
Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms




Abstract
:1. Introduction
2. Genomic and Nongenomic Mechanisms of Action of Thyroid Hormones
2.1. Genomic Actions
2.2. Nongenomic Actions
3. Fatty Acid Metabolism in Liver
- (i)
- can be synthesized directly within the hepatocytes through the involvement of de novo lipogenesis (DNL),
- (ii)
- can be taken up by liver from the pool of plasma FA released by the adipose tissue,
- (iii)
- can be generated in liver from the hydrolysis of chylomicrons coming from intestine.
4. Mitochondrial Fatty Acid β-Oxidation
5. De Novo Lipogenesis (DNL) and Its Regulation
6. Regulation of Enzyme Activities of DNL
7. Molecular Mechanisms of T3 Action on DNL
7.1. Effect of T3 on the Lipogenic Enzymes: ACC and FAS
7.2. Role of T3 in the Conversion of Carbohydrates into Fatty Acids
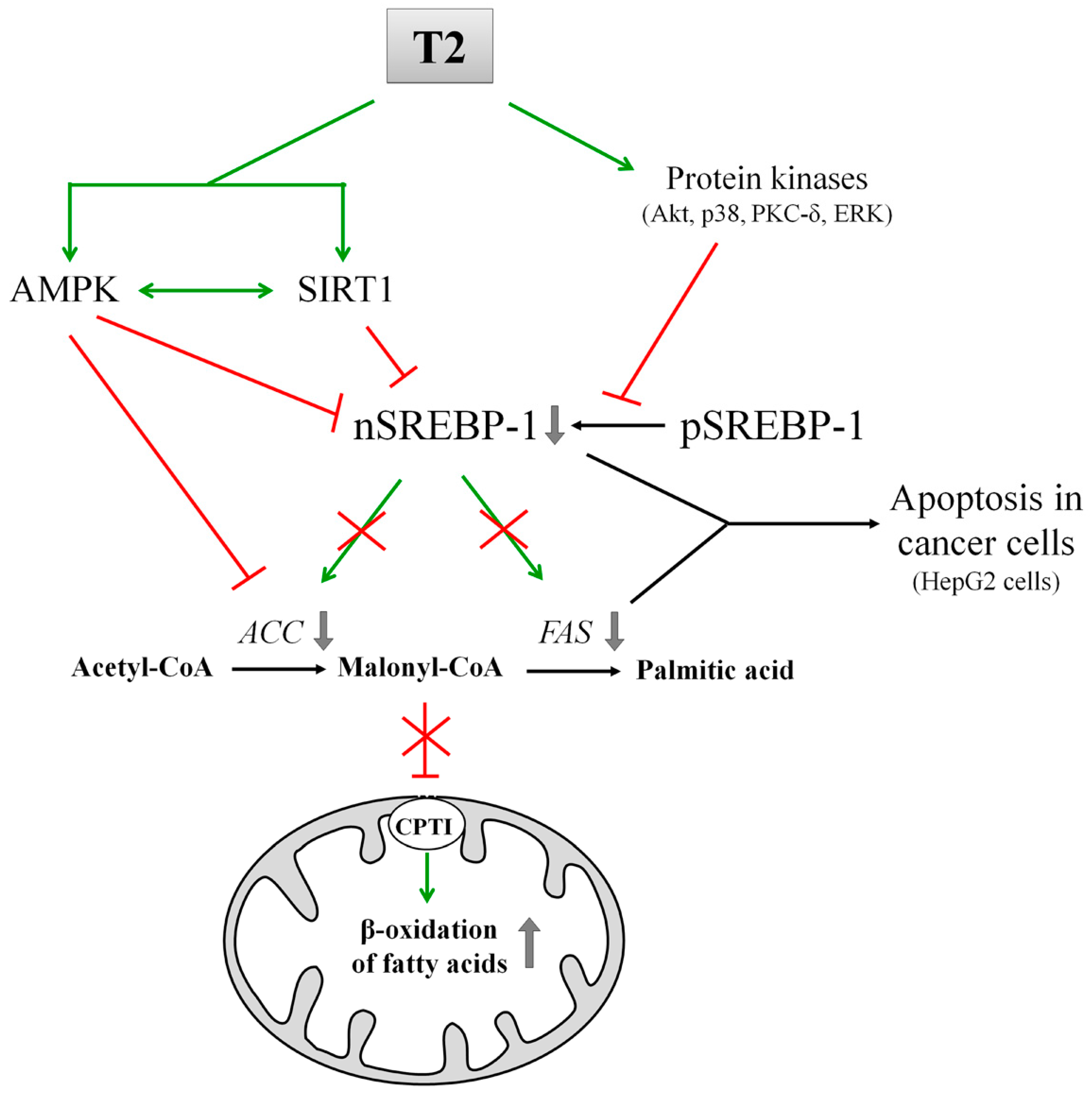
8. T2 and Its Effects on DNL
9. Conclusions and Future Perspectives
Conflicts of Interest
Abbreviations
| 6PGD | 6-Phosphogluconate dehydrogenase |
| ACC | Acetyl-CoA carboxylase |
| ACLY | ATP citrate lyase |
| AMPK | Protein kinase AMP-activated |
| BMR | Basal metabolic rate |
| ChREBP | Carbohydrate response element binding protein |
| CPT I | Carnitine palmitoyltransferase I |
| CPT II | Carnitine palmitoyltransferase II |
| CiC | Mitochondrial citrate carrier |
| DNL | De novo lipogenesis |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular-signal regulated kinases 1/2 |
| FA | Fatty acids |
| FAS | Fatty acid synthase |
| FoxO | Forkhead box O |
| G6PD | Glucose-6-phosphate dehydrogenase |
| IMM | Inner mitochondrial membrane |
| INSIG | Insulin-induced gene |
| IRES | Internal ribosome entry site |
| L-PK | Liver pyruvate kinase |
| LDs | Lipid droplets |
| MAPK | Mitogen-activated protein kinase |
| ME | Malic enzyme |
| MLX | Max-like protein X |
| MPC | Mitochondrial pyruvate carrier |
| mTOR | Mechanistic target of rapamycin |
| OAA | Oxalacetate |
| P38 | P38 mitogen-activated protein kinase |
| PGC1α | Peroxisome proliferative activated receptor γ coactivator 1 α |
| PI3K | Phosphoinositide 3-kinase |
| PKB/Akt | Protein kinase B/serine/threonine kinase 1 |
| PKC | Protein kinase C |
| PKM2 | M2 isoform of pyruvate kinase |
| RXR | Retinoid X receptor |
| S14 | Spot 14 |
| SCAP | SREBP-cleavage-activating protein |
| SIRT1 | Sirtuin 1 |
| SREBPs | Sterol regulatory element-binding proteins |
| TAGs | Triacylglycerols |
| TRE | T3 responsive element |
| T2 | 3,5-Diiodo-l-thyronine |
| T3 | 3,5,3′-Triiodo-l-thyronine |
| T4 | 3,3′,5,5′-Tetraiodo-l-thyronine |
| TCA | Tricarboxylic acid |
| THs | Thyroid hormones |
| TRα1 | Thyroid hormone receptor-α1 |
| TRβ1 | Thyroid hormone receptor-β1 |
| TRβ2 | Thyroid hormone receptor-β2 |
| TRs | Thyroid receptors |
| VLDL | Very low density lipoprotein |
References
