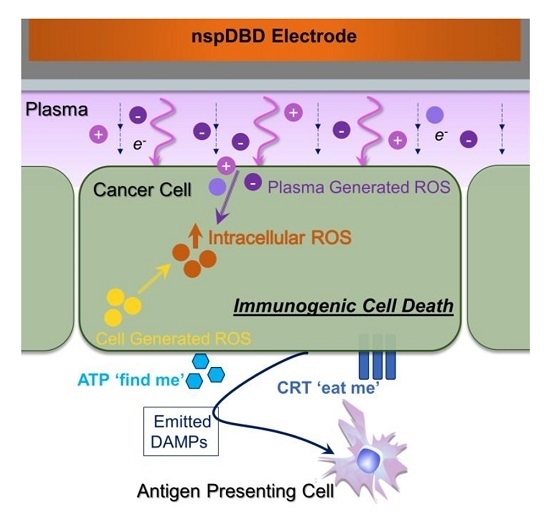
:
1. Introduction
2. Results
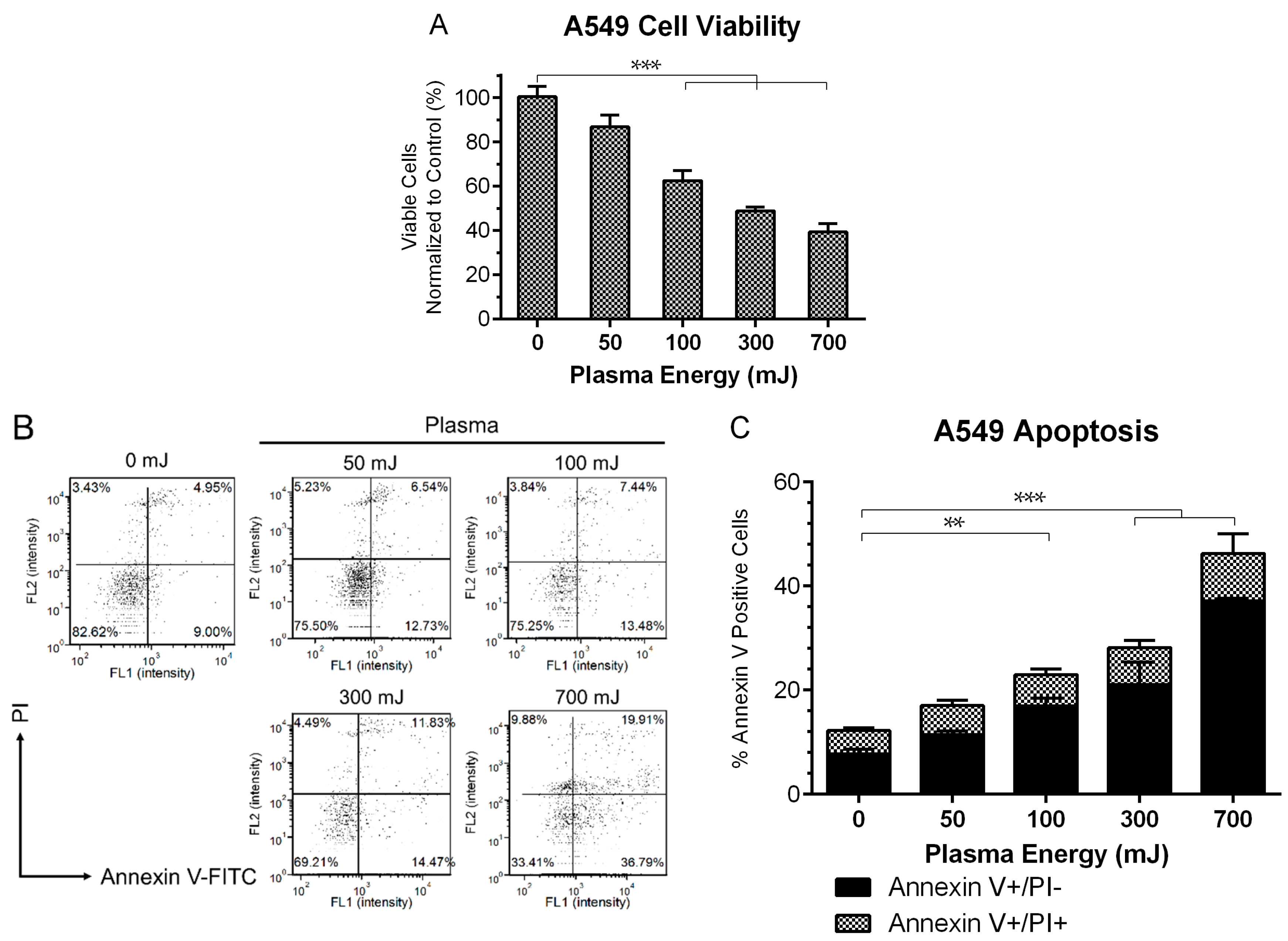
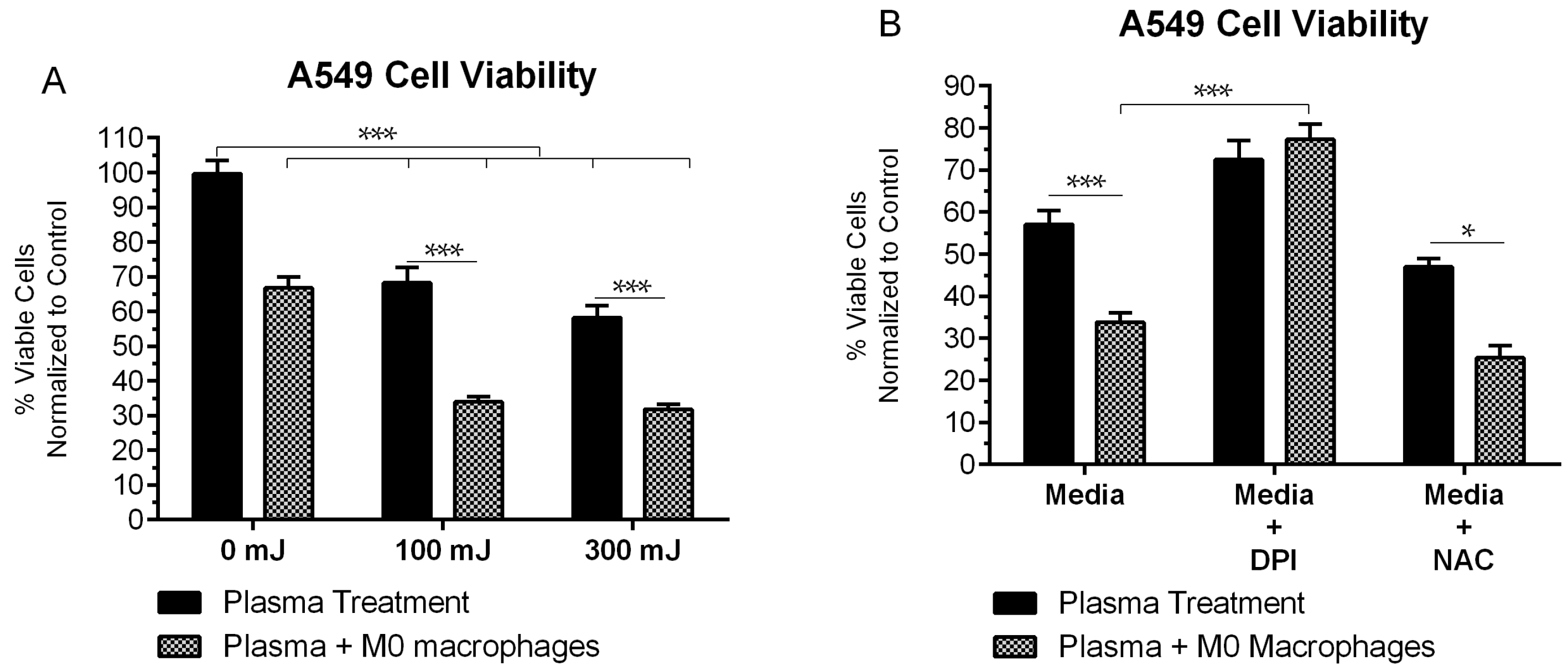
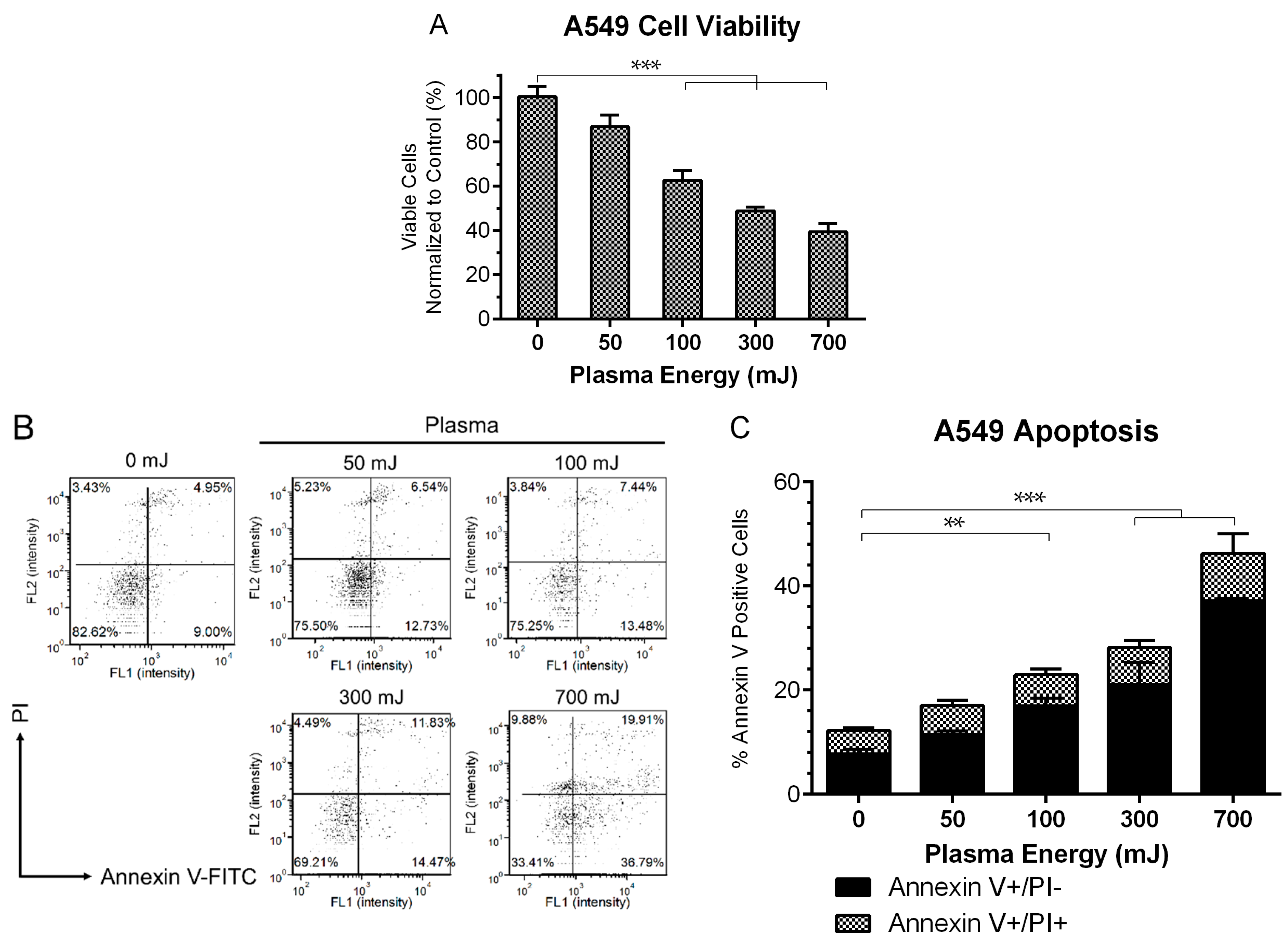
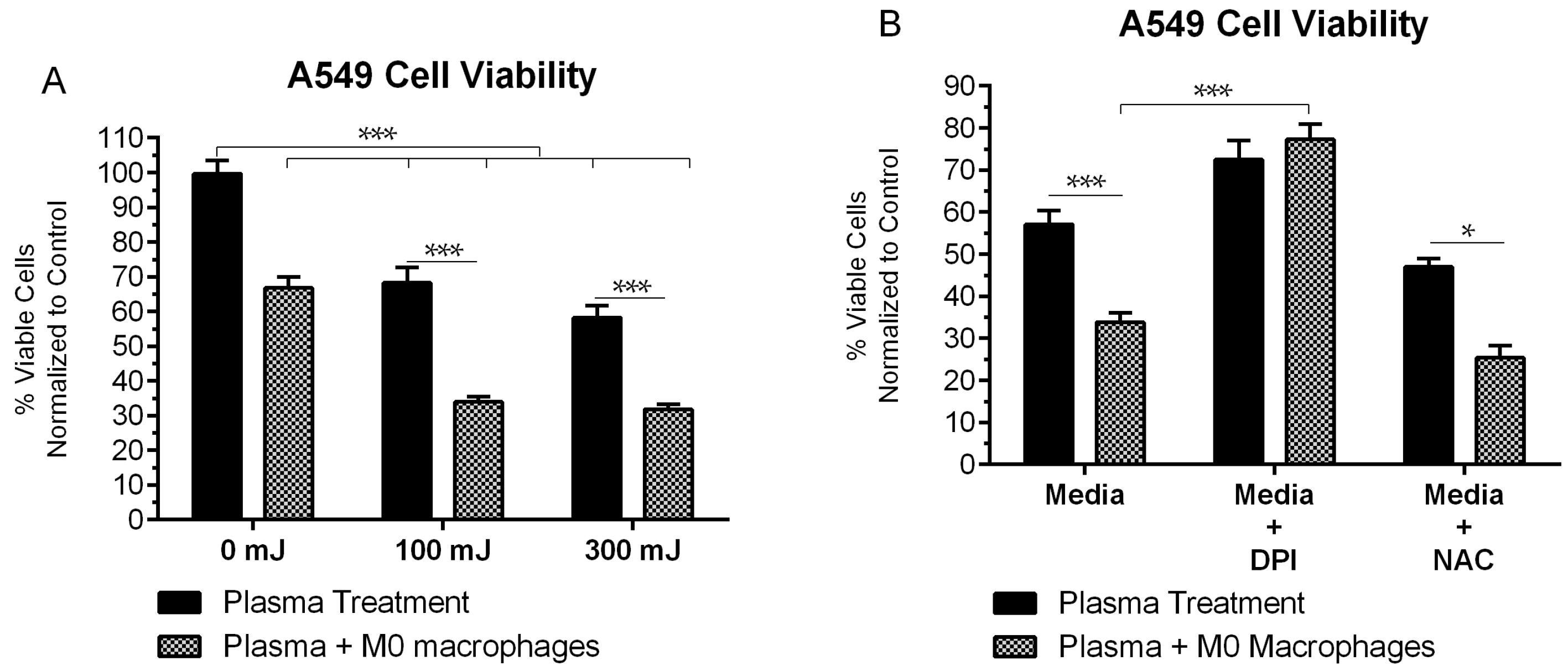
2.1. NspDBD Plasma Elicits Apoptosis in an Energy-Dependent Manner
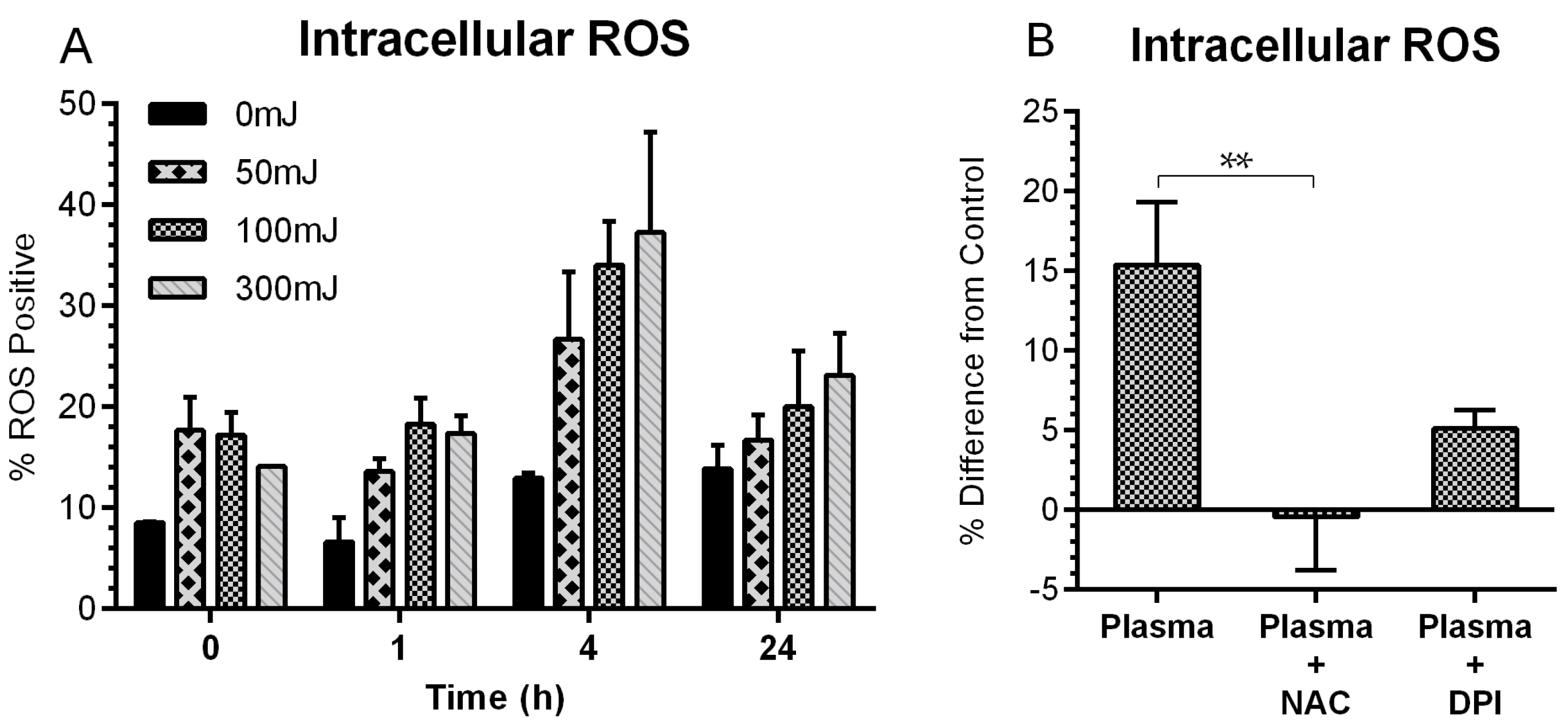
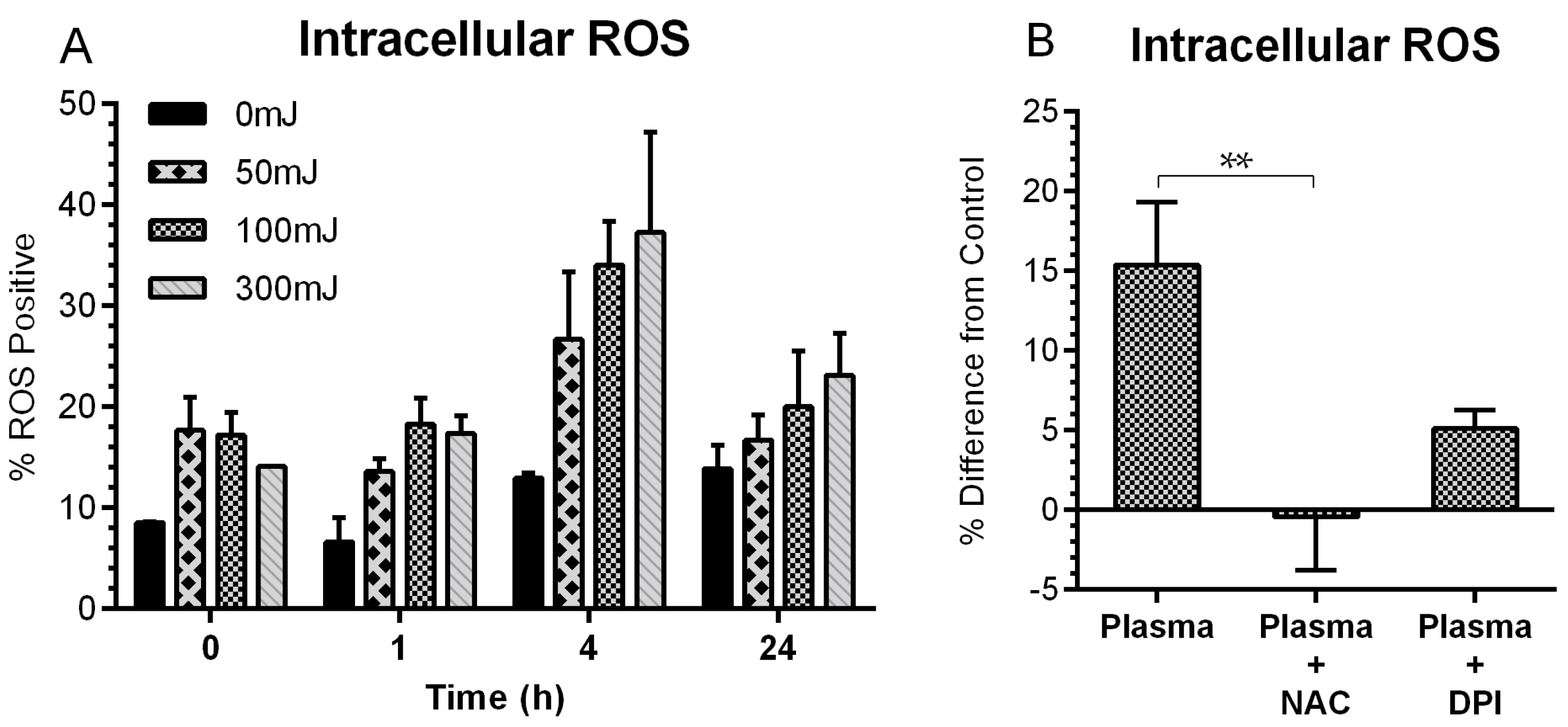
2.2. NspDBD Plasma Induces Oxidative Stress
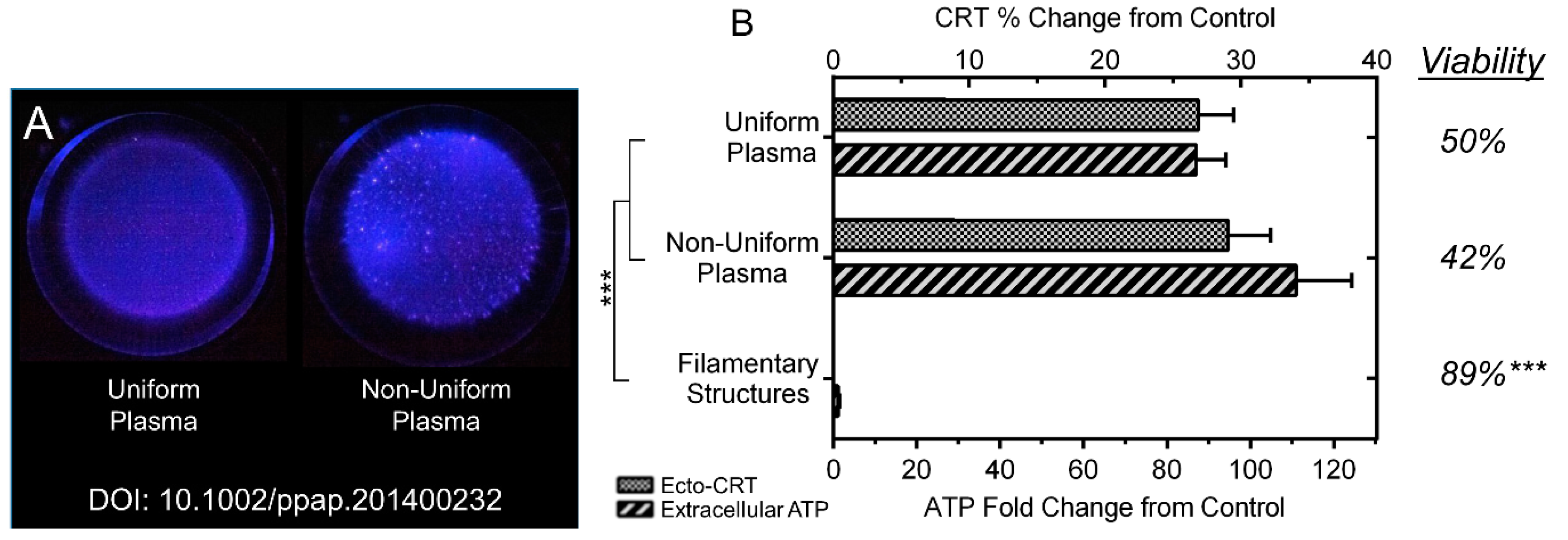
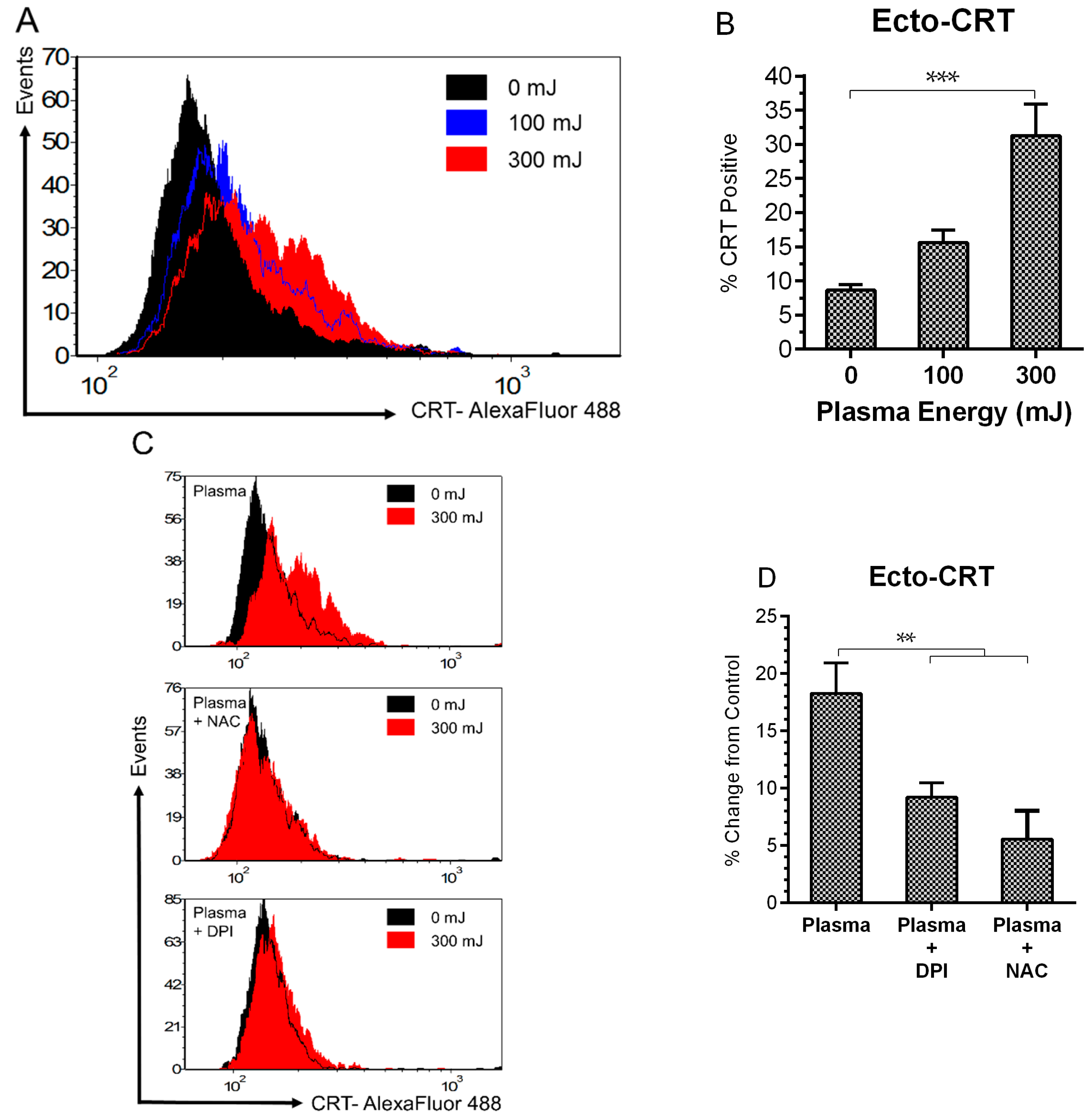
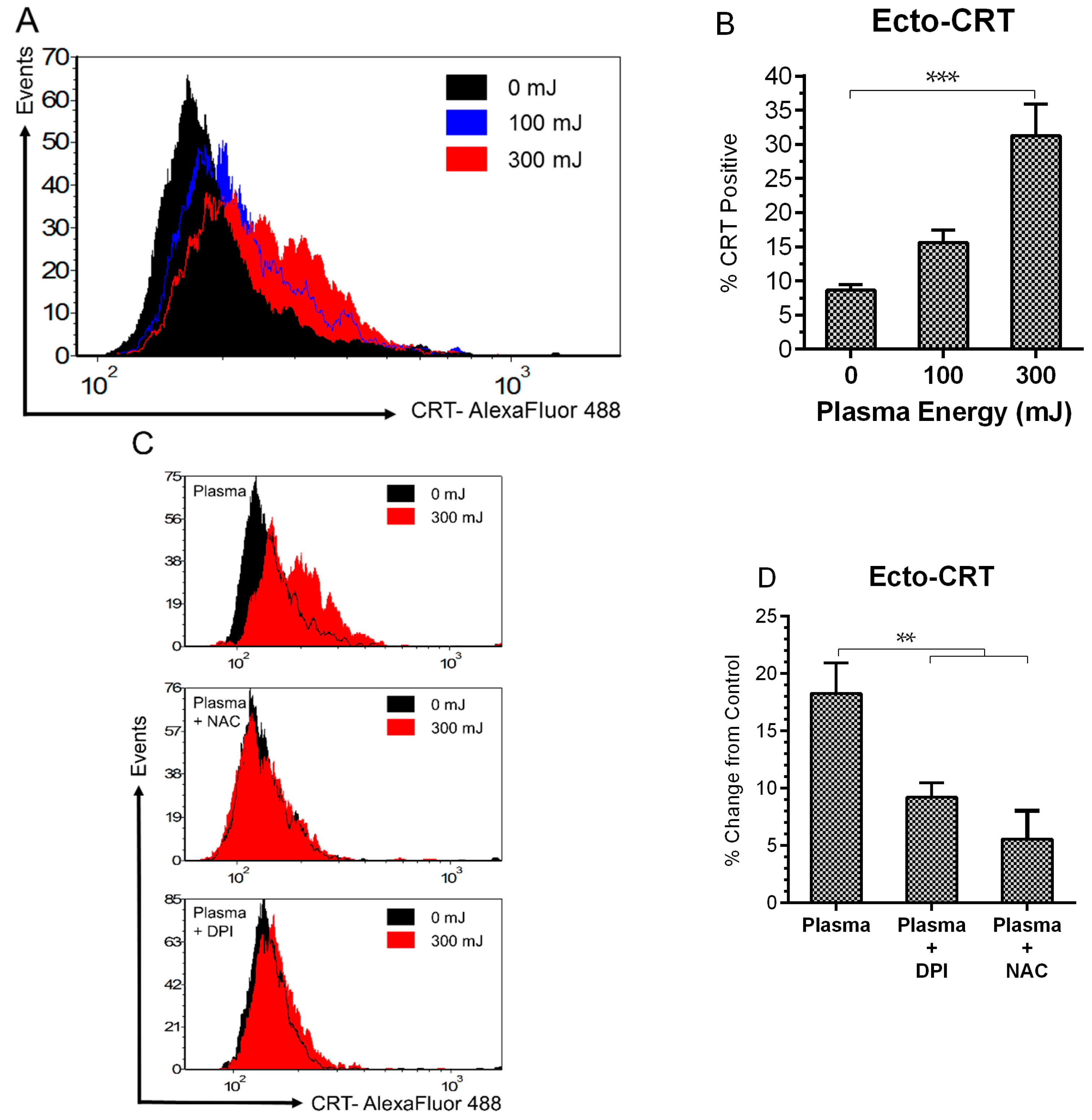
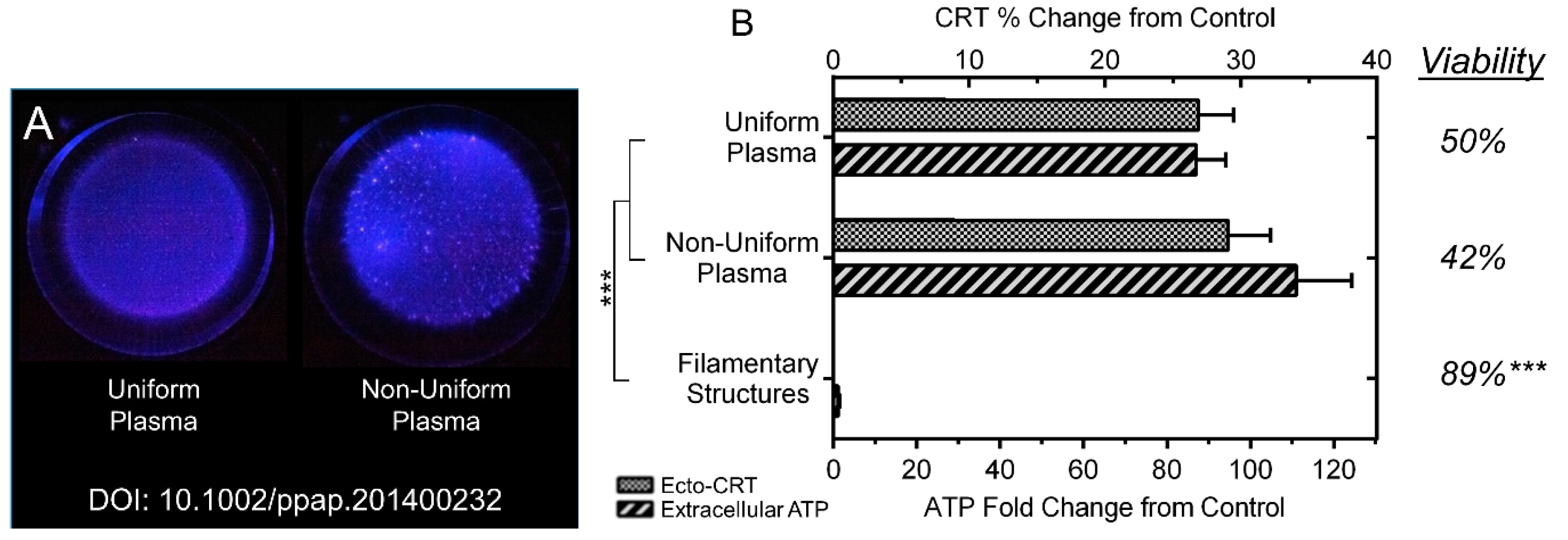
2.3. NspDBD Plasma Elicits Surface Exposure of CRT via Oxidative Stress
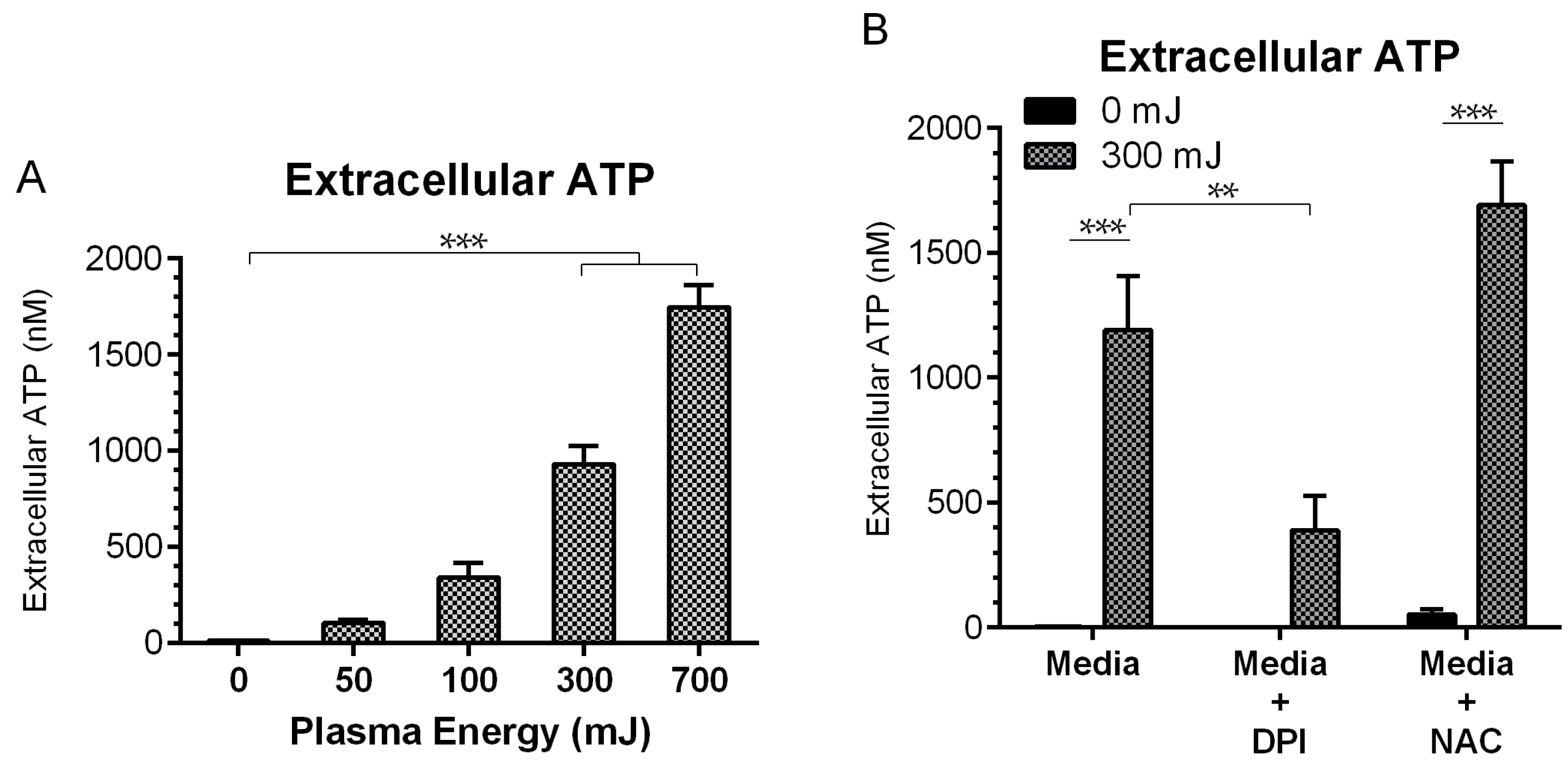
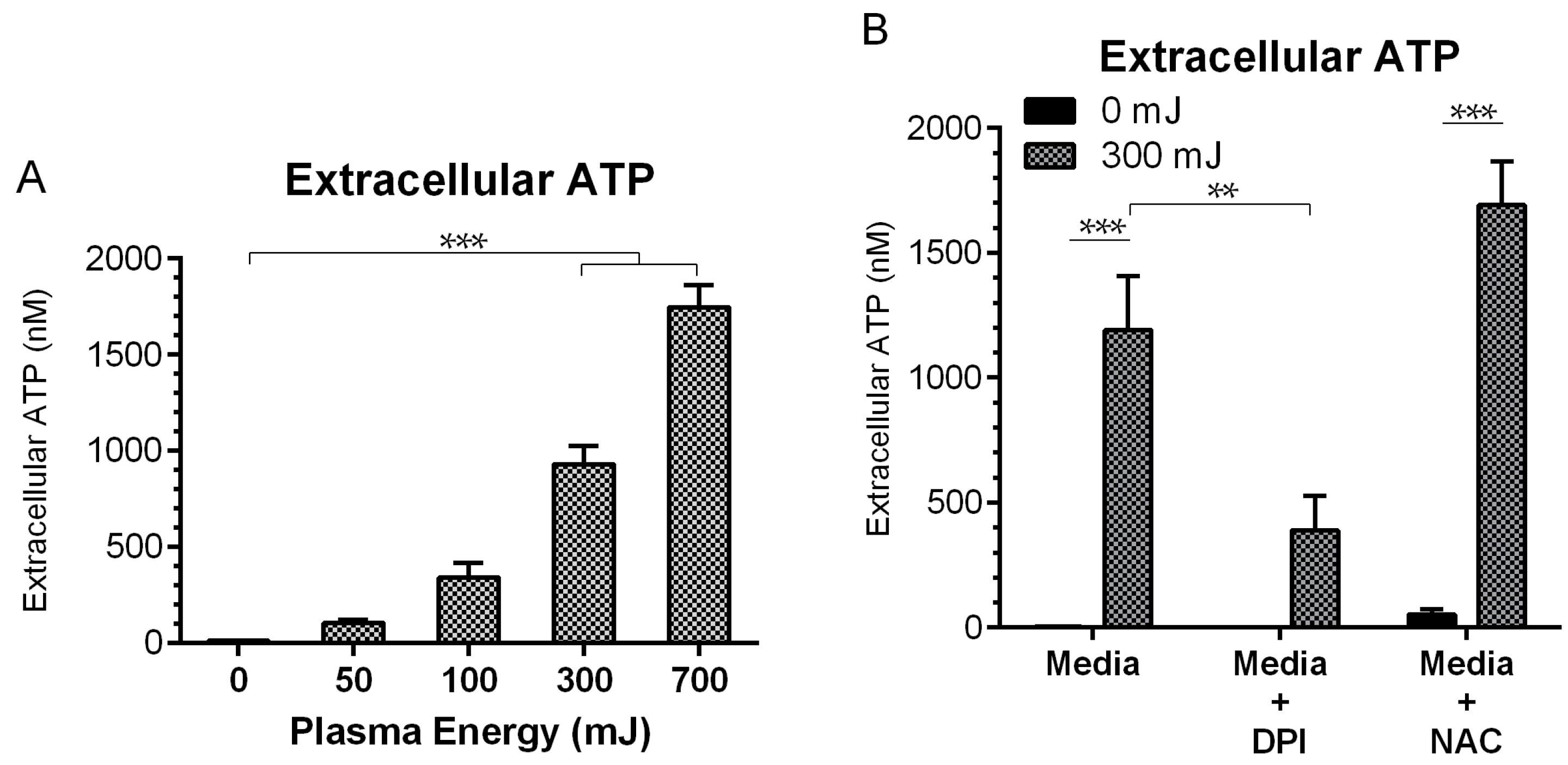
2.4. NspDBD Plasma Elicits Secretion of ATP via Oxidative Stress
2.5. NspDBD-Elicited ICD Enhances Anti-Tumor Activity of Macrophages
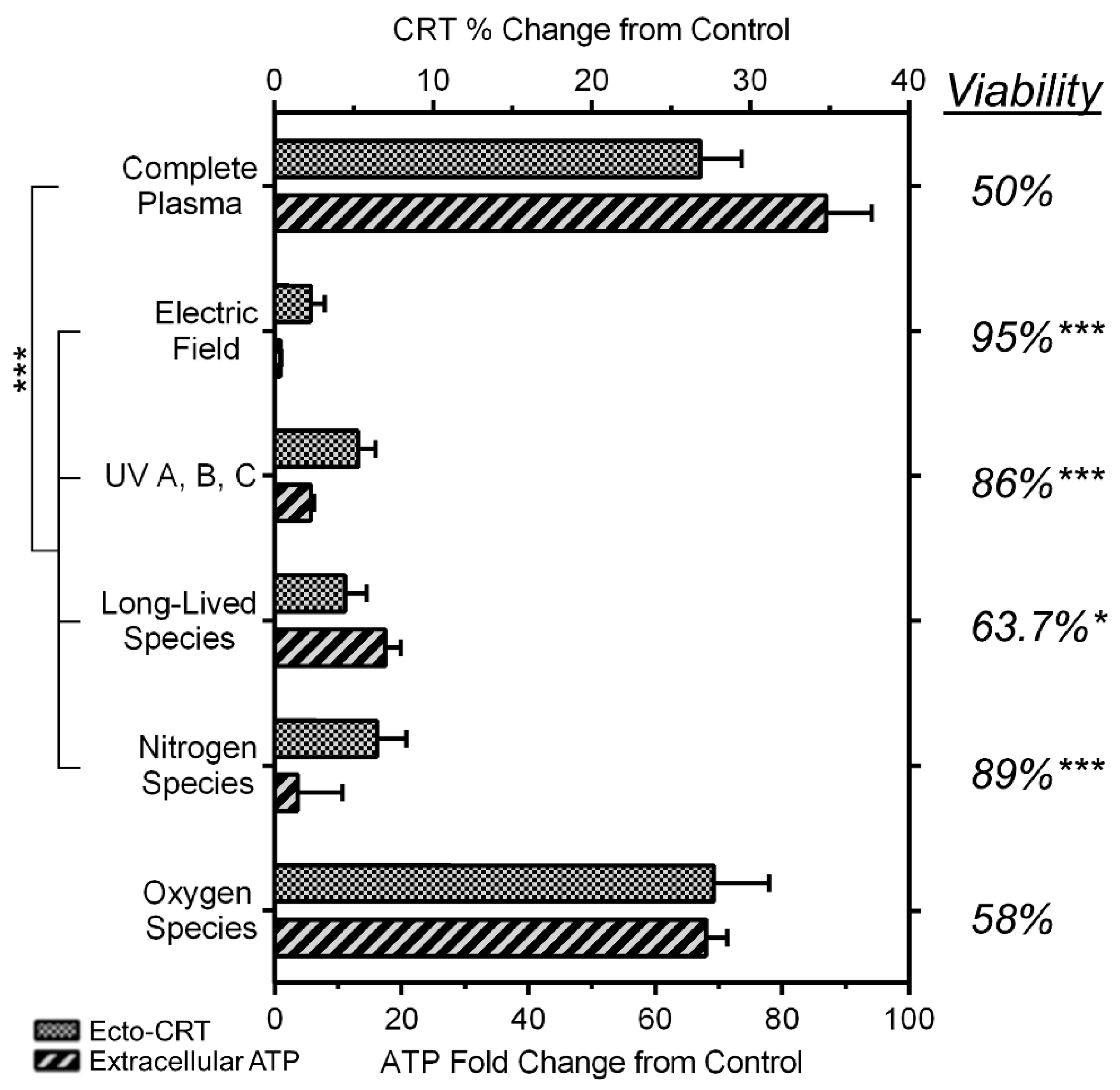
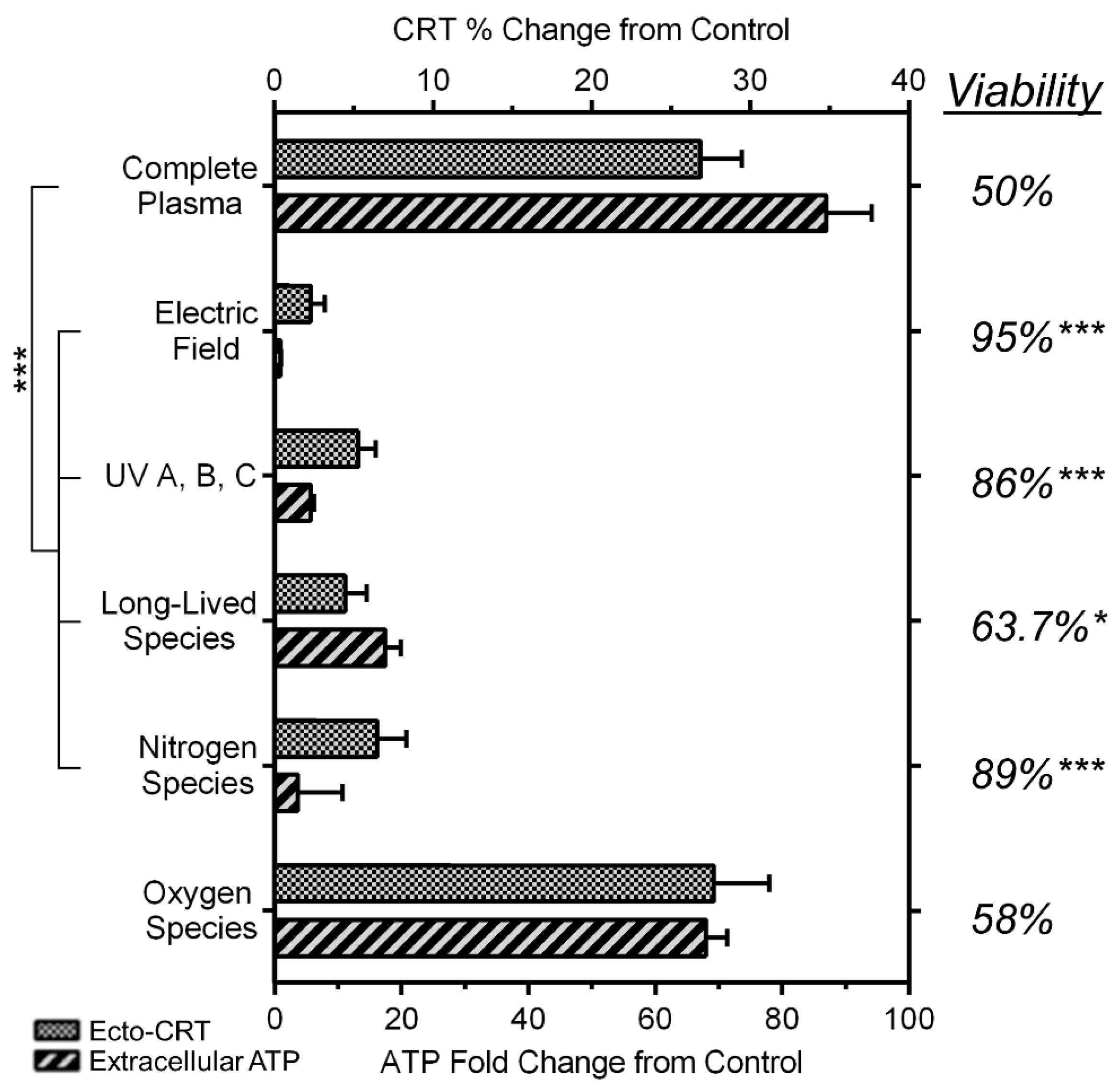
2.6. NspDBD Plasma-Generated ROS and Charges Are the Major Effectors of ICD
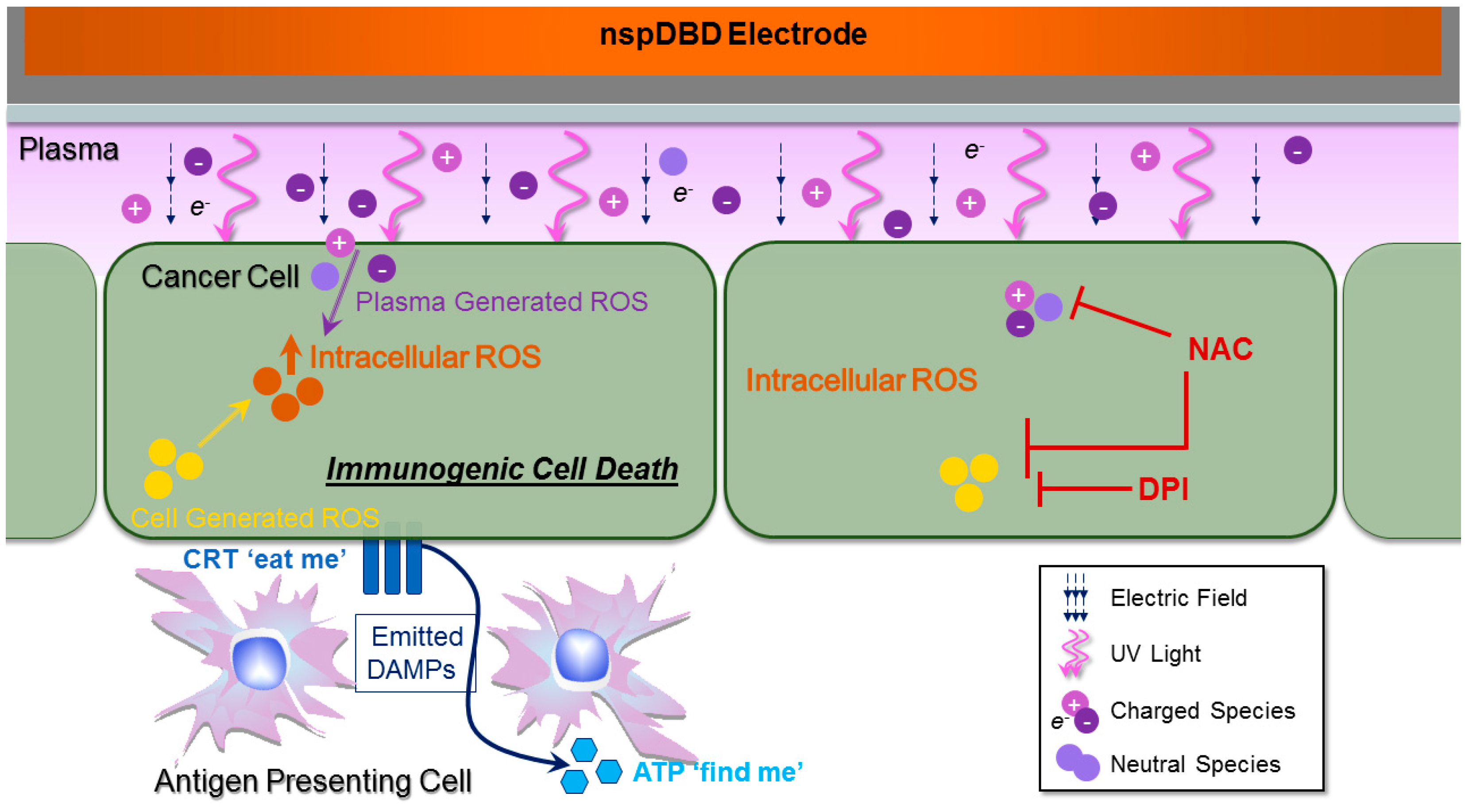
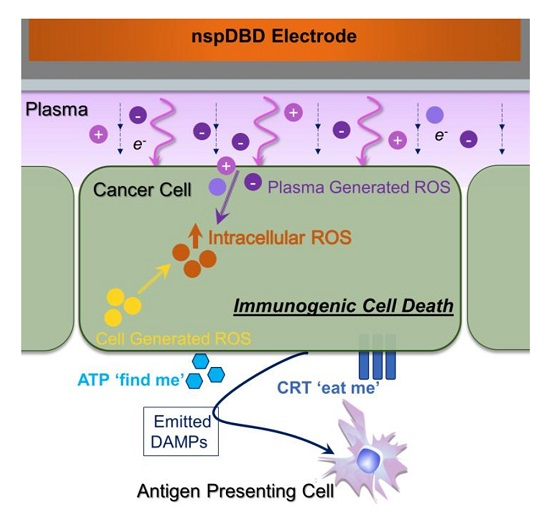
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Plating
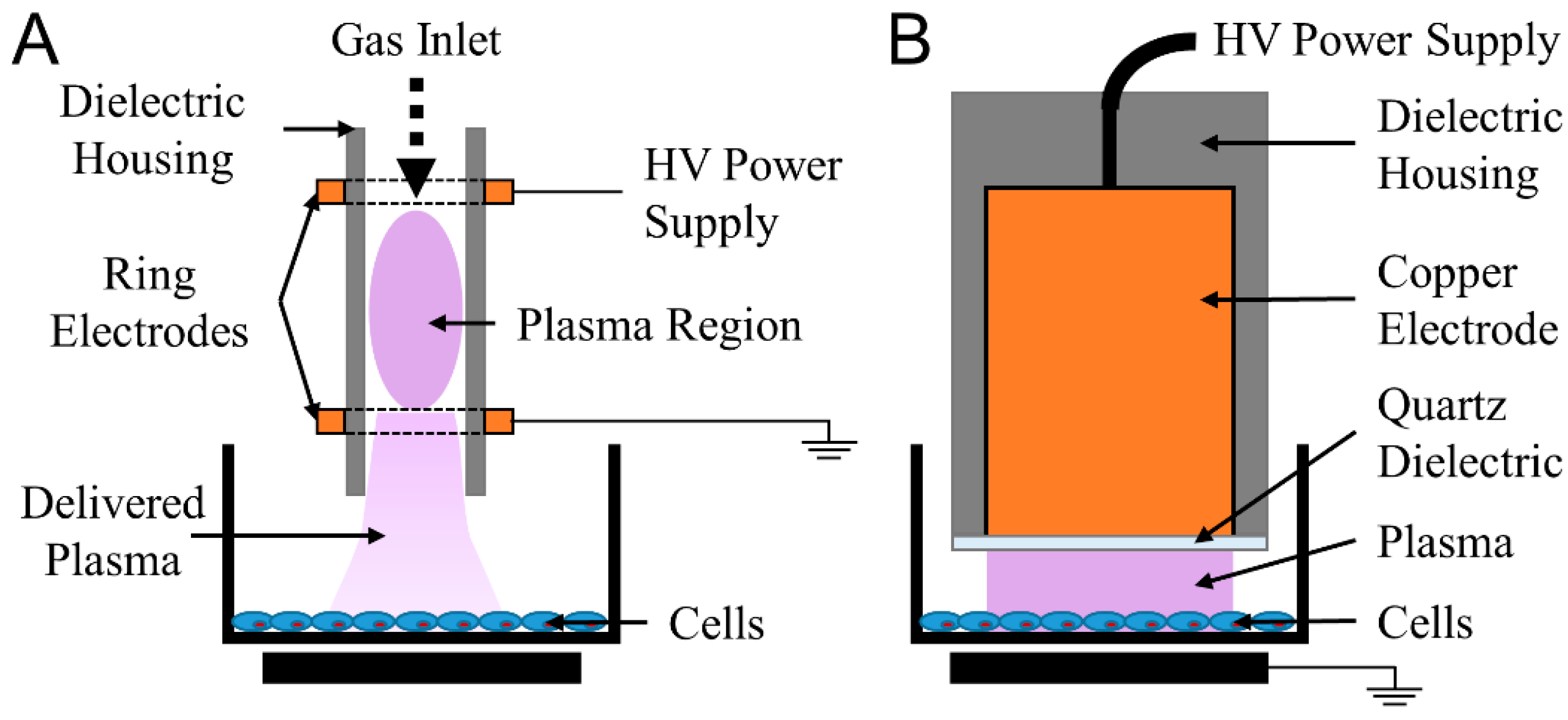
4.2. NspDBD Plasma Treatment Parameters
4.3. Removal of Plasma Effectors to Determine the Major Contributors of Plasma-Induced ICD
4.4. UV Power Measurements
4.5. Pre-Treatment with NAC and DPI to Attenuate Intracellular ROS
4.6. Quantification of Cell Viability
4.7. Quantification of Apoptotic and Necrotic Cells
4.8. Quantification of Extracellular ATP
4.9. Fluorescence Detection of Ecto-CRT
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ICD | Immunogenic cell death |
| APC | Antigen presenting cell |
| DAMP | Damage associated molecular pattern |
| ER | Endoplasmic reticulum |
| ROS | Reactive oxygen specie |
| CRT | Calreticulin |
| Ecto-CRT | Surface-exposed calreticulin |
| ATP | Adenosine triphosphate |
| HMGB1 | High mobility group protein B1 |
| HSP90 | Heat shock protein 90 |
| HSP70 | Heat shock protein 70 |
| HV | High voltage |
| RNS | Reactive nitrogen specie |
| DBD | Dielectric barrier discharge |
| ATF4 | Activating transcription factor 4 |
| STC2 | Stanniocalcin |
| NspDBD | Nanosecond-pulsed DBD |
| NAC | N-acetyl cysteine |
| DPI | Diphenyleneiodonium |
| PI | Propidium iodide |
| DCFDA | 2′,7′-dichlorofluorescein diacetate |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| DC | Dendritic cell |
| TNF-α | Tumor necrosis factor α |
| IL-1β | Interleukin 1β |
| IL-18 | Interleukin 18 |
| PMA | Phorbol 12-myristate 13-acetate |
Appendix A
References
