
SUMO-Specific Cysteine Protease 1 Promotes Epithelial Mesenchymal Transition of Prostate Cancer Cells via Regulating SMAD4 deSUMOylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
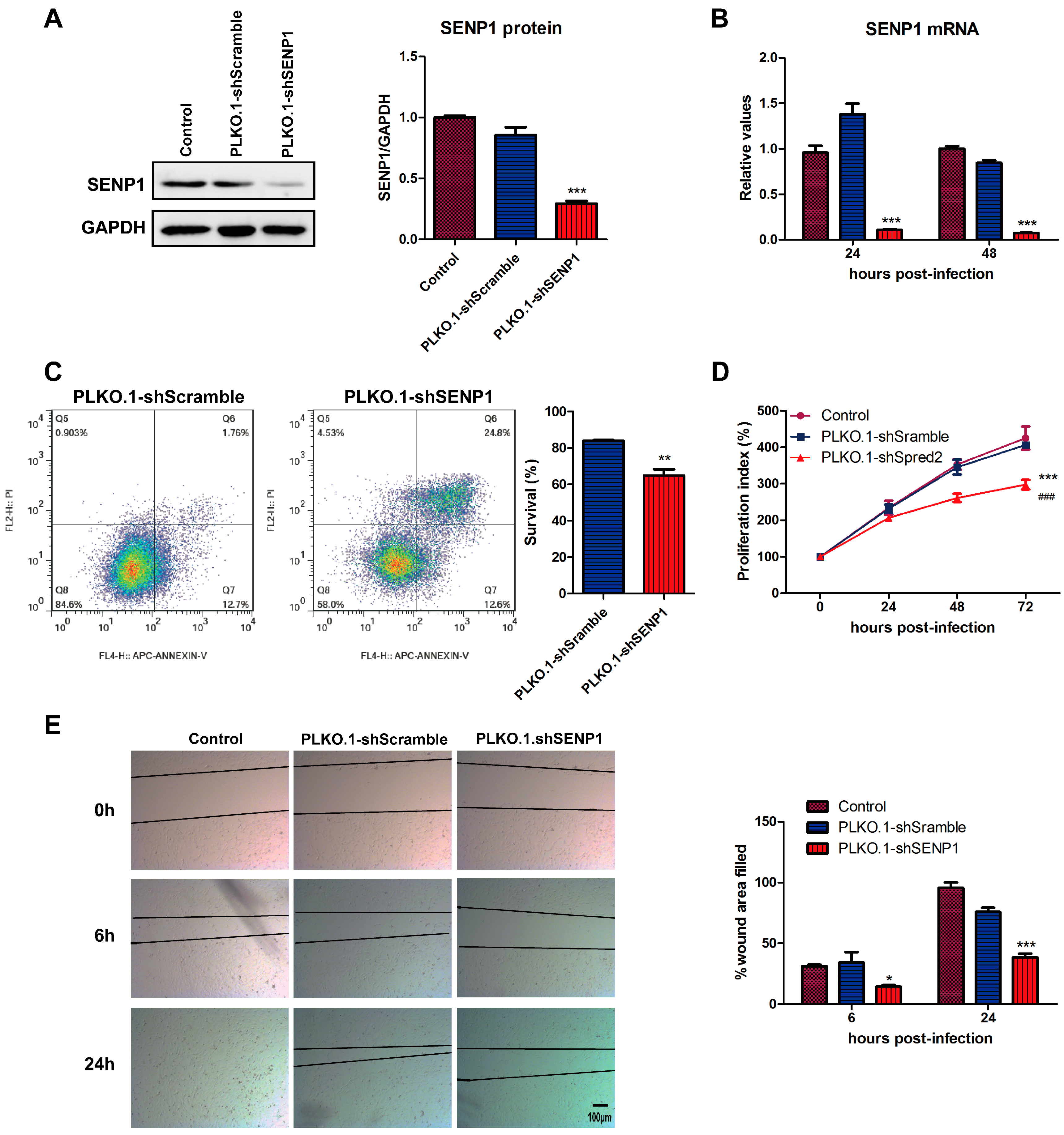
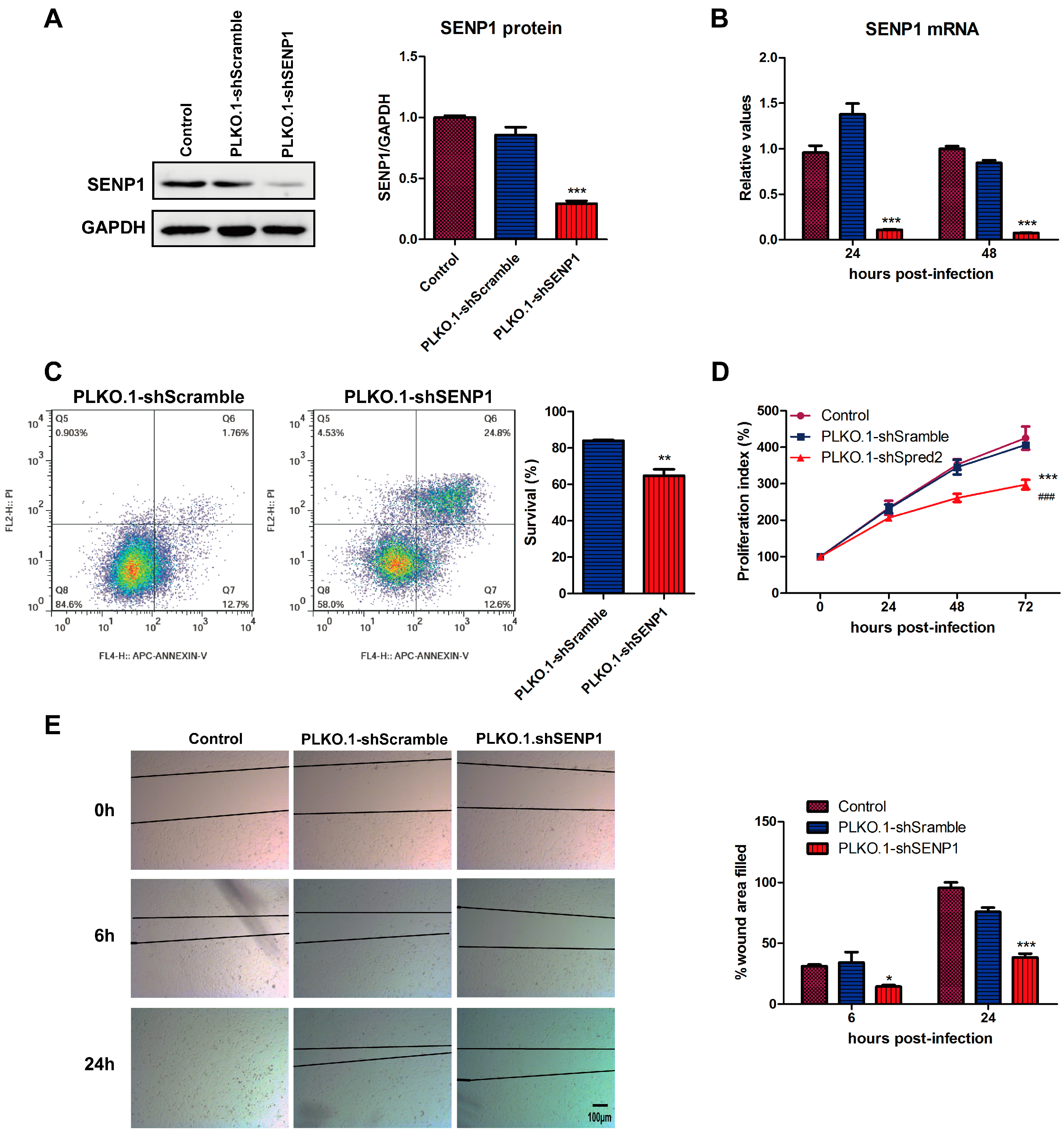
2.1. SENP1 Silencing Induces Apoptosis, Inhibits Cell