- Orozco, A.; Valverde, R.C.; Olvera, A.; García, G.C. Iodothyronine deiodinases: A functional and evolutionary perspective. J. Endocrinol. 2012, 215, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Gnocchi, D.; Steffensen, K.R.; Bruscalupi, G.; Parini, P. Emerging role of thyroid hormone metabolites. Acta Physiol. 2016, 217, 184–216. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.; Rudling, M.; Scanlan, T.S.; Lundasen, T.; Webb, P.; Baxter, J.; Angelin, B.; Parini, P. Selective thyroid receptor modulation by GC-1 reduces serum lipids and stimulates steps of reverse cholesterol transport in euthyroid mice. Proc. Natl. Acad. Sci. USA 2005, 102, 10297–10302. [Google Scholar] [CrossRef] [PubMed]
- Mörk, L.M.; Rehnmark, S.; Davoodpour, P.; Norata, G.D.; Larsson, L.; Witt, M.R.; Malm, J.; Parini, P. The thyroid receptor modulator KB3495 reduces atherosclerosis independently of total cholesterol in the circulation in ApoE deficient mice. PLoS ONE 2013, 8, e78534. [Google Scholar] [CrossRef] [PubMed]
- Brenta, G. Why can insulin resistance be a natural consequence of thyroid dysfunction? J. Thyroid Res. 2011, 2011, 152850. [Google Scholar] [CrossRef] [PubMed]
- Laville, M.; Khalfallah, Y.; Vidal, H.; Beylot, M.; Comte, B.; Riou, J.P. Hormonal control of glucose production and pyruvate kinase activity in isolated rat liver cells: Influence of hypothyroidism. Mol. Cell. Endocrinol. 1987, 50, 247–253. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.J.; Seitz, H.J. Thyroid hormone action on intermediary metabolism. Part III. Protein metabolism in hyper- and hypothyroidism. Klin. Wochenschr. 1984, 62, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F.; Moreno, M.; Lanni, A. Action of thyroid hormones at the cellular level: The mitochondrial target. FEBS Lett. 1999, 452, 115–120. [Google Scholar] [CrossRef]
- Senatore, V.; Cione, E.; Gnoni, A.; Genchi, G. Retinoylation reactions are inversely related to the cardiolipin level in testes mitochondria from hypothyroid rats. J. Bioenerg. Biomembr. 2010, 42, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Goglia, F. Biological effects of 3,5-diiodothyronine (T2). Biochemistry 2005, 70, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Horst, C.; Rokos, H.; Seitz, H.J. Rapid stimulation of hepatic oxygen consumption by 3,5-di-iodo-l-thyronine. Biochem. J. 1989, 261, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; Leo, M.; Geelen, M.J.; Gnoni, G.V. Short-term stimulation of lipogenesis by 3,5-l-diiodothyronine in cultured rat hepatocytes. Endocrinology 2005, 146, 3959–3966. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, A.; Gnoni, A.; Conte, E.; Siculella, L.; Zanotti, F.; Papa, S.; Gnoni, G.V. 3,5-Diiodo-l-thyronine increases FoF1-ATP synthase activity and cardiolipin level in liver mitochondria of hypothyroid rats. J. Bioenerg. Biomembr. 2011, 43, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, A.; Priore, P.; Gnoni, G.V.; Papa, S.; Zanotti, F.; Gnoni, A. 3,5-Diiodo-l-thyronine administration to hypothyroid rats rapidly enhances fatty acid oxidation rate and bioenergetic parameters in liver cells. PLoS ONE 2013, 8, e52328. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Lanni, A.; Goglia, F. Thyroid hormones, mitochondrial bioenergetics and lipid handling. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Cioffi, F.; de Lange, P.; Goglia, F.; Lanni, A. Thyroid: Biological actions of “nonclassical” thyroid hormones. J. Endocrinol. 2014, 221, R1–R12. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, A.; Taurino, F.; Damiano, F.; Siculella, L.; Sardanelli, A.M.; Gnoni, A. Acute administration of 3,5-diiodo-l-thyronine to hypothyroid rats stimulates bioenergetic parameters in liver mitochondria. J. Bioenerg. Biomembr. 2016, 48, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Lanni, A.; Moreno, M.; Lombardi, A.; de Lange, P.; Silvestri, E.; Ragni, M.; Farina, P.; Baccari, G.C.; Fallahi, P.; Antonelli, A.; et al. 3,5-Diiodothyronine powerfully reduces adiposity by increasing the burning of fats. FASEB J. 2005, 19, 1552–1554. [Google Scholar] [PubMed]
- Mollica, M.P.; Lionetti, L.; Moreno, M.; Lombardi, A.; de Lange, P.; Antonelli, A.; Lanni, A.; Cavaliere, G.; Barletta, A.; Goglia, F. 3,5-Diiodo-l-thyronine, by modulating mitochondrial functions, reverses hepatic fat accumulation in rats fed a high-fat diet. J. Hepatol. 2009, 51, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Canesi, L.; de Matteis, R.; Goglia, F.; Cioffi, F.; Fugassa, E.; Gallo, G.; Vergani, L. Direct effects of iodothyronines on excess fat storage in rat hepatocytes. J. Hepatol. 2011, 54, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, G.; Singh, R.; Kumar, A.; Kumar, S.; Kapoor, A.; Godbole, M.M. Severe hyperthyroidism induces mitochondria-mediated apoptosis in rat liver. Hepatology 2004, 39, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Vizarra, E.; Enriquez, J.A.; Pérez-Martos, A.; Montoya, J.; Fernández-Silva, P. Mitochondrial gene expression is regulated at multiple levels and differentially in the heart and liver by thyroid hormones. Curr. Genet. 2008, 54, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Mangiullo, R.; Gnoni, A.; Damiano, F.; Siculella, L.; Zanotti, F.; Papa, S.; Gnoni, G.V. 3,5-Diiodo-l-thyronine upregulates rat-liver mitochondrial FoF1-ATP synthase by GA-binding protein/nuclear respiratory factor-2. Biochim. Biophys. Acta 2010, 1797, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [PubMed]
- Hennemann, G.; Docter, R.; Friesema, E.C.; de Jong, M.; Krenning, E.P.; Visser, T.J. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr. Rev. 2001, 22, 451–476. [Google Scholar] [CrossRef] [PubMed]
- Radenne, A.; Akpa, M.; Martel, C.; Sawadogo, S.; Mauvoisin, D.; Mounier, C. Hepatic regulation of fatty acid synthase by insulin and T3: Evidence for T3 genomic and nongenomic actions. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E884–E894. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Leonard, J.L.; Davis, F.B. Mechanisms of nongenomic actions of thyroid hormone. Front. Neuroendocrinol. 2008, 29, 211–821. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, G.V.; Rochira, A.; Leone, A.; Damiano, F.; Marsigliante, S.; Siculella, L. 3,5,3′-Triiodo-l-thyronine induces SREBP-1 expression by non-genomic actions in human HEP G2 cells. J. Cell. Physiol. 2012, 227, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Goodman, S.L.; Xiong, J.P. Coming to grips with integrin binding to ligands. Curr. Opin. Cell Biol. 2002, 14, 641–651. [Google Scholar] [CrossRef]