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Johnson, C.M.; Mescher, M.F. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 2003, 171, 5165–5171. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008, 15, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, L.; Gorini, S.; Rosano, G.; la Sala, A. Immunoregulation through extracellular nucleotides. Blood 2012, 120, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Manfredi, A.A. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 2007, 220, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Aymeric, L.; Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Martins, I.; Kroemer, G.; Smyth, M.J.; Zitvogel, L. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010, 70, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Krysko, D.; Vandenabeele, P.; Agostinis, P. Extracellular ATP and P2X7 receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell Death Dis. 2016, 7, e2097. [Google Scholar] [CrossRef] [PubMed]
- Fridman, G.; Friedman, G.; Gutsol, A.; Shekhter, A.B.; Vasilets, V.N.; Fridman, A. Applied plasma medicine. Plasma Process. Polym. 2008, 5, 503–533. [Google Scholar] [CrossRef]
- Fridman, A.; Chirokov, A.; Gutsol, A. Non-thermal atmospheric pressure discharges. J. Phys. D Appl. Phys. 2005, 38, R1. [Google Scholar] [CrossRef]
- Lu, X.; Laroussi, M.; Puech, V. On atmospheric-pressure non-equilibrium plasma jets and plasma bullets. Plasma Sources Sci. Technol. 2012, 21, 034005. [Google Scholar] [CrossRef]
- Yousfi, M.; Eichwald, O.; Merbahi, N.; Jomaa, N. Analysis of ionization wave dynamics in low-temperature plasma jets from fluid modeling supported by experimental investigations. Plasma Sources Sci. Technol. 2012, 21, 045003. [Google Scholar] [CrossRef]
- Fridman, A.; Friedman, G. Plasma Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Lu, X.; Naidis, G.; Laroussi, M.; Reuter, S.; Graves, D.; Ostrikov, K. Reactive species in non-equilibrium atmospheric-pressure plasmas: Generation, transport, and biological effects. Phys. Rep. 2016, 630, 1–84. [Google Scholar] [CrossRef]
- Dobrynin, D.; Fridman, G.; Friedman, G.; Fridman, A. Physical and biological mechanisms of direct plasma interaction with living tissue. New J. Phys. 2009, 11, 115020. [Google Scholar] [CrossRef]
- Waskoenig, J.; Niemi, K.; Knake, N.; Graham, L.; Reuter, S.; Schulz-Von Der Gathen, V.; Gans, T. Atomic oxygen formation in a radio-frequency driven micro-atmospheric pressure plasma jet. Plasma Sources Sci. Technol. 2010, 19, 045018. [Google Scholar] [CrossRef]
- Xu, D.; Liu, D.; Wang, B.; Chen, C.; Chen, Z.; Li, D.; Yang, Y.; Chen, H.; Kong, M.G. In situ OH generation from O2− and H2O2 plays a critical role in plasma-induced cell death. PLoS ONE 2015, 10, e0128205. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.; Simms, M.; Mann, V.; Maitland, N.; O’Connell, D.; Frame, F. Low-temperature plasma treatment induces DNA damage leading to necrotic cell death in primary prostate epithelial cells. Br. J. Cancer 2015, 112, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; Niemi, K.; Cox, L.; Algwari, Q.T.; Gans, T.; O’Connell, D. Cold atmospheric pressure plasma jets as sources of singlet delta oxygen for biomedical applications. J. Appl. Phys. 2011, 109, 123302. [Google Scholar] [CrossRef]
