
Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis




Abstract
:1. Introduction
2. Cell Death in Drug-Induced Liver Injury (DILI)
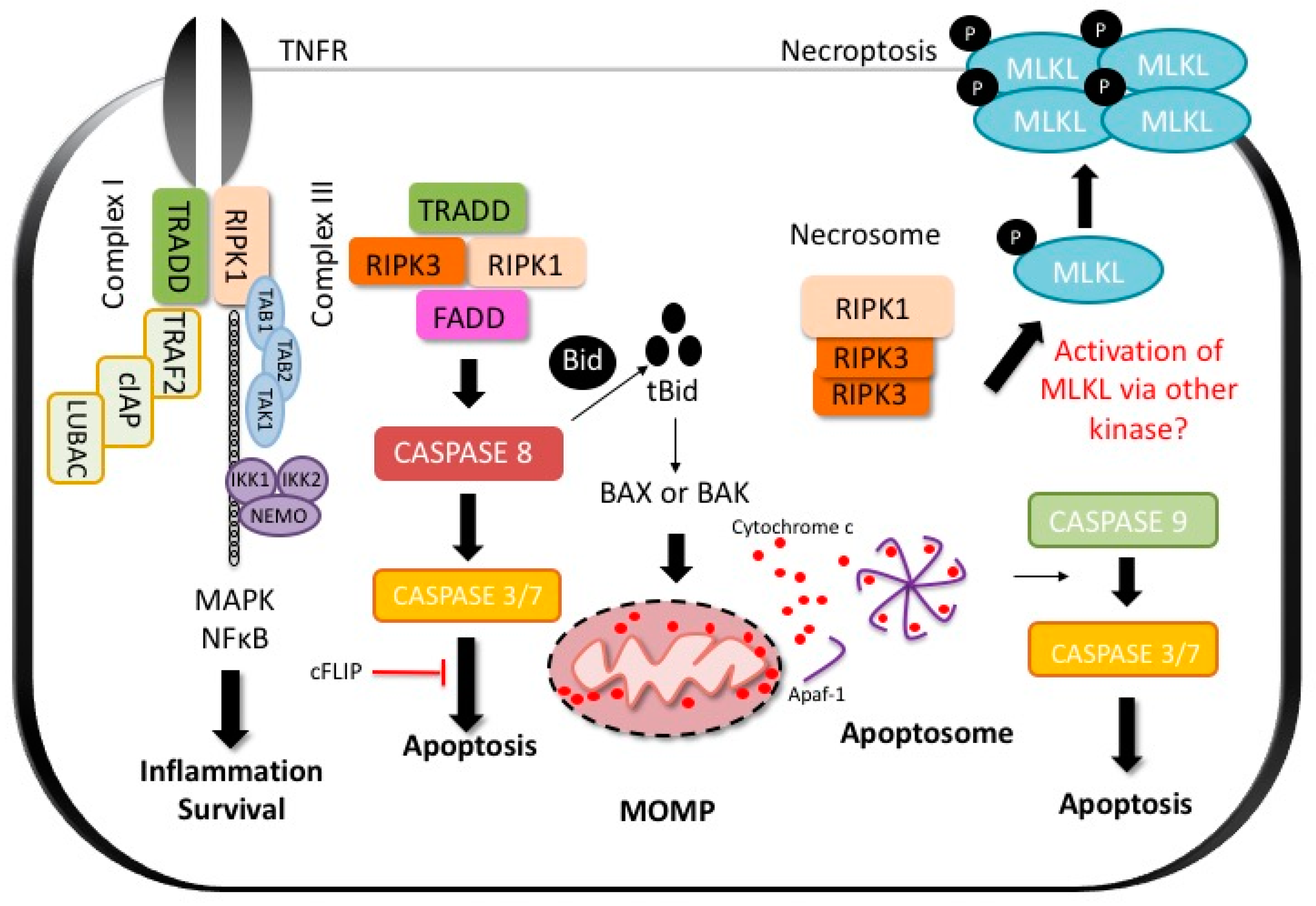
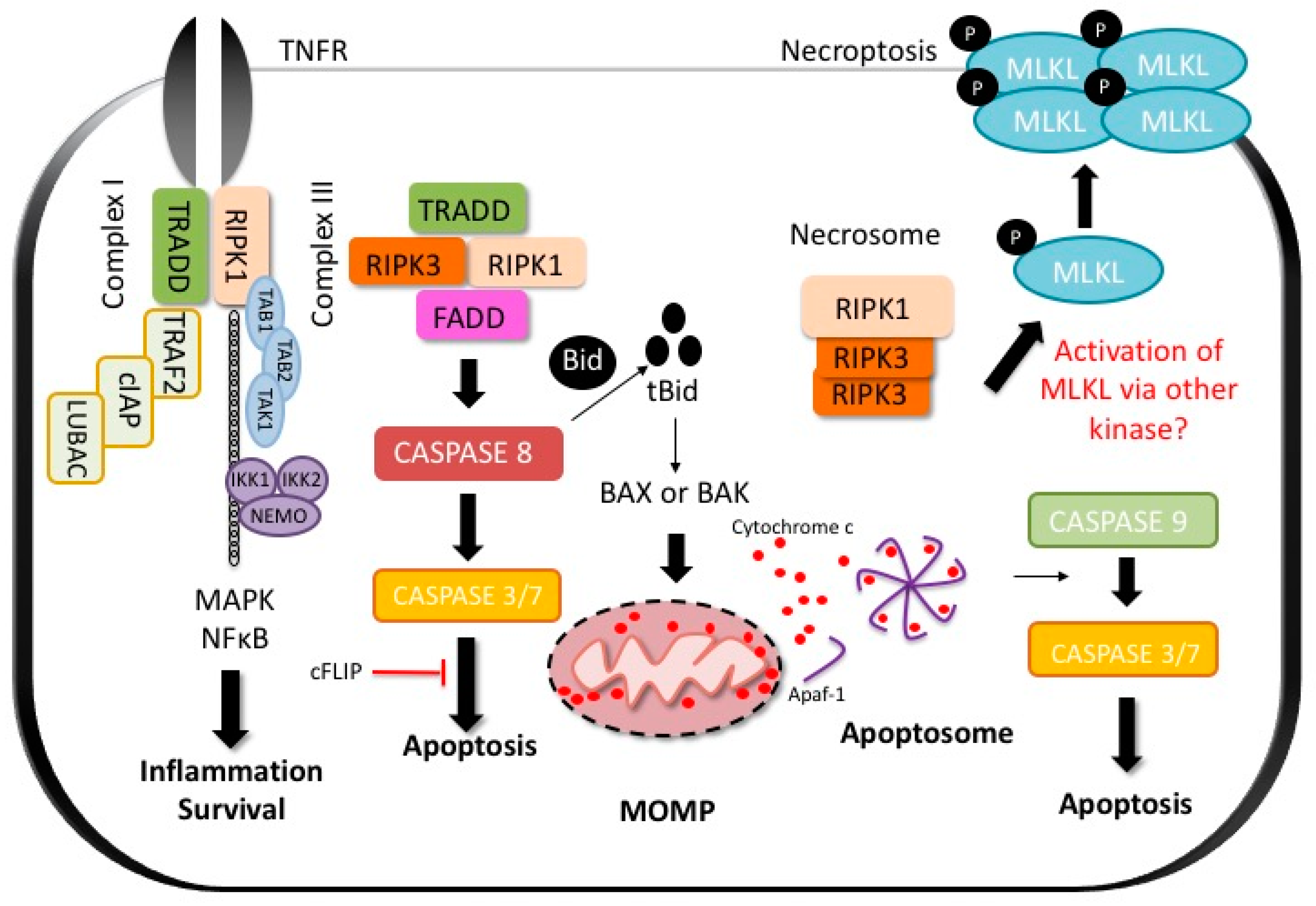
2.1. Apoptosis
2.2. Regulated Necrosis and Necroptosis
2.3. Autophagy
2.4. Other Forms of Cell Death: (Pyroptosis and Ferroptosis)
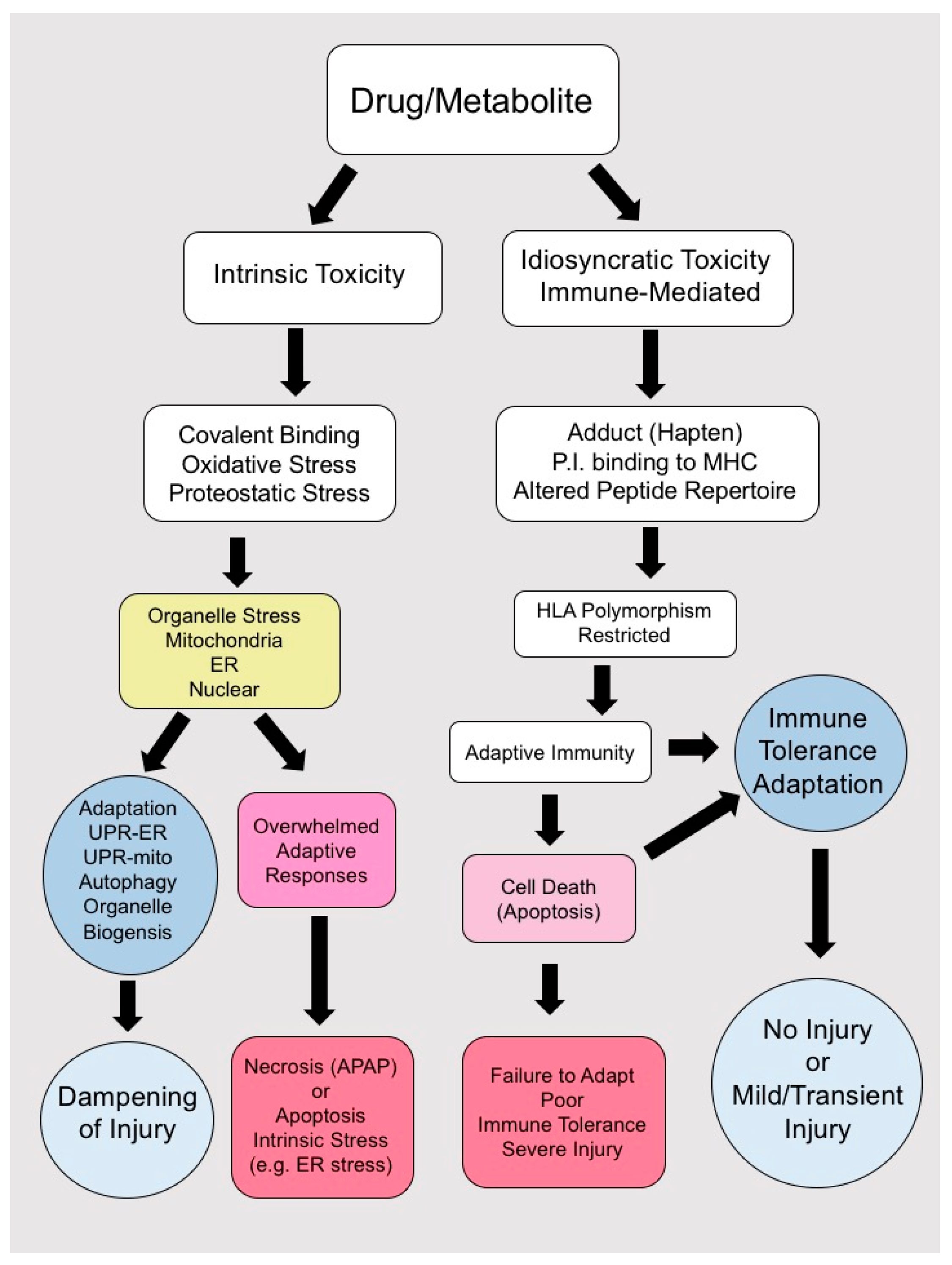
3. Pathogenesis of Idiosycratic Drug-Induced Liver Injury (IDILI)
3.1. Human Leukocyte Antigen (HLA) Associations
3.2. Receptor-Mediated Signaling and DILI
3.3. Hypothesis for Immune System Activation in IDILI
3.4. Immune-Tolerance and Adaptation
4. Direct Hepatocyte Toxicity
Acetaminophen Toxicity
5. Preclinical Models for Screening Drugs for IDILI
6. Clinical Aspects
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| α-GalCer | α-Galactosylceramide |
| AIH | Autoimmune hepatitis |
| ALF | Acute liver failure |
| APAF-1 | Apoptotic peptidase activating factor-1 |
| APAP | Acetaminophen |
| AQ | Amodiaquine |
| ASK1 | Apoptosis signal regulating kinase 1 |
| ATG | Autophagy |
| ATP | Adenosine triphosphate |
| Bax | Bcl-2-like protein 4 |
| Bcl-2 | B-cell lymphoma 2 |
| c-FLIP | FLICE inhibitory protein |
| CD4 | Cluster of differentiation 4 |
| CD8 | Cluster of differentiation 8 |
| CD95 | Cluster of differentiation 95, also known as the FAS receptor |
| cIAP1/2 | Cellular inhibitor of apoptosis 1/2 |
| Con A | Concanavalin A |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CYP | Cytochrome P450 |
| DILI | Drug-induced liver injury |
| DILIN | Drug-Induced Liver Injury Network |
| DNA | Deoxyribonucleic acid |
| DOK4 | Docking protein 4 |
| DR | Death receptors |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| FADD | Fas-associated protein with death domain |
| FasL | Fas ligand |
| Fer-1 | Ferrostatins |
| FLICE | Fas-associating protein with death domain-like interleukin-1 β-converting enzyme |
| GalN | D-galactosamine |
| GPX4 | Glutathione peroxidase 4 |
| GSDMD | Gasdermin D |
| GSH | Glutathione |
| GSK3β | Glycogen synthase kinase 3 β |
| GST1 | Glutathione-S-transferase |
| GWAS | Genome-wide association studies |
| HDS | Herbal and dietary supplements |
| HLA | Human leukocyte antigen |
| HSC | Hepatic stellate cells |
| IDILI | Idiosycratic drug-induced liver injury |
| IFNγ | Interferon γ |
| IL-10 | Interleukin 10 |
| INH | Isoniazide |
| iNOS | Inducible nitric oxide synthase |
| iPSC | Induced pluripotent stem cell |
| JAK | Janus kinase |
| JNK | c-Jun terminal kinase |
| KC | Kupffer cells |
| LPO | Lipid peroxidation |
| LPS | Lipopolysaccharide |
| LSECs | Liver sinusoidal endothelial cells |
| MAPK | Mitogen-activated protein kinases |
| MHC | Major histocompatibility complex |
| MiR | micro RNA |
| MKK4 | Mitogen-activated protein kinase kinase 4 |
| MLK3 | Mixed-lineage kinase 3 |
| MLKL | Mixed lineage domain like |
| MOMP | Mitochondrial outer membrane permeabilization |
| MPT | Membrane permeability transition |
| NAPQI | N-acetyl-p-benzoquinoneimine |
| Nec | Necrostatins |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cells |
| NKT | Natural killer T cells |
| NOS | Nitric oxide synthase |
| NPC | Non-parenchymal cells |
| PD1 | Programmed cell death protein 1 |
| PKCα | Protein kinase C α |
| PHH | Primary human hepatocytes |
| PMH | Primary mouse hepatocytes |
| PTPN6 | Protein tyrosine phosphatase type 6 |
| RHIM | RIP homology interaction motif |
| RIPK1/RIP1 | Receptor interacting serine/threonine kinase 1 |
| RIPK3/RIP3 | Receptor interacting serine/threonine kinase 3 |
| ROS | Reactive oxygen species |
| Sab | SH3 binding protein 5 |
| SEB | Super-antigen staphylococcal enterotoxin B |
| SJS | Stevens–Johnson syndrome |
| STAT | Signal transducer and activator of transcription |
| STAT1 | Signal transducer and activator of transcription 1 |
| SULT2A1 | sulfotransferase 2A1 |
| T-regs | Regulatory T cells |
| TAB2/3 | TAK1-binding protein 2/3 |
| TAK1 | Transforming growth factor-β-activated kinase 1 |
| tBid | Cleaved Bid |
| TCR | T cell receptor |
| TEN | Toxic epidermal necrosis |
| TGFβ | Transforming growth factor β |
| TNF | Tumor necrosis factor |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TNFα | Tumor necrosis factor α |
| TRADD | TNFR-associated death domain |
| TRAF2/5 | TNFR-associated factor 2/ 5 |
| TRAIL-R1/R2 | TNF-related apoptosis-inducing ligand receptor 1/2 |
| UPR | Unfolded protein response |
| WT | Wild type |
References