- Bergh, J.J.; Lin, H. Y.; Lansing, L.; Mohamed, S.N.; Davis, F.B.; Mousa, S.; Davis, P.J. Integrin αVβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 2005, 146, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Shih, A.; Lin, H.Y.; Martino, L.J.; Davis, F.B. Thyroxine promotes association of mitogen-activated protein kinase and nuclear thyroid hormone receptor (TR) and causes serine phosphorylation of TR. J. Biol. Chem. 2000, 275, 38032–38039. [Google Scholar] [CrossRef] [PubMed]
- De Lange, P.; Senese, R.; Cioffi, F.; Moreno, M.; Lombardi, A.; Silvestri, E.; Goglia, F.; Lanni, A. Rapid activation by 3,5,3′-l-triiodothyronine of adenosine 5′-monophosphate-activated protein kinase/acetyl-coenzyme a carboxylase and Akt/protein kinase B signaling pathways: Relation to changes in fuel metabolism and myosin heavy-chain protein content in rat gastrocnemius muscle in vivo. Endocrinology 2008, 149, 6462–6470. [Google Scholar] [PubMed]
- Cao, X.; Kambe, F.; Moeller, L.C.; Refetoff, S.; Seo, H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol. Endocrinol. 2005, 19, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Kavok, N.S.; Krasilnikova, O.A.; Babenko, N.A. Thyroxine signal transduction in liver cells involves phospholipase C and phospholipase D activation. Genomic independent action of thyroid hormone. BMC Cell Biol. 2001, 2, 5. [Google Scholar] [CrossRef]
- Eaton, S. Control of mitochondrial β-oxidation flux. Prog. Lipid. Res. 2002, 41, 197–239. [Google Scholar] [CrossRef]
- Paradies, G.; Papa, S. On the kinetics and substrate specificity of the pyruvate translocator in rat liver mitochondria. Biochim. Biophys. Acta 1977, 462, 333–346. [Google Scholar] [CrossRef]
- Glerum, D.M.; Claeys, D.; Mertens, W.; Azzi, A. The tricarboxylate carrier from rat liver mitochondria. Purification, reconstitution and kinetic characterization. Eur. J. Biochem. 1990, 194, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, G.V.; Priore, P.; Geelen, M.J.; Siculella, L. The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 2009, 61, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H. Regulation of mammalian acetyl coenzyme A carboxylase. Annu. Rev. Nutr. 1997, 17, 77–99. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Urstad, A.P.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: Housekeeper or messenger? Biochim. Biophys. Acta 2012, 182, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Giudetti, A.M. Dietary long-chain unsaturated fatty acids acutely and differently reduce the activities of lipogenic enzymes and of citrate carrier in rat liver. J. Physiol. Biochem. 