- Heinlin, J.; Morfill, G.; Landthaler, M.; Stolz, W.; Isbary, G.; Zimmermann, J.L.; Shimizu, T.; Karrer, S. Plasma medicine: Possible applications in dermatology. JDDG J. Deutsch. Dermatol. Ges. 2010, 8, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, P.; Kushner, M.J.; Locke, B.R.; Gardeniers, J.; Graham, W.; Graves, D.B.; Hofman-Caris, R.; Maric, D.; Reid, J.P.; Ceriani, E. Plasma–liquid interactions: A review and roadmap. Plasma Sources Sci. Technol. 2016, 25, 053002. [Google Scholar] [CrossRef]
- Manda, G.; Nechifor, M.T.; Neagu, T.M. Reactive oxygen species, cancer and anti-cancer therapies. Curr. Chem. Biol. 2009, 3, 342–366. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef] [PubMed]
- Van der Most, R.G.; Currie, A.J.; Robinson, B.; Lake, R.A. Decoding dangerous death: How cytotoxic chemotherapy invokes inflammation, immunity or nothing at all. Cell Death Differ. 2008, 15, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Renschler, M.F. The emerging role of reactive oxygen species in cancer therapy. Eur. J. Cancer 2004, 40, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; MacGill, R.S.; Miller, G.C.; Pardini, R.S. Photoactivation of hypericin generates singlet oxygen in mitochondria and inhibits succinoxidase. Photochem. Photobiol. 1992, 55, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Fridman, G.; Brooks, A.D.; Balasubramanian, M.; Fridman, A.; Gutsol, A.; Vasilets, V.N.; Ayan, H.; Friedman, G. Comparison of direct and indirect effects of non-thermal atmospheric-pressure plasma on bacteria. Plasma Process. Polym. 2007, 4, 370–375. [Google Scholar] [CrossRef]
- Babaeva, N.Y.; Kushner, M.J. Reactive fluxes delivered by dielectric barrier discharge filaments to slightly wounded skin. J. Phys. D Appl. Phys. 2013, 46, 025401. [Google Scholar] [CrossRef]
- Yonemori, S.; Ono, R. Flux of OH and O radicals onto a surface by an atmospheric-pressure helium plasma jet measured by laser-induced fluorescence. J. Phys. D Appl. Phys. 2014, 47, 125401. [Google Scholar] [CrossRef]
- Darny, T.; Pouvesle, J.M.; Puech, V.; Douat, C.; Dozias, S.; Robert, E. Analysis of conductive target influence in plasma jet experiments through helium metastable and electric field measurements. Plasma Sources Sci. Technol. 2017, 26, 045008. [Google Scholar] [CrossRef]
- Lin, A.; Chernets, N.; Han, J.; Alicea, Y.; Dobrynin, D.; Fridman, G.; Freeman, T.A.; Fridman, A.; Miller, V. Non-equilibrium dielectric barrier discharge treatment of mesenchymal stem cells: Charges and reactive oxygen species play the major role in cell death. Plasma Process. Polym. 2015, 12, 1117–1127. [Google Scholar] [CrossRef]
- Kirson, E.D.; Gurvich, Z.; Schneiderman, R.; Dekel, E.; Itzhaki, A.; Wasserman, Y.; Schatzberger, R.; Palti, Y. Disruption of cancer cell replication by alternating electric fields. Cancer 2004, 64, 3288–3295. [Google Scholar] [CrossRef]