- Motamedi, N.D.L.; Kaplowitz, N. Clinical considerations of drug-induced hepatotoxicity. In Comprehensive Toxicology, 3rd ed.; McQueen, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Kaplowitz, N. Mechanisms of drug-induced liver injury. Clin. Liver Dis. 2013, 17, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Han, D.; Kaplowitz, N. Mechanisms of Cell Death and Relevance to Drug Toxicity, 3rd ed.; Academic Press (an imprint of Elsevier): Cambridge, MA, USA, 2012. [Google Scholar]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiodt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a united states multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, R.A.; Kaplowitz, N. Drug hepatotoxicity. Clin. Liver Dis. 2006, 10, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Senior, J.R. Drug hepatotoxicity from a regulatory perspective. Clin. Liver Dis. 2007, 11, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N.; Win, S.; Than, T.A.; Liu, Z.X.; Dara, L. Targeting signal transduction pathways which regulate necrosis in acetaminophen hepatotoxicity. J. Hepatol. 2015, 63, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Andrade, R.J.; Kaplowitz, N.; Garcia-Cortes, M.; Fernandez, M.C.; Romero-Gomez, M.; Bruguera, M.; Hallal, H.; Robles-Diaz, M.; Rodriguez-Gonzalez, J.F.; et al. Phenotypic characterization of idiosyncratic drug-induced liver injury: The influence of age and sex. Hepatology 2009, 49, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Denk, H. Drug-induced liver injury. Verh. Dtsch. Ges. Pathol. 2002, 86, 120–125. [Google Scholar] [PubMed]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Avoiding idiosyncratic DILI: Two is better than one. Hepatology 2013, 58, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Chalasani, N. Epidemiology of idiosyncratic drug-induced liver injury. Semin. Liver Dis. 2009, 29, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.L.Z.; Kaplowitz, N. Cell death in drug induced liver injury. In Cell Death in Liver Disease; Ding, W.X., Yin, X.M., Eds.; Springer: Berlin, Germany, 2017. [Google Scholar]
- Odin, J.A.; Huebert, R.C.; Casciola-Rosen, L.; LaRusso, N.F.; Rosen, A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-e2, a primary biliary cirrhosis autoantigen, during apoptosis. J. Clin. Investig. 2001, 108, 223–232. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Wang, X.; Kaplowitz, N.; Shulman, H.M.; Bart, J.A.; van der Hoek, A. Sinusoidal endothelial cells as a target for acetaminophen toxicity: Direct action versus requirement for hepatocyte activation in different mouse strains. Biochem. Pharmacol. 1997, 53, 1339–1345. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Kuhlenkamp, J.F.; Kaplowitz, N. Toxicity of azathioprine and monocrotaline in murine sinusoidal endothelial cells and hepatocytes: The role of glutathione and relevance to hepatic venoocclusive disease. Hepatology 1996, 23, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Salvesen, G. Regulated cell death: Signaling and mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NFκB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NFκB signals induce the expression of c-flip. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Matsumaru, K.; Feng, G.; Kaplowitz, N. Reduced glutathione depletion causes necrosis and sensitization to tumor necrosis factor-α-induced apoptosis in cultured mouse hepatocytes. Hepatology 2002, 36, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.H.; Campbell, J.S.; Stephenson, A.B.; Franklin, C.C.; Chaisson, M.; Poot, M.; Kavanagh, T.J.; Rabinovitch, P.S.; Fausto, N. Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am. J. Pathol. 2000, 157, 221–236. [Google Scholar] [CrossRef]
- Lou, H.; Kaplowitz, N. Glutathione depletion down-regulates tumor necrosis factor α-induced NFκB activity via IκB kinase-dependent and -independent mechanisms. J. Biol. Chem. 2007, 282, 29470–29481. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic. Biol. Med. 2006, 41, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on APAF-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. APAF-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Bratton, S.B.; Salvesen, G.S. Regulation of the APAF-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Manning, G. Necroptosis and inflammation. Annu. Rev. Biochem. 2016, 85, 743–763. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; He, G.W.; Kremer, A.E.; Murphy, J.M.; Petrie, E.J.; Amann, K.; Vandenabeele, P.; Linkermann, A.; Poremba, C.; Schleicher, U.; et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Investig. 2016, 126, 4346–4360. [Google Scholar] [CrossRef] [PubMed]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015, 129, 721–739. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Liu, Z.X.; Kaplowitz, N. Questions and controversies: The role of necroptosis in liver disease. Cell Death Discov. 2016, 2, 16089. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N.; de Almagro, M.C.; Vucic, D.; Komuves, L.; Ferrando, R.E.; French, D.M.; Webster, J.; et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Zhou, W.; Maki, J.L.; Yuan, J. Assays for necroptosis and activity of RIP kinases. Methods Enzymol. 2014, 545, 1–33. [Google Scholar] [PubMed]