2016, 72, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Glerum, M.; Koller, R.; Mertens, W.; Spycher, S. The mitochondrial tricarboxylate carrier. J. Bioenerg. Biomembr. 1993, 25, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; Sabetta, S.; di Summa, R.; Leo, M.; Damiano, F.; Siculella, L.; Gnoni, G.V. Differential effects of coconut oil- and fish oil-enriched diets on tricarboxylate carrier in rat liver mitochondria. J. Lipid Res. 2003, 44, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Siculella, L.; Damiano, F.; Sabetta, S.; Gnoni, G.V. n-6 PUFAs downregulate expression of the tricarboxylate carrier in rat liver by transcriptional and posttranscriptional mechanisms. J. Lipid Res. 2004, 45, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Siculella, L.; Sabetta, S.; Damiano, F.; Giudetti, A.M.; Gnoni, G.V. Different dietary fatty acids have dissimilar effects on activity and gene expression of mitochondrial tricarboxylate carrier in rat liver. FEBS Lett. 2004, 578, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Functional analysis of rat liver citrate carrier promoter: Differential responsiveness to polyunsaturated fatty acids. Biochem. J. 2009, 417, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Damiano, F.; Mercuri, E.; Stanca, E.; Gnoni, G.V.; Siculella, L. Streptozotocin-induced diabetes affects in rat liver citrate carrier gene expression by transcriptional and posttranscriptional mechanisms. Int. J. Biochem. Cell Biol. 2011, 43, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Giudetti, A.M.; Leo, M.; Siculella, L.; Gnoni, G.V. Hypothyroidism down-regulates mitochondrial citrate carrier activity and expression in rat liver. Biochim. Biophys. Acta 2006, 1761, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Siculella, L.; Sabetta, S.; Giudetti, A.M.; Gnoni, G.V. Hypothyroidism reduces tricarboxylate carrier activity and expression in rat liver mitochondria by reducing nuclear transcription rate and splicing efficiency. J. Biol. Chem. 2006, 281, 19072–19080. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, G.V.; Giudetti, A.M.; Mercuri, E.; Damiano, F.; Stanca, E.; Priore, P.; Siculella, L. Reduced activity and expression of mitochondrial citrate carrier in streptozotocin-induced diabetic rats. Endocrinology 2010, 151, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Damiano, F.; Tocci, R.; Gnoni, G.V.; Siculella, L. Expression of citrate carrier gene is activated by ER stress effectors XBP1 and ATF6α, binding to an UPRE in its promoter. Biochim. Biophys. Acta 2015, 1849, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Nakamoto, T.; Mori, Y.; Kawakami, M.; Mabuchi, H.; Ohishi, Y.; Ichikawa, N.; Koike, A.; Masuda, K. Comparative effects of dietary fat types on hepatic enzyme activities related to the synthesis and oxidation of fatty acid and to lipogenesis in rats. Biosci. Biotechnol. Biochem. 2001, 65, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K. Recent progress on the role of ChREBP in glucose and lipid metabolism. Endocr. J. 2013, 60, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.I.; Osborne, T.F. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, G.V.; Geelen, M.J.; Bijleveld, C.; Quagliariello, E.; van den Bergh, S.G. Short-term stimulation of lipogenesis by triiodothyronine in maintenance cultures of rat hepatocytes. Biochem. Biophys. Res. Commun. 1985, 128, 525–530. [Google Scholar] [CrossRef]