Growth and Migration in PC3M, an Androgen-Independent Prostate Cancer Cell Line
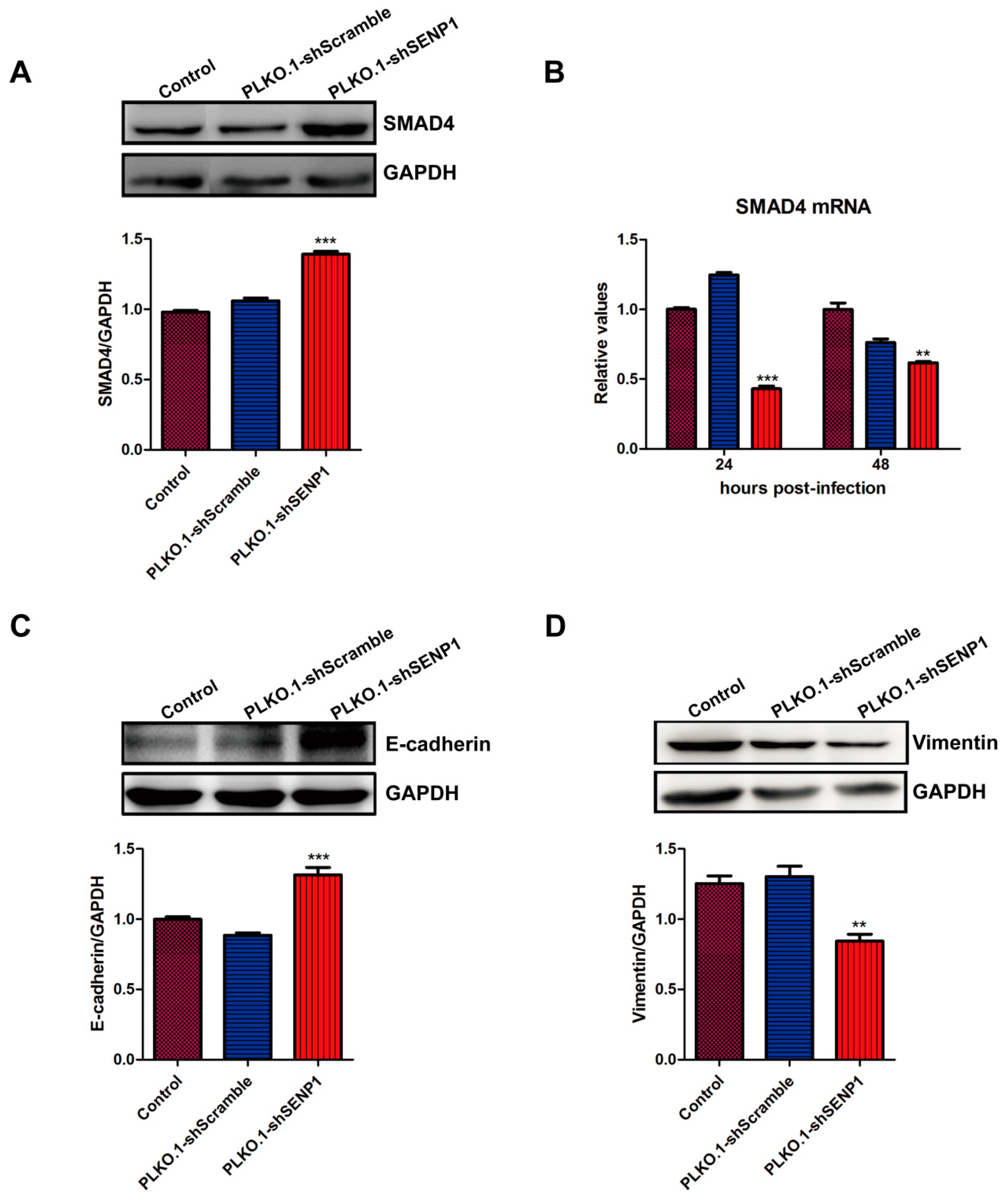
2.2. SENP1 Interference Enhances TGF-β/Smads Signaling and Inhibits EMT in PC3M Cells
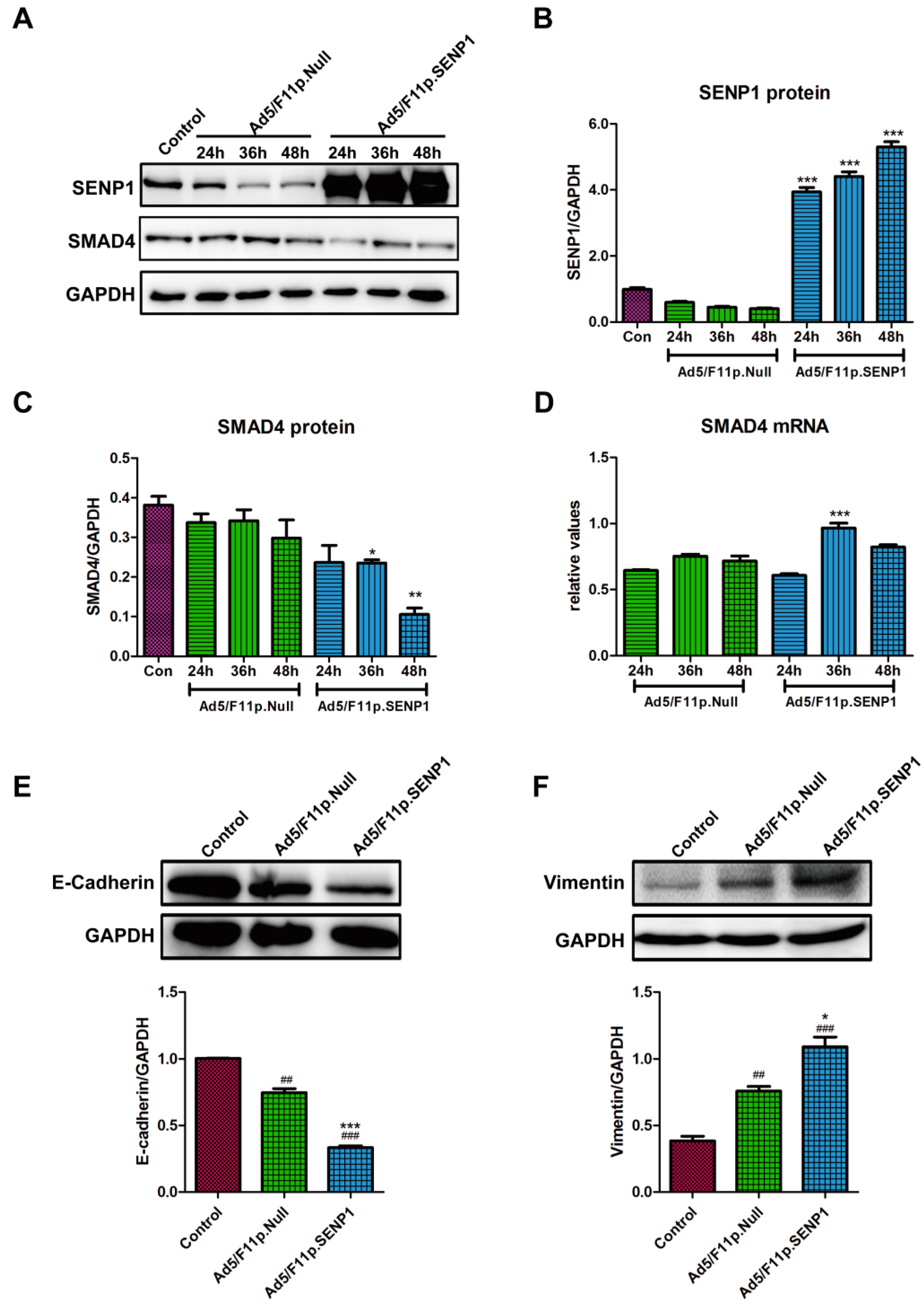
2.3. SENP1 Over-Expression Impairs TGF-β/Smads Signaling and Promotes EMT of Androgen-Dependent Prostate Cancer Cells, LNCaP