- Sun, X.; Lee, J.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 1999, 274, 16871–16875. [Google Scholar] [CrossRef] [PubMed]
- Kasof, G.M.; Prosser, J.C.; Liu, D.; Lorenzi, M.V.; Gomes, B.C. The RIP-like kinase, RIP3, induces apoptosis and NFκB nuclear translocation and localizes to mitochondria. FEBS Lett. 2000, 473, 285–291. [Google Scholar] [CrossRef]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Mehrhof, F.; Harms, C.; Lattig-Tunnemann, G.; Lee, S.L.; Endres, M.; Li, M.; Sellge, G.; Mandic, A.D.; Trautwein, C.; et al. ARC is a novel therapeutic approach against acetaminophen-induced hepatocellular necrosis. J. Hepatol. 2013, 58, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Barilla, R.; et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015, 6, e1759. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Dueber, E.C. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator GPX4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Monshi, M.M.; Faulkner, L.; Gibson, A.; Jenkins, R.E.; Farrell, J.; Earnshaw, C.J.; Alfirevic, A.; Cederbrant, K.; Daly, A.K.; French, N.; et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology 2013, 57, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Terano, Y.; Sakagami, C.; Hasegawa, I.; Mizoguchi, Y.; Morisawa, S. Drug-specific T cells derived from patients with drug-induced allergic hepatitis. J. Immunol. 1992, 149, 706–716. [Google Scholar] [CrossRef]
- Dara, L.; Liu, Z.X.; Kaplowitz, N. Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int. 2015, 36, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Ingelman-Sundberg, M. The importance of patient-specific factors for hepatic drug response and toxicity. Int. J. Mol. Sci. 2016, 17, 1714. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Fullerton, A.M.; Semple, K.; Chea, L.S.; Proctor, W.R.; Bourdi, M.; Kleiner, D.E.; Zeng, X.; Ryan, P.M.; Dagur, P.K.; et al. Drug-induced allergic hepatitis developed in mice when myeloid-derived suppressor cells were depleted prior to halothane treatment. Hepatology 2015, 62, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and outcomes of 899 patients with drug-induced liver injury: The dilin prospective study. Gastroenterology 2015, 148, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Donaldson, P.T.; Bhatnagar, P.; Shen, Y.; Pe’er, I.; Floratos, A.; Daly, M.J.; Goldstein, D.B.; John, S.; Nelson, M.R.; et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet. 2009, 41, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Wuillemin, N.; Adam, J.; Fontana, S.; Krahenbuhl, S.; Pichler, W.J.; Yerly, D. HLA haplotype determines hapten or p-i t cell reactivity to flucloxacillin. J. Immunol. 2013, 190, 4956–4964. [Google Scholar] [CrossRef] [PubMed]
- Bhogaraju, A.; Nazeer, S.; Al-Baghdadi, Y.; Rahman, M.; Wrestler, F.; Patel, N. Diclofenac-associated hepatitis. South. Med. J. 1999, 92, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.K.; Aithal, G.P.; Leathart, J.B.; Swainsbury, R.A.; Dang, T.S.; Day, C.P. Genetic susceptibility to diclofenac-induced hepatotoxicity: Contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology 2007, 132, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Mallal, S.; Phillips, E.; Carosi, G.; Molina, J.M.; Workman, C.; Tomazic, J.; Jagel-Guedes, E.; Rugina, S.; Kozyrev, O.; Cid, J.F.; et al. HLA-B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 2008, 358, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Hautekeete, M.L.; Horsmans, Y.; van Waeyenberge, C.; Demanet, C.; Henrion, J.; Verbist, L.; Brenard, R.; Sempoux, C.; Michielsen, P.P.; Yap, P.S.; et al. HLA association of amoxicillin-clavulanate--induced hepatitis. Gastroenterology 1999, 117, 1181–1186. [Google Scholar] [CrossRef]
- Kindmark, A.; Jawaid, A.; Harbron, C.G.; Barratt, B.J.; Bengtsson, O.F.; Andersson, T.B.; Carlsson, S.; Cederbrant, K.E.; Gibson, N.J.; Armstrong, M.; et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008, 8, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Molokhia, M.; Shen, Y.; Urban, T.J.; Aithal, G.P.; Andrade, R.J.; Day, C.P.; Ruiz-Cabello, F.; Donaldson, P.T.; Stephens, C.; et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Balamurugan, A.; Saha, P.K.; Pandey, R.M.; Mehra, N.K. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment. Am. J. Respir. Crit. Care Med. 2002, 166, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, Y.; Tang, S.; Lv, X.; Wu, S.; Sun, F.; Xia, Y.; Zhan, S.Y. The association between HLA-DQB1 polymorphism and antituberculosis drug-induced liver injury: A case-control study. J. Clin. Pharm. Ther. 2015, 40, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Petros, Z.; Kishikawa, J.; Makonnen, E.; Yimer, G.; Habtewold, A.; Aklillu, E. HLA-B*57 allele is associated with concomitant anti-tuberculosis and antiretroviral drugs induced liver toxicity in ethiopians. Front. Pharmacol. 