- Beebe, S.J.; Fox, P.M.; Rec, L.J.; Willis, E.L.K.; Schoenbach, K.H. Nanosecond, high-intensity pulsed electric fields induce apoptosis in human cells. FASEB J. 2003, 17, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.H.; Schoenbach, K.H.; Beebe, S.J. Nanosecond pulsed electric fields induce apoptosis in p53-wildtype and p53-null HCT116 colon carcinoma cells. Apoptosis 2007, 12, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Caricchio, R.; McPhie, L.; Cohen, P.L. Ultraviolet B radiation-induced cell death: Critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J. Immunol. 2003, 171, 5778–5786. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Sage, E.; Douki, T. Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 571, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Panaretakis, T.; Joza, N.; Tufi, R.; Tesniere, A.; van Endert, P.; Zitvogel, L.; Kroemer, G. Calreticulin exposure is required for the immunogenicity of γ-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007, 14, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Kogelschatz, U. Dielectric-barrier discharges: Their history, discharge physics, and industrial applications. Plasma Chem. Plasma Process. 2003, 23, 1–46. [Google Scholar] [CrossRef]
- Steinbeck, M.J.; Chernets, N.; Zhang, J.; Kurpad, D.S.; Fridman, G.; Fridman, A.; Freeman, T.A. Skeletal cell differentiation is enhanced by atmospheric dielectric barrier discharge plasma treatment. PLoS ONE 2013, 8, e82143. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Kim, K.I.; Hoan, N.N.; Kim, C.H.; Moon, E.; Choi, K.S.; Yang, S.S.; Lee, J.S. Targeting cancer cells with reactive oxygen and nitrogen species generated by atmospheric-pressure air plasma. PLoS ONE 2014, 9, e86173. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Burdo, R.H.; Rice-Evans, C. Free radicals and the regulation of mammalian cell proliferation. Free Radic. Res. Commun. 1989, 6, 345–358. [Google Scholar] [CrossRef]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J., Jr.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Truong, B.; Pappas, A.; Kirifides, L.; Oubarri, A.; Chen, S.; Lin, S.; Dobrynin, D.; Fridman, G.; Fridman, A. Uniform nanosecond pulsed dielectric barrier discharge plasma enhances anti-tumor effects by induction of immunogenic cell death in tumors and stimulation of macrophages. Plasma Process. Polym. 2015, 12, 1392–1399. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kaufman, R.J. All roads lead to ATF4. Dev. Cell 2003, 4, 442–444. [Google Scholar] [CrossRef]
- Ito, D.; Walker, J.R.; Thompson, C.S.; Moroz, I.; Lin, W.; Veselits, M.L.; Hakim, A.M.; Fienberg, A.A.; Thinakaran, G. Characterization of stanniocalcin 2, a novel target of the mammalian unfolded protein response with cytoprotective properties. Mol. Cell. Biol. 2004, 24, 9456–9469. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Sun, C.; Wang, H.; Mao, S.; Liu, J.; Li, S.; Wang, J. Reactive oxygen species involved in CT26 immunogenic cell death induced by Clostridium difficile toxin B. Immunol. Lett. 2015, 164, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.B. Reactive species from cold atmospheric plasma: Implications for cancer therapy. Plasma Process. Polym. 2014, 11, 1120–1127. [Google Scholar] [CrossRef]