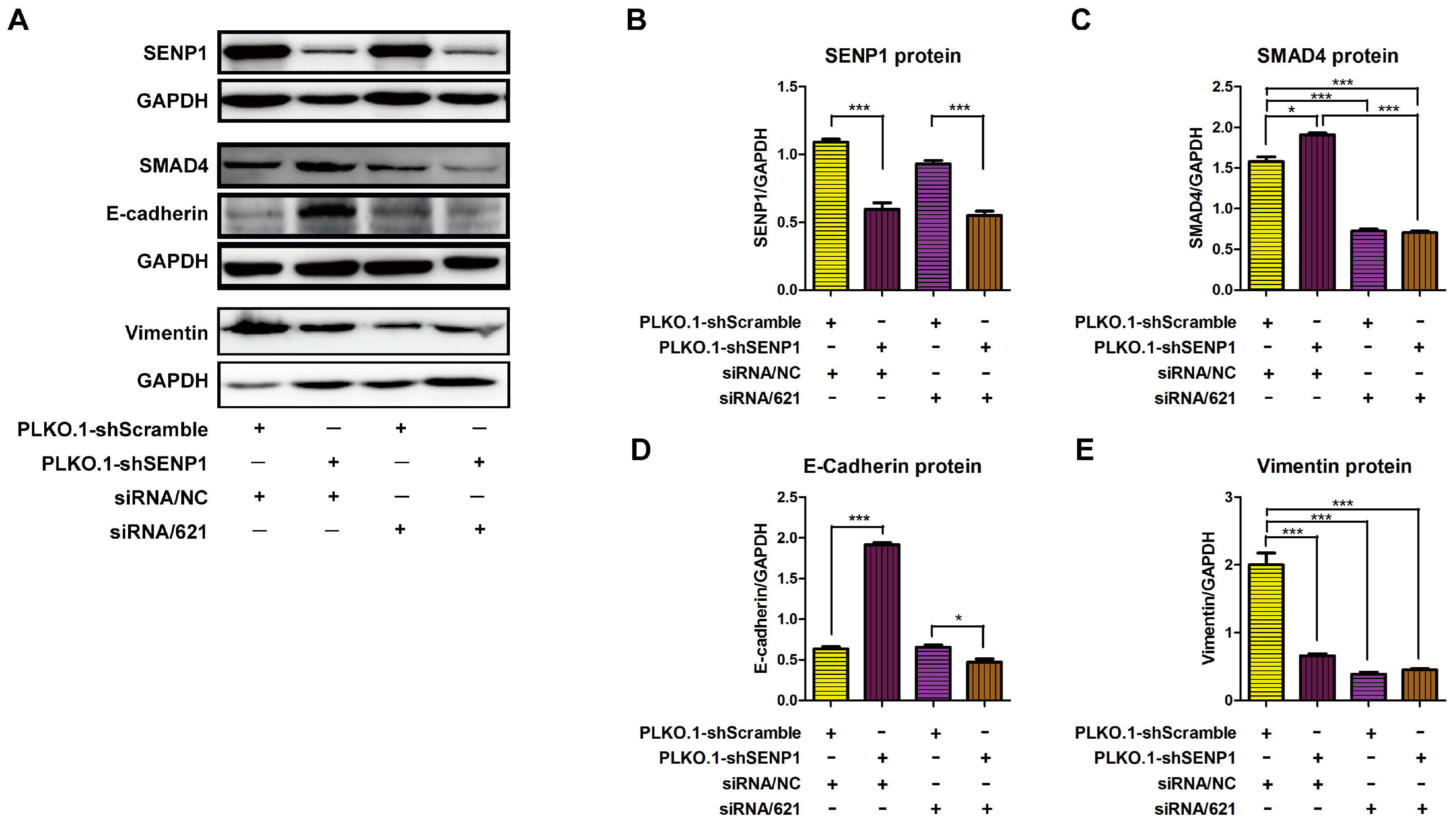
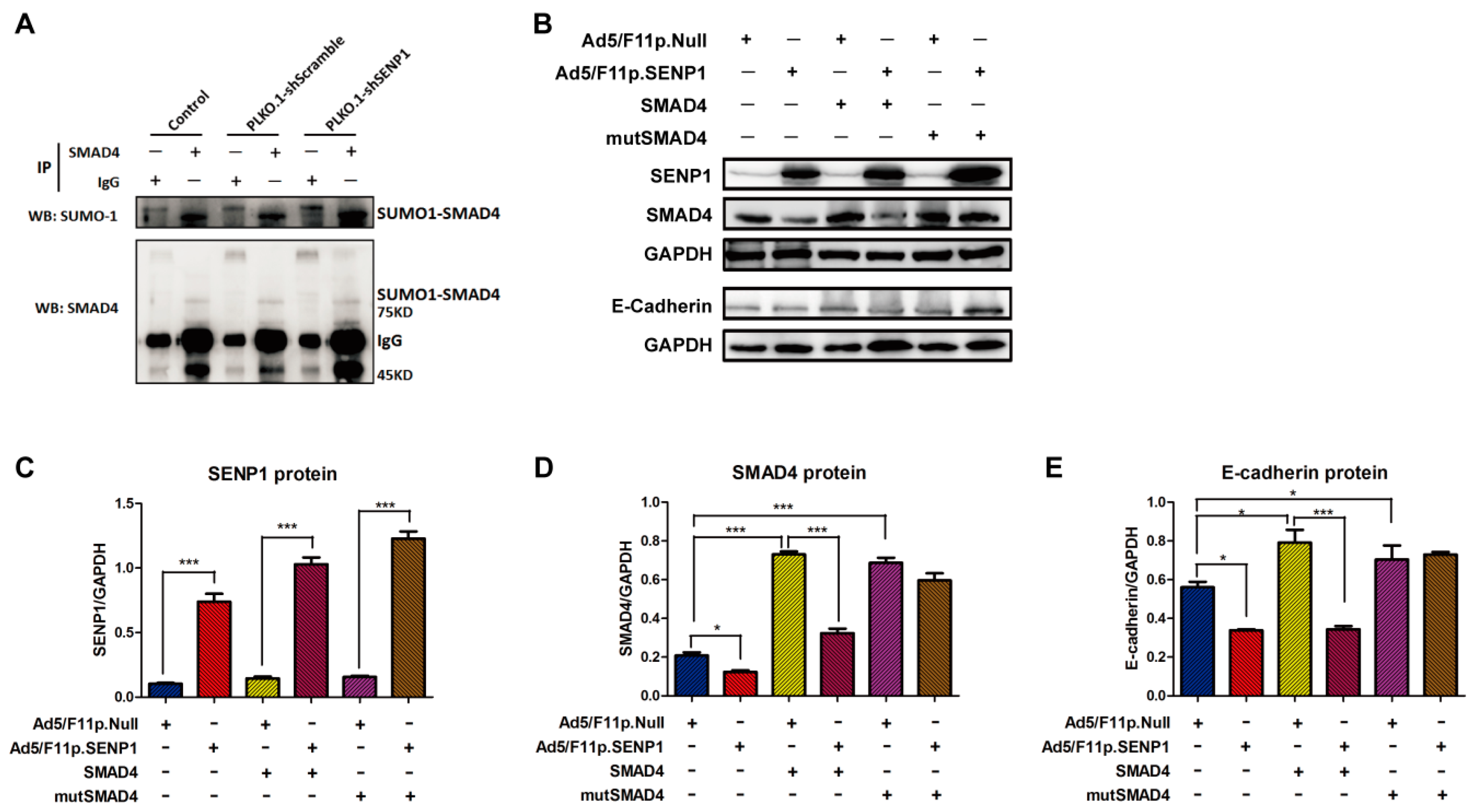
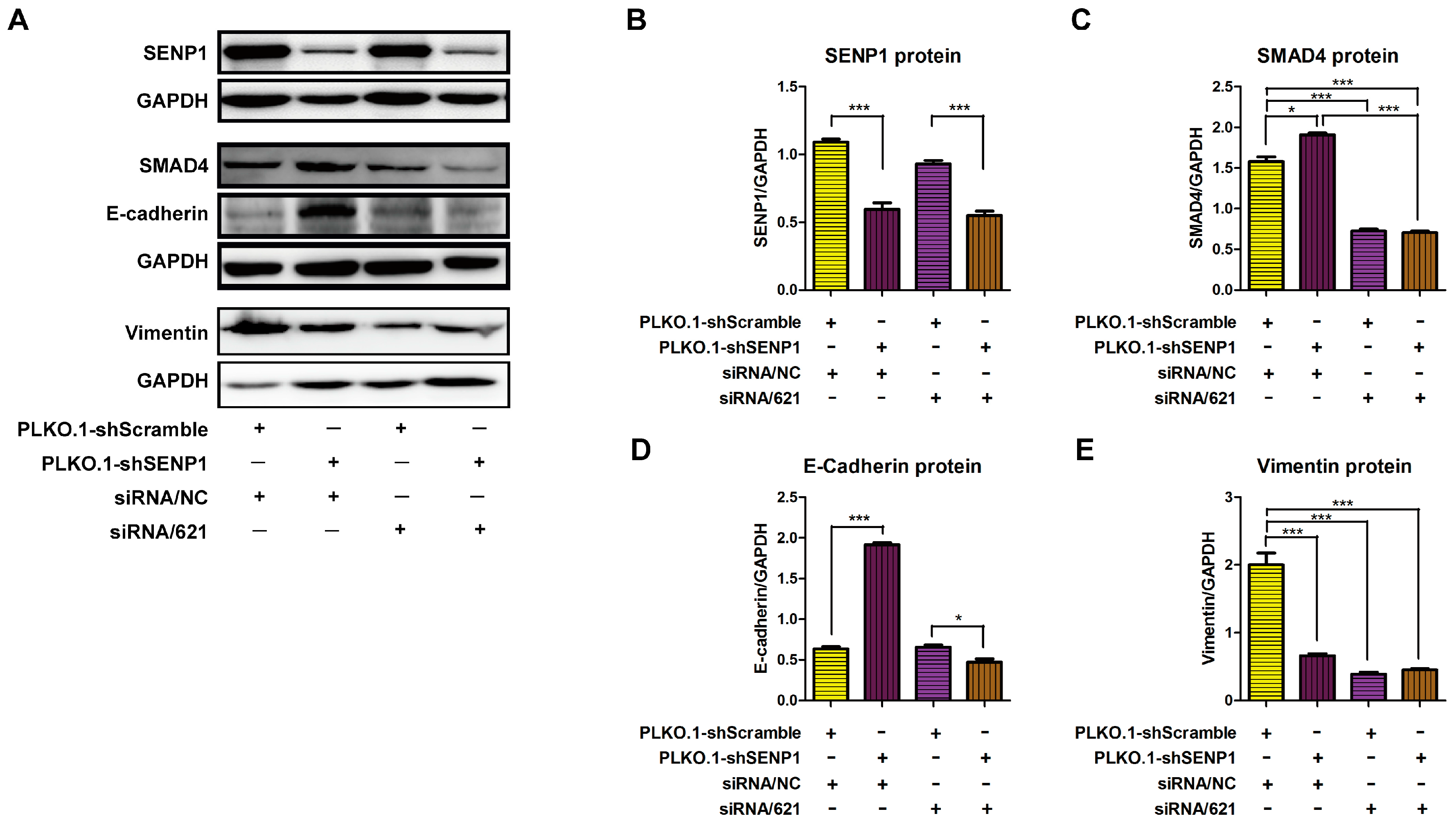
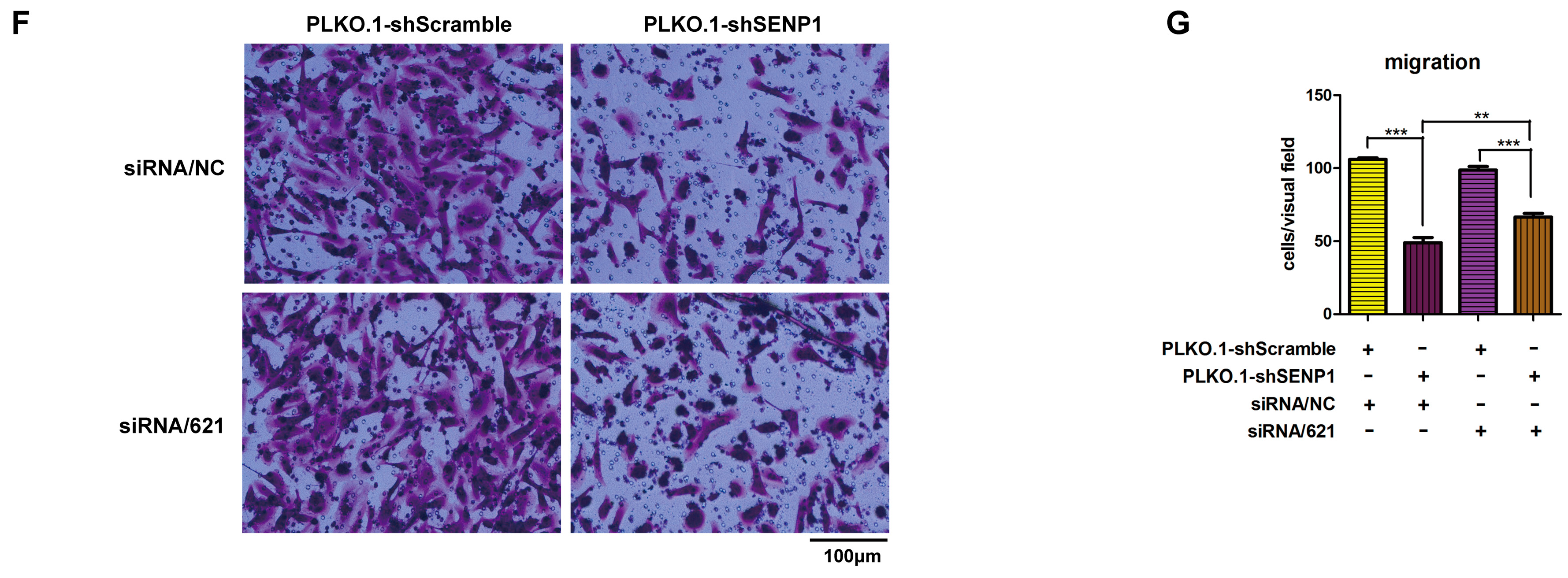
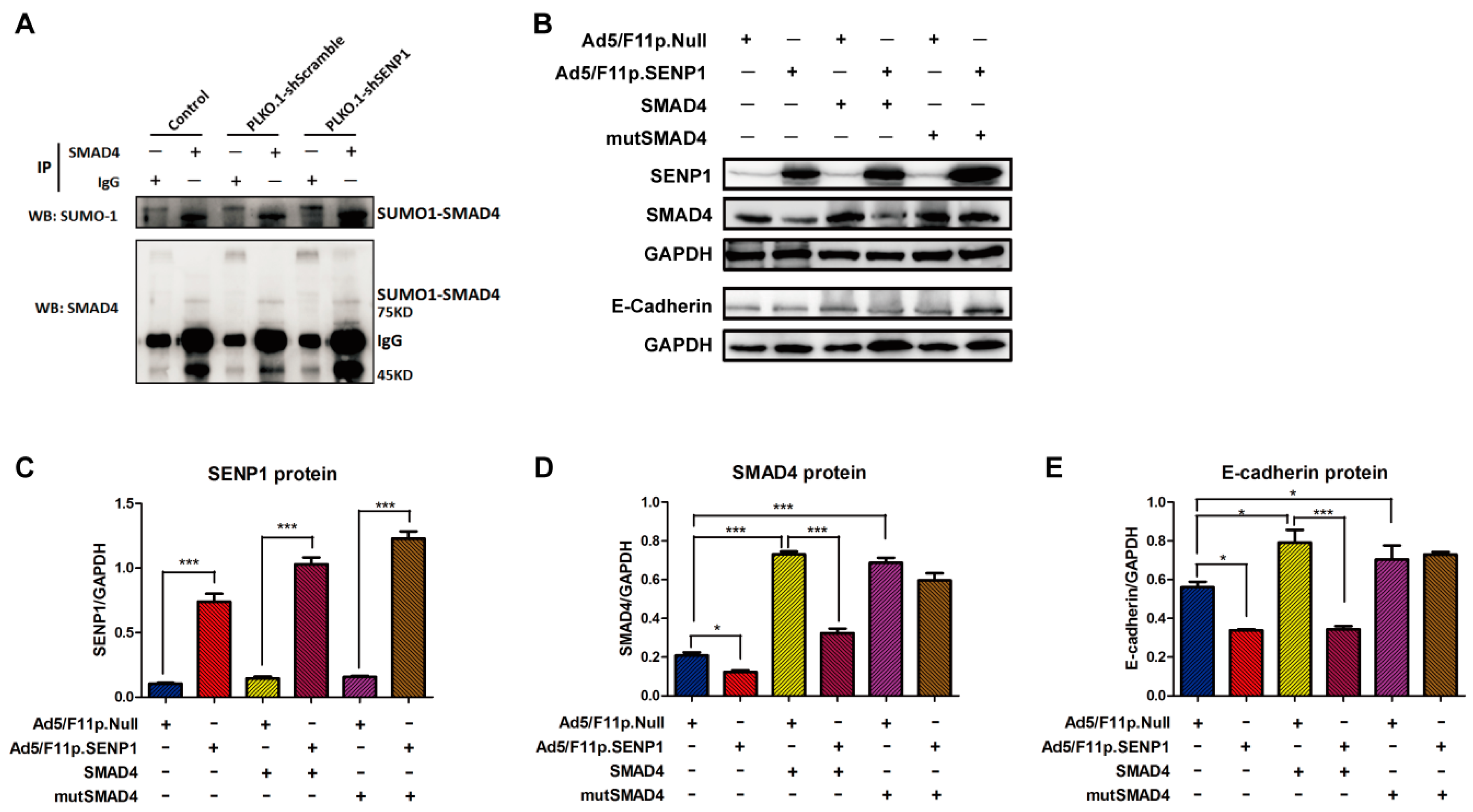
2.4. SENP1 Regulates deSUMOylation of SMAD4 and Promotes EMT of Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Vectors
4.3. Biological Analysis of PC3M Cells after Infection with Lentiviruses
4.3.1. Apoptosis Analysis
4.3.2. Proliferation Assay
4.3.3. Migration Assay
4.4. Western-Blotting and Immunoprecipitation
4.5. Real-Time Reverse Transcript Polymerase Chain Reaction (RT-PCR)
4.6. Statistical Analysis
5. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Sutinen, P.; Malinen, M.; Heikkinen, S.; Palvimo, J.J. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res. 2014, 42, 8310–8319. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.E.; Huang, T.T. How SUMOylation fine-tunes the fanconi anemia DNA repair pathway. Front. Genet. 2016, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Bawa-Khalfe, T.; Yeh, E.T. SUMO losing balance: SUMO proteases disrupt SUMO homeostasis to facilitate cancer development and progression. Genes Cancer 2010, 1, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, J.; Zhang, J.; Wang, L.; Cao, J. Depletion of SENP1 suppresses the proliferation and invasion of triple-negative breast cancer cells. Oncol. Rep. 2016, 36, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Inc. Cancer Facts & Figures 2015. Available online: http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf (accessed on 12 April 2017).