2017, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Spraggs, C.F.; Budde, L.R.; Briley, L.P.; Bing, N.; Cox, C.J.; King, K.S.; Whittaker, J.C.; Mooser, V.E.; Preston, A.J.; Stein, S.H.; et al. HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J. Clin. Oncol. 2011, 29, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Urban, T.J.; Nicoletti, P.; Chalasani, N.; Serrano, J.; Stolz, A.; Daly, A.; Aithal, G.; Dillon, J.; Navarro, V.; Odin, J.; et al. Minocycline hepatotoxicity: Clinical characterization and identification of HLA-B*35:02 as a risk factor. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, P.; Aithal, G.P.; Bjornsson, E.S.; Andrade, R.J.; Sawle, A.; Arrese, M.; Barnhart, H.X.; Bondon-Guitton, E.; Hayashi, P.H.; Bessone, F.; et al. Association of liver injury from specific drugs, or groups of drugs, with polymorphisms in HLA and other genes in a genome-wide association study. Gastroenterology 2017, 152, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Phillips, E.; Bartlett, J.A.; Sanne, I.; Lederman, M.M.; Hinkle, J.; Rousseau, F.; Dunn, D.; Pavlos, R.; James, I.; Mallal, S.A.; et al. Associations between HLA-DRB1*0102, HLA-B*5801, and hepatotoxicity during initiation of nevirapine-containing regimens in south africa. J. Acquir. Immune Defic. Syndr. 2013, 62, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- Uetrecht, J. Idiosyncratic drug reactions: Past, present, and future. Chem. Res. Toxicol. 2008, 21, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kodama, S.; Sugiyama, E.; Nakamura, R. Predictive genomic markers for severe adverse drug reactions. Yakugaku Zasshi 2015, 135, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, P.; Werk, A.N.; Sawle, A.; Shen, Y.; Urban, T.J.; Coulthard, S.A.; Bjornsson, E.S.; Cascorbi, I.; Floratos, A.; Stammschulte, T.; et al. HLA-DRB1*16:01-DQB1*05:02 is a novel genetic risk factor for flupirtine-induced liver injury. Pharmacogenet. Genom. 2016, 26, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Freneaux, E.; Larrey, D.; Lepage, V.; Douay, C.; Mallet, C.; Fromenty, B.; Benhamou, J.P.; Pessayre, D. Possible role of HLA in hepatotoxicity: An exploratory study in 71 patients with drug-induced idiosyncratic hepatitis. J. Hepatol. 1994, 20, 336–342. [Google Scholar] [CrossRef]
- Daly, A.K.; Day, C.P. Genetic association studies in drug-induced liver injury. Semin. Liver Dis. 2009, 29, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Russmann, S.; Kaye, J.A.; Jick, S.S.; Jick, H. Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: Cohort study using data from the UK general practice research database. Br. J. Clin. Pharmacol. 2005, 60, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.B.; Lewitzky, S.; Leroy, E.; Yang, F.; Zhao, X.; Klickstein, L.; Wright, T.M.; Meyer, J.; Paulding, C.A. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat. Genet. 2010, 42, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Johnson, T.; Wang, X.; Carpenter, C.; Graves, A.; Warren, L.; Xue, Z.; King, K.S.; Fraser, D.J.; Stinnett, S.; et al. HLA-B*57:01 confers susceptibility to pazopanib-associated liver injury in patients with cancer. Clin. Cancer Res. 2016, 22, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Takagi, H.; Yamamoto, M.; Matsumoto, T.; Nishiya, T.; Mori, K.; Shimizu, S.; Masumoto, H.; Okutani, Y. Ticlopidine-induced hepatotoxicity is associated with specific human leukocyte antigen genomic subtypes in japanese patients: A preliminary case-control study. Pharmacogenom. J. 2008, 8, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, M.; Takagi, H.; Mori, M. HLA-A33/B44/DR6 is highly related to intrahepatic cholestasis induced by tiopronin. Dig. Dis. Sci. 2000, 45, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Lucena, M.I.; Fernandez, M.C.; Pelaez, G.; Pachkoria, K.; Garcia-Ruiz, E.; Garcia-Munoz, B.; Gonzalez-Grande, R.; Pizarro, A.; Duran, J.A.; et al. Drug-induced liver injury: An analysis of 461 incidences submitted to the spanish registry over a 10-year period. Gastroenterology 2005, 129, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.S. Drug-induced liver injury: An overview over the most critical compounds. Arch. Toxicol. 2015, 89, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Bessone, F.; Hernandez, N.; Lucena, M.I.; Andrade, R.J.; Latin DILI Network; Spanish DILI Registry. The Latin American DILI registry experience: A successful ongoing collaborative strategic initiative. Int. J. Mol. Sci. 2016, 17, 313. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, E.S.; Bergmann, O.M.; Bjornsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, H.; Murata, Y.; Horiike, N.; Fukui, H.; Onji, M. Drug-induced liver injury in japan: An analysis of 1676 cases between 1997 and 2006. Hepatol. Res. 2009, 39, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A.; Maiuri, A.R.; Ganey, P.E. Idiosyncratic drug-induced liver injury: Is drug-cytokine interaction the linchpin? J. Pharmacol. Exp. Ther. 2017, 360, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Kusters, S.; Gantner, F.; Kunstle, G.; Tiegs, G. Interferon γ plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996, 111, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Watanabe, Y.; Akaike, T. Protective effect of hepatocyte growth factor on interferon-γ-induced cytotoxicity in mouse hepatocytes. Hepatology 1995, 21, 1585–1593. [Google Scholar] [PubMed]