- Spagnuolo, G.; D’Antò, V.; Cosentino, C.; Schmalz, G.; Schweikl, H.; Rengo, S. Effect of N-acetyl-l-cysteine on ROS production and cell death caused by HEMA in human primary gingival fibroblasts. Biomaterials 2006, 27, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic. Biol. Med. 2005, 38, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Trush, M.A. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem. Biophys. Res. Commun. 1998, 253, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.C.; Lozier, A.; Flament, C.; Ricciardi-Castagnoli, P.; Bellet, D.; Suter, M.; Perricaudet, M.; Tursz, T.; Maraskovsky, E.; Zitvogel, L. Dendritic cells directly trigger NK cell functions: Cross-talk relevant in innate anti-tumor immune responses in vivo. Nat. Med. 1999, 5, 405–411. [Google Scholar] [PubMed]
- La Sala, A.; Ferrari, D.; di Virgilio, F.; Idzko, M.; Norgauer, J.; Girolomoni, G. Alerting and tuning the immune response by extracellular nucleotides. J. Leukoc. Biol. 2003, 73, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Gough, M.J.; Melcher, A.A.; Ahmed, A.; Crittenden, M.R.; Riddle, D.S.; Linardakis, E.; Ruchatz, A.N.; Emiliusen, L.M.; Vile, R.G. Macrophages orchestrate the immune response to tumor cell death. Cancer Res. 2001, 61, 7240–7247. [Google Scholar] [PubMed]
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP triggers tumor necrosis factor-α release from rat microglia. J. Neurochem. 2000, 75, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Kusner, D.J.; Barton, J.A. ATP stimulates human macrophages to kill intracellular virulent Mycobacterium tuberculosis via calcium-dependent phagosome-lysosome fusion. J. Immunol. 2001, 167, 3308–3315. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. Atp activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Reichner, J.S.; Mateo, R.B.; Albina, J.E. Activated murine macrophages induce apoptosis in tumor cells through nitric oxide-dependent or-independent mechanisms. Cancer Res. 1994, 54, 2462–2467. [Google Scholar] [PubMed]
- Laroussi, M.; Leipold, F. Evaluation of the roles of reactive species, heat, and UV radiation in the inactivation of bacterial cells by air plasmas at atmospheric pressure. Int. J. Mass Spectrom. 2004, 233, 81–86. [Google Scholar] [CrossRef]
- Dobrynin, D.; Friedman, G.; Fridman, A.; Starikovskiy, A. Inactivation of bacteria using dc corona discharge: Role of ions and humidity. New J. Phys. 2011, 13, 103033. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a source of reactive oxygen and nitrogen species: From molecular mechanisms to human health. Antioxid. Redox Signal. 2013, 18, 2029–2074. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A. Plasma Chemistry; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Chen, X.; Kolb, J.F.; Swanson, R.J.; Schoenbach, K.H.; Beebe, S.J. Apoptosis initiation and angiogenesis inhibition: Melanoma targets for nanosecond pulsed electric fields. Pigment Cell Melanoma Res. 2010, 23, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Beebe, S.J. An apoptosis targeted stimulus with nanosecond pulsed electric fields (nsPEFs) in E4 squamous cell carcinoma. Apoptosis 2011, 16, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xiong, Z.; Liu, Y.; Yao, C.; Li, C. Low voltage irreversible electroporation induced apoptosis in HeLa cells. J. Cancer Res. Ther. 2012, 8, 80. [Google Scholar] [PubMed]