- Kaikkonen, S.; Jaaskelainen, T.; Karvonen, U.; Rytinki, M.M.; Makkonen, H.; Gioeli, D.; Paschal, B.M.; Palvimo, J.J. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol. Endocrinol. 2009, 23, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Bawa-Khalfe, T.; Cheng, J.; Lin, S.H.; Ittmann, M.M.; Yeh, E.T. SENP1 induces prostatic intraepithelial neoplasia through multiple mechanisms. J. Biol. Chem. 2010, 285, 25859–25866. [Google Scholar] [CrossRef] [PubMed]
- Burdelski, C.; Menan, D.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Melling, N.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; et al. The prognostic value of SUMO1/Sentrin specific peptidase 1 (SENP1) in prostate cancer is limited to ERG-fusion positive tumors lacking PTEN deletion. BMC Cancer 2015, 15, 538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Dai, A.; Jiang, Y.; Tan, X.; Zhang, X. SENP1 enhances hypoxiainduced proliferation of rat pulmonary artery smooth muscle cells by regulating hypoxiainducible factor1α. Mol. Med. Rep. 2016, 13, 3482–3490. [Google Scholar] [PubMed]
- Ao, Q.; Su, W.; Guo, S.; Cai, L.; Huang, L. SENP1 desensitizes hypoxic ovarian cancer cells to cisplatin by up-regulating HIF-1α. Sci. Rep. 2015, 5, 16396. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-β signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [CrossRef]
- Ding, Z.; Wu, C.J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wu, S.P.; Creighton, C.J.; Dai, F.; Xie, X.; Cheng, C.M.; Frolov, A.; Ayala, G.; Lin, X.; Feng, X.H.; et al. COUP-TFII inhibits TGF-β-induced growth barrier to promote prostate tumorigenesis. Nature 2013, 493, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Melchior, F.; Feng, X.H.; Lin, X. Regulation of Smad4 sumoylation and transforming growth factor-β signaling by protein inhibitor of activated STAT1. J. Biol. Chem. 2004, 279, 22857–22865. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, M.; Liang, Y.Y.; Brunicardi, F.C.; Melchior, F.; Feng, X.H. Activation of transforming growth factor-β signaling by SUMO-1 modification of tumor suppressor Smad4/DPC4. J. Biol. Chem. 2003, 278, 18714–18719. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Cannata, D.H.; Kirschenbaum, A.; Levine, A.C. Androgen deprivation therapy as primary treatment for prostate cancer. J. Clin. Endocrinol. Metab. 2012, 97, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R. The use of bisphosphonates in cancer treatment. Ann. N. Y. Acad. Sci. 2011, 1218, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damiao, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Zhu, M.L.; Partin, J.V.; Bruckheimer, E.M.; Strup, S.E.; Kyprianou, N. TGF-β signaling and androgen receptor status determine apoptotic cross-talk in human prostate cancer cells. Prostate 2008, 68, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Gallo-Oller, G.; Vollmann-Zwerenz, A.; Melendez, B.; Rey, J.A.; Hau, P.; Dotor, J.; Castresana, J.S. P144, a transforming growth factor β inhibitor peptide, generates antitumoral effects and modifies SMAD7 and SKI levels in human glioblastoma cell lines. Cancer Lett. 2016, 381, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-β signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Xu, W.; Zhang, Z.; Yang, Y.; Hu, Z.; Wang, C.H.; Morgan, M.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.; et al. Ad5/48 hexon oncolytic virus expressing sTGFβRIIFc produces reduced hepatic and systemic toxicities and inhibits prostate cancer bone metastases. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Gerseny, H.; Zhang, Z.; Chen, Y.J.; Berg, A.; Stock, S.; Seth, P. Oncolytic adenovirus expressing soluble TGFβ receptor II-Fc-mediated inhibition of established bone metastases: A safe and effective systemic therapeutic approach for breast cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Wang, G.; He, D.; Liu, F. Repression of Smad4 transcriptional activity by SUMO modification. Biochem. J. 2004, 379, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Inui, M.; Newfeld, S.J. Regulation of TGF-β signal transduction by mono- and deubiquitylation of Smads. FEBS Lett. 2012, 586, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Liu, X.; Han, Z.; Zhou, L.; Sui, H.; Yan, L.; Jiang, H.; Ren, J.; Cai, J.; Li, Q. Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-β1/Smads signaling pathway mediated Snail/E-cadherin expression. BMC Cancer 2015, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-β-induced epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.B.; Wu, C.T.; Wang, H.; Zhang, Q.W.; Wang, L.; Wang, R.L.; Lu, Z.Z.; Wang, L.S. A simplified system for generating oncolytic adenovirus vector carrying one or two transgenes. Cancer Gene Ther. 2008, 15, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Neill, T.; Yang, Y.; Hu, Z.; Cleveland, E.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.R.; Shevrin, D.; et al. The systemic delivery of an oncolytic adenovirus expressing decorin inhibits bone metastasis in a mouse model of human prostate cancer. Gene Ther. 2015, 22, 247–256. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wang, H.; Wang, H.; Xiao, F.; Seth, P.; Xu, W.; Jia, Q.; Wu, C.; Yang, Y.; Wang, L. SUMO-Specific Cysteine Protease 1 Promotes Epithelial Mesenchymal Transition of Prostate Cancer Cells via Regulating SMAD4 deSUMOylation. Int. J. Mol. Sci. 2017, 18, 808. https://doi.org/10.3390/ijms18040808
Zhang X, Wang H, Wang H, Xiao F, Seth P, Xu W, Jia Q, Wu C, Yang Y, Wang L. SUMO-Specific Cysteine Protease 1 Promotes Epithelial Mesenchymal Transition of Prostate Cancer Cells via Regulating SMAD4 deSUMOylation. International Journal of Molecular Sciences. 2017; 18(4):808. https://doi.org/10.3390/ijms18040808
Chicago/Turabian StyleZhang, Xiaoyan, Hao Wang, Hua Wang, Fengjun Xiao, Prem Seth, Weidong Xu, Qinghua Jia, Chutse Wu, Yuefeng Yang, and Lisheng Wang. 2017. "SUMO-Specific Cysteine Protease 1 Promotes Epithelial Mesenchymal Transition of Prostate Cancer Cells via Regulating SMAD4 deSUMOylation" International Journal of Molecular Sciences 18, no. 4: 808. https://doi.org/10.3390/ijms18040808
APA StyleZhang, X., Wang, H., Wang, H., Xiao, F., Seth, P., Xu, W., Jia, Q., Wu, C., Yang, Y., & Wang, L. (2017). SUMO-Specific Cysteine Protease 1 Promotes Epithelial Mesenchymal Transition of Prostate Cancer Cells via Regulating SMAD4 deSUMOylation. International Journal of Molecular Sciences, 18(4), 808. https://doi.org/10.3390/ijms18040808