- Vodovotz, Y.; Kim, P.K.; Bagci, E.Z.; Ermentrout, G.B.; Chow, C.C.; Bahar, I.; Billiar, T.R. Inflammatory modulation of hepatocyte apoptosis by nitric oxide: In vivo, in vitro, and in silico studies. Curr. Mol. Med. 2004, 4, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, M.; Muto, Y.; Ohnishi, H.; Yasuda, S.; Sano, K.; Naito, T.; Maeda, T.; Yamada, T.; Moriwaki, H. Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology 1994, 106, 450–458. [Google Scholar] [CrossRef]
- Gantner, F.; Leist, M.; Jilg, S.; Germann, P.G.; Freudenberg, M.A.; Tiegs, G. Tumor necrosis factor-induced hepatic DNA fragmentation as an early marker of T cell-dependent liver injury in mice. Gastroenterology 1995, 109, 166–176. [Google Scholar] [CrossRef]
- Gantner, F.; Leist, M.; Lohse, A.W.; Germann, P.G.; Tiegs, G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: The role of tumor necrosis factor. Hepatology 1995, 21, 190–198. [Google Scholar] [PubMed]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin a. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Gantner, F.; Naumann, H.; Bluethmann, H.; Vogt, K.; Brigelius-Flohe, R.; Nicotera, P.; Volk, H.D.; Wendel, A. Tumor necrosis factor-induced apoptosis during the poisoning of mice with hepatotoxins. Gastroenterology 1997, 112, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Liu, Z.; Kaplowitz, N. A murder mystery in the liver: Who done it and how? J. Clin. Investig. 2016, 126, 4068–4071. [Google Scholar] [CrossRef] [PubMed]
- Suda, J.; Dara, L.; Yang, L.; Aghajan, M.; Song, Y.; Kaplowitz, N.; Liu, Z.X. Knockdown of RIPK1 markedly exacerbates murine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of nf-kappab. J. Immunol. 2016, 197, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Filliol, A.; Piquet-Pellorce, C.; Le Seyec, J.; Farooq, M.; Genet, V.; Lucas-Clerc, C.; Bertin, J.; Gough, P.J.; Dimanche-Boitrel, M.T.; Vandenabeele, P.; et al. RIPK1 protects from TNF-α-mediated liver damage during hepatitis. Cell Death Dis. 2016, 7, e2462. [Google Scholar] [CrossRef] [PubMed]
- Von Greyerz, S.; Bultemann, G.; Schnyder, K.; Burkhart, C.; Lotti, B.; Hari, Y.; Pichler, W.J. Degeneracy and additional alloreactivity of drug-specific human αβ+ T cell clones. Int. Immunol. 2001, 13, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Von Greyerz, S.; Zanni, M.P.; Frutig, K.; Schnyder, B.; Burkhart, C.; Pichler, W.J. Interaction of sulfonamide derivatives with the TCR of sulfamethoxazole-specific human αβ+ T cell clones. J. Immunol. 1999, 162, 595–602. [Google Scholar] [PubMed]
- Pichler, W.J. Pharmacological interaction of drugs with antigen-specific immune receptors: The p-i concept. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.I.; Aithal, G.P. Human leukocyte antigen genetic risk factors of drug-induced liver toxicology. Expert Opin. Drug Metab. Toxicol. 2015, 11, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, D.A.; Grant, B.J.; Pompeu, Y.A.; Sidney, J.; Harndahl, M.; Southwood, S.; Oseroff, C.; Lu, S.; Jakoncic, J.; de Oliveira, C.A.; et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc. Natl. Acad. Sci. USA 2012, 109, 9959–9964. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.Y.; Chung, W.H.; Huang, H.W.; Chen, Y.T.; Hung, S.I. Direct interaction between HLA-B and carbamazepine activates T cells in patients with stevens-johnson syndrome. J. Allergy Clin. Immunol. 2012, 129, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P. A review of the common properties of drugs with idiosyncratic hepatotoxicity and the “multiple determinant hypothesis” for the manifestation of idiosyncratic drug toxicity. Chem. Biol. Interact. 2002, 142, 7–23. [Google Scholar] [CrossRef]
- Ulrich, R.G. Idiosyncratic toxicity: A convergence of risk factors. Annu. Rev. Med. 2007, 58, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.C.; Drummond, G.B. Halothane hepatitis. Br. J. Anaesth. 1991, 67, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Dugan, C.M.; MacDonald, A.E.; Roth, R.A.; Ganey, P.E. A mouse model of severe halothane hepatitis based on human risk factors. J. Pharmacol. Exp. Ther. 2010, 333, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Khouri, M.R.; Saul, S.H.; Dlugosz, A.A.; Soloway, R.D. Hepatocanalicular injury associated with vitamin a derivative etretinate. An idiosyncratic hypersensitivity reaction. Dig. Dis. Sci. 1987, 32, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Fukano, M.; Amano, S.; Sato, J.; Yamamoto, K.; Adachi, H.; Okabe, H.; Fujiyama, Y.; Bamba, T. Subacute hepatic failure associated with a new antidiabetic agent, troglitazone: A case report with autopsy examination. Hum. Pathol. 2000, 31, 250–253. [Google Scholar] [CrossRef]