- Vernier, P.T.; Levine, Z.A.; Wu, Y.-H.; Joubert, V.; Ziegler, M.J.; Mir, L.M.; Tieleman, D.P. Electroporating fields target oxidatively damaged areas in the cell membrane. PLoS ONE 2009, 4, e7966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhuang, J.; von Woedtke, T.; Kolb, J.F.; Zhang, J.; Fang, J.; Weltmann, K.D. Synergistic antibacterial effects of treatments with low temperature plasma jet and pulsed electric fields. Appl. Phys. Lett. 2014, 105, 104103. [Google Scholar] [CrossRef]
- Gorbanev, Y.; O’Connell, D.; Chechik, V. Non-thermal plasma in contact with water: The origin of species. Chem. Eur. J. 2016, 22, 3496–3505. [Google Scholar] [CrossRef] [PubMed]
- Jaquet, V.; Scapozza, L.; Clark, R.A.; Krause, K.H.; Lambeth, J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 2009, 11, 2535–2552. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2016, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Bundscherer, L.; Wende, K.; Ottmüller, K.; Barton, A.; Schmidt, A.; Bekeschus, S.; Hasse, S.; Weltmann, K.-D.; Masur, K.; Lindequist, U. Impact of non-thermal plasma treatment on MAPK signaling pathways of human immune cell lines. Immunobiology 2013, 218, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Yonetamari, K.; Shirakawa, Y.; Akiyama, T.; Ono, R. Anti-tumor immune response induced by nanosecond pulsed streamer discharge in mice. J. Phys. D Appl. Phys. 2017, 50, 12LT01. [Google Scholar] [CrossRef]
- Keidar, M.; Walk, R.; Shashurin, A.; Srinivasan, P.; Sandler, A.; Dasgupta, S.; Ravi, R.; Guerrero-Preston, R.; Trink, B. Cold plasma selectivity and the possibility of a paradigm shift in cancer therapy. Br. J. Cancer 2011, 105, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Keidar, M.; Shashurin, A.; Volotskova, O.; Stepp, M.A.; Srinivasan, P.; Sandler, A.; Trink, B. Cold atmospheric plasma in cancer therapy. Phys. Plasmas 2013, 20, 057101. [Google Scholar] [CrossRef]
- Schlegel, J.; Köritzer, J.; Boxhammer, V. Plasma in cancer treatment. Clin. Plasma Med. 2013, 1, 2–7. [Google Scholar] [CrossRef]
- Vandamme, M.; Robert, E.; Pesnel, S.; Barbosa, E.; Dozias, S.; Sobilo, J.; Lerondel, S.; Le Pape, A.; Pouvesle, J.M. Antitumor effect of plasma treatment on U87 glioma xenografts: Preliminary results. Plasma Process. Polym. 2010, 7, 264–273. [Google Scholar] [CrossRef]
- Vandamme, M.; Robert, E.; Lerondel, S.; Sarron, V.; Ries, D.; Dozias, S.; Sobilo, J.; Gosset, D.; Kieda, C.; Legrain, B. ROS implication in a new antitumor strategy based on non-thermal plasma. Int. J. Cancer 2012, 130, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Hirst, A.M.; Frame, F.M.; Arya, M.; Maitland, N.J.; O’Connell, D. Low temperature plasmas as emerging cancer therapeutics: The state of play and thoughts for the future. Tumor Biol. 2016, 37, 7021–7031. [Google Scholar] [CrossRef] [PubMed]
- Chernets, N.; Kurpad, D.S.; Alexeev, V.; Rodrigues, D.B.; Freeman, T.A. Reaction chemistry generated by nanosecond pulsed dielectric barrier discharge treatment is responsible for the tumor eradication in the B16 melanoma mouse model. Plasma Process. Polym. 2015, 12, 1400–1409. [Google Scholar] [CrossRef]