- Deng, X.; Luyendyk, J.P.; Ganey, P.E.; Roth, R.A. Inflammatory stress and idiosyncratic hepatotoxicity: Hints from animal models. Pharmacol. Rev. 2009, 61, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Ganey, P.E.; Roth, R.A. Idiosyncratic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity. Toxicol. Sci. 2010, 118, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Uetrecht, J. The role of CD8 T cells in amodiaquine-induced liver injury in PD1−/− mice cotreated with anti-CTLA-4. Chem. Res. Toxicol. 2015, 28, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Metushi, I.G.; Hayes, M.A.; Uetrecht, J. Treatment of PD-1−/− mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015, 61, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Calne, R.Y.; Sells, R.A.; Pena, J.R.; Davis, D.R.; Millard, P.R.; Herbertson, B.M.; Binns, R.M.; Davies, D.A. Induction of immunological tolerance by porcine liver allografts. Nature 1969, 223, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Gerken, G. Local control of the immune response in the liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Sanchez-Fueyo, A.; Samuel, D. From immunosuppression to tolerance. J. Hepatol. 2015, 62, S170–S185. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Long, M.W.; Thorgeirsson, U.P.; Jollow, D.J. Acetylation rates and monthly liver function tests during one year of isoniazid preventive therapy. Chest 1975, 68, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Uetrecht, J.; Kaplowitz, N. Inhibition of immune tolerance unmasks drug-induced allergic hepatitis. Hepatology 2015, 62, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E.; de Zanger, R.B.; Charels, K.; van der Smissen, P.; McCuskey, R.S. The liver sieve: Considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of disse. Hepatology 1985, 5, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.; Dobbs, B.R.; Rogers, G.W. Lipoproteins and the liver sieve: The role of the fenestrated sinusoidal endothelium in lipoprotein metabolism, atherosclerosis, and cirrhosis. Hepatology 1995, 21, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Knolle, P.A.; Bilo, K.; Uhrig, A.; Waldmann, C.; Ibe, M.; Schmitt, E.; Gerken, G.; Meyer Zum Buschenfelde, K.H. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and kupffer cells. Gastroenterology 1996, 110, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Germann, T.; Treichel, U.; Uhrig, A.; Schmitt, E.; Hegenbarth, S.; Lohse, A.W.; Gerken, G. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J. Immunol. 1999, 162, 1401–1407. [Google Scholar] [PubMed]
- Bissell, D.M.; Wang, S.S.; Jarnagin, W.R.; Roll, F.J. Cell-specific expression of transforming growth factor-β in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J. Clin. Investig. 1995, 96, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Uhrig, A.; Protzer, U.; Trippler, M.; Duchmann, R.; Meyer zum Buschenfelde, K.H.; Gerken, G. Interleukin-10 expression is autoregulated at the transcriptional level in human and murine kupffer cells. Hepatology 1998, 27, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.; Schlaak, J.; Uhrig, A.; Kempf, P.; zum Buschenfelde, K.H.M.; Gerken, G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J. Hepatol. 1995, 22, 226–229. [Google Scholar] [CrossRef]
- Rieder, H.; Ramadori, G.; Allmann, K.H.; zum Buschenfelde, K.H.M. Prostanoid release of cultured liver sinusoidal endothelial cells in response to endotoxin and tumor necrosis factor: Comparison with umbilical vein endothelial cells. J. Hepatol. 1990, 11, 359–366. [Google Scholar] [CrossRef]
- Dieter, P.; Schulze-Specking, A.; Karck, U.; Decker, K. Prostaglandin release but not superoxide production by rat Kupffer cells stimulated in vitro depends on Na+/H+ exchange. Eur. J. Biochem. 1987, 170, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Johnston, A.; Uetrecht, J. Effects of immunization and checkpoint inhibition on amodiaquine-induced liver injury. J. Immunotoxicol. 2017, 14, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Acetaminophen and the U.S. acute liver failure study group: Lowering the risks of hepatic failure. Hepatology 2004, 40, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Major, J.M.; Zhou, E.H.; Wong, H.L.; Trinidad, J.P.; Pham, T.M.; Mehta, H.; Ding, Y.; Staffa, J.A.; Iyasu, S.; Wang, C.; et al. Trends in rates of acetaminophen-related adverse events in the United States. Pharmacoepidemiol. Drug Saf. 2016, 25, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Nourjah, P.; Ahmad, S.R.; Karwoski, C.; Willy, M. Estimates of acetaminophen (paracetomal)-associated overdoses in the United States. Pharmacoepidemiol. Drug Saf. 2006, 15, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Rowden, A.K.; Norvell, J.; Eldridge, D.L.; Kirk, M.A. Updates on acetaminophen toxicity. Med. Clin. N. Am. 2005, 89, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.F. Gastrointestinal absorption of drugs. Med. Clin. N. Am. 1974, 58, 907–916. [Google Scholar] [CrossRef]
- Josting, D.; Winne, D.; Bock, K.W. Glucuronidation of paracetamol, morphine and 1-naphthol in the rat intestinal loop. Biochem. Pharmacol. 1976, 25, 613–616. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185–194. [Google Scholar] [PubMed]
- Mitchell, J.R.; Thorgeirsson, S.S.; Potter, W.Z.; Jollow, D.J.; Keiser, H. Acetaminophen-induced hepatic injury: Protective role of glutathione in man and rationale for therapy. Clin. Pharmacol. Ther. 1974, 16, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Van de Straat, R.; de Vries, J.; Kulkens, T.; Debets, A.J.; Vermeulen, N.P. Paracetamol, 3-monoalkyl- and 3,5-dialkyl derivatives: Comparison of their microsomal cytochrome P-450 dependent oxidation and toxicity in freshly isolated hepatocytes. Biochem. Pharmacol. 1986, 35, 3693–3699. [Google Scholar] [CrossRef]
- Moldeus, P. Paracetamol metabolism and toxicity in isolated hepatocytes from rat and mouse. Biochem. Pharmacol. 1978, 27, 2859–2863. [Google Scholar] [CrossRef]
- Gibson, J.D.; Pumford, N.R.; Samokyszyn, V.M.; Hinson, J.A. Mechanism of acetaminophen-induced hepatotoxicity: Covalent binding versus oxidative stress. Chem. Res. Toxicol. 1996, 9, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Pumford, N.R.; Hinson, J.A.; Benson, R.W.; Roberts, D.W. Immunoblot analysis of protein containing 3-(cystein-S-yl) acetaminophen adducts in serum and subcellular liver fractions from acetaminophen-treated mice. Toxicol. Appl. Pharmacol. 1990, 104, 521–532. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Mkrtchian, S.; Ingelman-Sundberg, M. The role of micrornas in liver injury at the crossroad between hepatic cell death and regeneration. Biochem. Biophys. Res. Commun. 2017, 482, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Marzolf, B.; Troisch, P.; Brightman, A.; Hu, Z.; Hood, L.E.; Galas, D.J. Circulating micrornas, potential biomarkers for drug-induced liver injury. Proc. Natl. Acad. Sci. USA 2009, 106, 4402–4407. [Google Scholar] [CrossRef] [PubMed]
- Szkolnicka, D.; Lucendo-Villarin, B.; Moore, J.K.; Simpson, K.J.; Forbes, S.J.; Hay, D.C. Reducing hepatocyte injury and necrosis in response to paracetamol using noncoding RNAs. Stem Cells Transl. Med. 2016, 5, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 173–179. [Google Scholar] [CrossRef] [PubMed]
- Gujral, J.S.; Knight, T.R.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Mode of cell death after acetaminophen overdose in mice: Apoptosis or oncotic necrosis? Toxicol. Sci. 2002, 67, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Gujral, J.S.; Bajt, M.L. Apoptosis and necrosis in liver disease. Liver Int. 2004, 24, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.A.; Fisher, M.A.; Simmons, C.A.; Farhood, A.; Jaeschke, H. Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol. Appl. Pharmacol. 1999, 156, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Bajt, M.L.; Cover, C.; Lemasters, J.J.; Jaeschke, H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol. Sci. 2006, 94, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Williams, C.D.; Farhood, A. No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatology 2011, 53, 718–719. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Gadang, V.; Jaeschke, A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol. Pharmacol. 2012, 82, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S.; Hikiba, Y.; Ohmae, T.; Shibata, W.; Yanai, A.; Sakamoto, K.; Ogura, K.; Noguchi, T.; Karin, M.; et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 2008, 135, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. C-jun n-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Win, S.; Than, T.A.; Yin, S.; Ye, M.; Hu, H.; Kaplowitz, N. Antcin h protects against acute liver injury through disruption of the interaction of c-Jun N-terminal kinase with mitochondria. Antioxid. Redox Signal. 2017, 26, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed]