- Fridman, G.; Shereshevsky, A.; Jost, M.M.; Brooks, A.D.; Fridman, A.; Gutsol, A.; Vasilets, V.; Friedman, G. Floating electrode dielectric barrier discharge plasma in air promoting apoptotic behavior in melanoma skin cancer cell lines. Plasma Chem. Plasma Process. 2007, 27, 163–176. [Google Scholar] [CrossRef]
- Miller, V.; Lin, A.; Fridman, G.; Dobrynin, D.; Fridman, A. Plasma stimulation of migration of macrophages. Plasma Process. Polym. 2014, 11, 1193–1197. [Google Scholar] [CrossRef]
- Miller, V.; Lin, A.; Fridman, A. Why target immune cells for plasma treatment of cancer. Plasma Chem. Plasma Process. 2015, 36, 259–268. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Min, B.; Choi, K.H.; Hong, Y.J.; Miller, V.; Fridman, A.; Choi, E.H. Cytotoxic macrophage-released tumour necrosis factor-α (TNF-α) as a killing mechanism for cancer cell death after cold plasma activation. J. Phys. D Appl. Phys. 2016, 49, 084001. [Google Scholar] [CrossRef]
- Lee, A.; Lin, A.; Shah, K.; Singh, H.; Miller, V.; Rao, S.G. Optimization of non-thermal plasma treatment in an in vivo model organism. PLoS ONE 2016, 11, e0160676. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, P.; Shrivastav, R.; Wang, M.; Lin, A.; Fridman, G.; Fridmam, A.; Han, L.; Miller, V. Nanosecond pulsed dielectric barrier discharge induced anti-tumor effects propagate through the depth of tissue via intracellular signaling. Plasma Med. 2017, in press. [Google Scholar]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kolb, J.; Xiao, S.; Laroussi, M.; Schoenbach, K.; Schamiloglu, E. In Dielectric strength of sub-millimeter water gaps subjected to microsecond and sub-microsecond voltage pulses. In Proceedings of the 2005 IEEE Pulsed Power Conference, Monterey, CA, USA, 13–17 June 2005. [Google Scholar]
- De Flora, S.; D’Agostini, F.; Masiello, L.; Giunciuglio, D.; Albini, A. Synergism between N-acetylcysteine and doxorubicin in the prevention of tumorigenicity and metastasis in murine models. Int. J. Cancer 1996, 67, 842–848. [Google Scholar] [CrossRef]
- Ayan, H.; Staack, D.; Fridman, G.; Gutsol, A.; Mukhin, Y.; Starikovskii, A.; Fridman, A.; Friedman, G. Application of nanosecond-pulsed dielectric barrier discharge for biomedical treatment of topographically non-uniform surfaces. J. Phys. D Appl. Phys. 2009, 42, 125202. [Google Scholar] [CrossRef]
- Ayan, H.; Fridman, G.; Gutsol, A.F.; Vasilets, V.N.; Fridman, A.; Friedman, G. Nanosecond-pulsed uniform dielectric-barrier discharge. IEEE Trans. Plasma Sci. 2008, 36, 504–508. [Google Scholar] [CrossRef]
- Liu, C.; Dobrynin, D.; Fridman, A. Uniform and non-uniform modes of nanosecond-pulsed dielectric barrier discharge in atmospheric air: Fast imaging and spectroscopic measurements of electric fields. J. Phys. D Appl. Phys. 2014, 47, 252003. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, A.; Darny, T.; Pechereau, F.; Pouvesle, J.M.; Viegas, P.; Iséni, S.; Robert, E. Numerical and experimental study of the dynamics of a µs helium plasma gun discharge with various amounts of N2 admixture. Plasma Sources Sci. Technol. 2016, 25, 035002. [Google Scholar] [CrossRef]
- Olszewski, P.; Wagenaars, E.; McKay, K.; Bradley, J.; Walsh, J. Measurement and control of the streamer head electric field in an atmospheric-pressure dielectric barrier plasma jet. Plasma Sources Sci. Technol. 2014, 23, 015010. [Google Scholar] [CrossRef]
- Babaeva, N.Y.; Kushner, M.J. Intracellular electric fields produced by dielectric barrier discharge treatment of skin. J. Phys. D Appl. Phys. 2010, 43, 185206. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Excitation | Nanosecond-pulsed |
| Voltage | 29 kV |
| Rise Time | 2 ns |
| Pulse Width | 20 ns |
| Treatment Time | 10 s |
| Plasma Discharge in 24-Well Plate (Energy per Pulse: 0.9 mJ/pulse) | |
| Gap Distance | 1 mm |
| Frequency | 5, 15, 30, 75 Hz |
| Total Plasma Energies | 50, 100, 300, 700 mJ |
| Plasma Discharge on Copper Mesh Barrier (Energy per Pulse: 1.9 mJ/pulse) | |
| Gap Distance | 1 mm |
| Frequency | 15 Hz |
| Total Plasma Energy | 300 mJ |
| Treatment Condition | Effectors Delivered | Effectors Removed | ||
|---|---|---|---|---|
| 1. | NspDBD in Air | 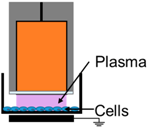 | E-Field, UV, Charges, Neutrals | - |
| 2. | Electrode Dipped in Media | 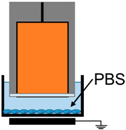 | E-Field | UV, Charges, Neutrals |
| 3. | NspDBD w/Quartz Barrier | 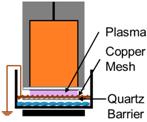 | UV | E-Field, Charges, Neutrals |
| 4. | NspDBD w/Mesh Barrier | 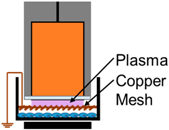 | Long-Lived Neutrals, UV | Global E-Field, Charges, Short-Lived Neutrals |
| 5. | NspDBD in Oxygen |  | ROS, Charges, E-Field, UV | Other Neutral Species |
| 6. | NspDBD in Nitrogen | 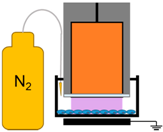 | Nitrogen Species, Charges, E-Field, UV | Other Neutral Species |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, A.; Truong, B.; Patel, S.; Kaushik, N.; Choi, E.H.; Fridman, G.; Fridman, A.; Miller, V. Nanosecond-Pulsed DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress. Int. J. Mol. Sci. 2017, 18, 966. https://doi.org/10.3390/ijms18050966
Lin A, Truong B, Patel S, Kaushik N, Choi EH, Fridman G, Fridman A, Miller V. Nanosecond-Pulsed DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress. International Journal of Molecular Sciences. 2017; 18(5):966. https://doi.org/10.3390/ijms18050966
Chicago/Turabian StyleLin, Abraham, Billy Truong, Sohil Patel, Nagendra Kaushik, Eun Ha Choi, Gregory Fridman, Alexander Fridman, and Vandana Miller. 2017. "Nanosecond-Pulsed DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress" International Journal of Molecular Sciences 18, no. 5: 966. https://doi.org/10.3390/ijms18050966
APA StyleLin, A., Truong, B., Patel, S., Kaushik, N., Choi, E. H., Fridman, G., Fridman, A., & Miller, V. (2017). Nanosecond-Pulsed DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress. International Journal of Molecular Sciences, 18(5), 966. https://doi.org/10.3390/ijms18050966