- Uzi, D.; Barda, L.; Scaiewicz, V.; Mills, M.; Mueller, T.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Iwawaki, T.; Nahmias, Y.; Xavier, R.; et al. Chop is a critical regulator of acetaminophen-induced hepatotoxicity. J. Hepatol. 2013, 59, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Kon, K.; Kim, J.S.; Jaeschke, H.; Lemasters, J.J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic. Res. 2011, 45, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Loguidice, A.; Boelsterli, U.A. Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin d-regulated permeability transition. Hepatology 2011, 54, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Khetani, S.R. Advances in engineered liver models for investigating drug-induced liver injury. BioMed Res. Int. 2016, 2016, 1829148. [Google Scholar] [CrossRef] [PubMed]
- Porceddu, M.; Buron, N.; Roussel, C.; Labbe, G.; Fromenty, B.; Borgne-Sanchez, A. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol. Sci. 2012, 129, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Gerbal-Chaloin, S.; Funakoshi, N.; Caillaud, A.; Gondeau, C.; Champon, B.; Si-Tayeb, K. Human induced pluripotent stem cells in hepatology: Beyond the proof of concept. Am. J. Pathol. 2014, 184, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R.; Schulze, J.; Eickhoff, A.; Danan, G. Drug induced liver injury: Can biomarkers assist RUCAM in causality assessment? Int. J. Mol. Sci. 2017, 18, 803. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J.; Watkins, P.B.; Bonkovsky, H.L.; Chalasani, N.; Davern, T.; Serrano, J.; Rochon, J.; Group, D.S. Drug-induced liver injury network (DILIN) prospective study: Rationale, design and conduct. Drug Saf. 2009, 32, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Lucena, M.I.; Camargo, R.; Andrade, R.J.; Perez-Sanchez, C.J.; de la Cuesta, F.S. Comparison of two clinical scales for causality assessment in hepatotoxicity. Hepatology 2001, 33, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Teschke, R. Rucam in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2016, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Causality assessment versus guilt-by-association in drug hepatotoxicity. Hepatology 2001, 33, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Russmann, S.; Kullak-Ublick, G.A.; Grattagliano, I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr. Med. Chem. 2009, 16, 3041–3053. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Drug | Reference | Human Leukocyte Antigen Associations |
|---|---|---|
| Abacavir | [70] | B*57:01 |
| Allopurinol | [81] | B*58:01 ^ |
| Amoxicillin-Clavulanate | [71,72,73] | A*02:01, B*18:01, DRB1*1501, DQB1*0602, DRB1*07 ‡, A*3002, DQB1*0402 |
| Anti-Tuberculous Drugs | [74,75,76] | DQB1*02:01, DQB1*05:02, DQA1*01:02 ‡, B*57 #, DRB1*03 |
| Antiretroviral Drugs | [76] | B*57 # |
| Carbamazepine | [82] | B*15:11 ^, A*31:01 ^ |
| Clometacin | [84] | B*08 |
| Diclofenac | [85] | DRB1*13 |
| Fenofibrate | [79] | A*33:01 |
| Flucloxacillin | [66,67,86] | B*57:01, DRB1*07:01, DQB1*03:03, DRB1*15 ‡ |
| Flupirtine | [83] | DRB1*16:01, DQB1*05:02 |
| Lapatinib | [77] | DQA1*02:01, DQB1*02:02, DRB1*07:01 |
| Lumiracoxib | [87] | DRB1*15:01, DQB1*06:02, DRB5*01:01, DQA1*01:02 |
| Minocyclin | [78] | HLA B*35:02, B*35:02 |
| Nevirapine | [80] | DRB1*01:02, DRB1*01, B*58:01 |
| Pazopanib | [88] | B*57:01 |
| Phenobarbital | [82] | B*51:01 ^ |
| Terbinafine | [80] | A*33:01 |
| Ticlopidine | [89] | A*33:03, A*33:01, B*44:03, Cw*1403, DRB1*1302, DQB1*0604 |
| Tiopronine | [90] | A*33 B44 DR ^ |
| Ximelagatran | [72] | DRB1*0701, DQA1*0201 |
| Zonisamide | [82] | A*02:07 ^ |
| Name of Hypothesis | Definition |
|---|---|
| Hapten hypothesis | Reactive metabolites are generated from drugs that can bind to endogenous proteins and form neoantigens, activating the immune system. |
| Pharmacological Interaction (p-i) Hypothesis | Certain drugs can act like small molecules and directly form non-covalent interactions with MHC molecules altering their binding pocket. |
| The altered peptide repertoire hypothesis | Drugs induce mistargeting of endogenous peptides to the wrong HLA leading to autoimmunity. |
| Multiple determinant hypothesis | Multiple risk factors (i.e., polymorphisms, age, gender), are necessary and overlap together to induce DILI. |
| Inflammatory stress hypothesis | A small inflammation occurring during drug therapy could interact with the action of the drug and escalate into liver injury. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018. https://doi.org/10.3390/ijms18051018
Iorga A, Dara L, Kaplowitz N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. International Journal of Molecular Sciences. 2017; 18(5):1018. https://doi.org/10.3390/ijms18051018
Chicago/Turabian StyleIorga, Andrea, Lily Dara, and Neil Kaplowitz. 2017. "Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis" International Journal of Molecular Sciences 18, no. 5: 1018. https://doi.org/10.3390/ijms18051018
APA StyleIorga, A., Dara, L., & Kaplowitz, N. (2017). Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. International Journal of Molecular Sciences, 18(5), 1018. https://doi.org/10.3390/ijms18051018