- Clarke, S.D.; Hembree, J. Inhibition of triiodothyronine’s induction of rat liver lipogenic enzymes by dietary fat. J. Nutr. 1990, 120, 625–630. [Google Scholar] [PubMed]
- Blennemann, B.; Moon, Y.K.; Freake, H.C. Tissue-specific regulation of fatty acid synthesis by thyroid hormone. Endocrinology 1992, 130, 637–643. [Google Scholar] [PubMed]
- Huang, C.; Freake, H.C. Thyroid hormone regulates the acetyl-CoA carboxylase PI promoter. Biochem. Biophys. Res. Commun. 1998, 249, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Hillgartner, F.B.; Charron, T.; Chesnut, K.A. Triiodothyronine stimulates and glucagon inhibits transcription of the acetyl-CoA carboxylase gene in chick embryo hepatocytes: Glucose and insulin amplify the effect of triiodothyronine. Arch. Biochem. Biophys. 1997, 337, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, L.; Hillgartner, F.B. Thyroid hormone stimulates acetyl-CoA carboxylase-α transcription in hepatocytes by modulating the composition of nuclear receptor complexes bound to a thyroid hormone response element. J. Biol. Chem. 2001, 276, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Sasaki, S.; Morita, H.; Ito, T.; Suzuki, S.; Misawa, H.; Nakamura, H. Unliganded thyroid hormone receptor-β1 represses liver X receptor α/oxysterol-dependent transactivation. Endocrinology 2004, 145, 5515–5524. [Google Scholar] [CrossRef] [PubMed]
- Blennemann, B.; Leahy, P.; Kim, T.S.; Freake, H.C. Tissue-specific regulation of lipogenic mRNAs by thyroid hormone. Mol. Cell Endocrinol. 1995, 110, 1–8. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, Y.; Hillgartner, F.B. Sterol regulatory element-binding protein-1 interacts with the nuclear thyroid hormone receptor to enhance acetyl-CoA carboxylase-α transcription in hepatocytes. J. Biol. Chem. 2002, 277, 19554–19565. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, S.R.; Mitchell, D.A.; Salati, L.M.; Goodridge, A.G. Triiodothyronine stimulates transcription of the fatty acid synthase gene in chick embryo hepatocytes in culture. Insulin and insulin-like growth factor amplify that effect. J. Biol. Chem. 1990, 265, 18442–18446. [Google Scholar] [PubMed]
- Magaña, M.M.; Osborne, T.F. Two tandem binding sites for sterol regulatory element binding proteins are required for sterol regulation of fatty-acid synthase promoter. J. Biol. Chem. 1996, 271, 32689–32694. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.M.; Bennett, M.K.; Sanchez, H.B.; Rosenfeld, J.M.; Osborne, T.F. Sterol regulation of acetyl coenzyme A carboxylase: A mechanism for coordinate control of cellular lipid. Proc. Natl. Acad. Sci. USA 1996, 93, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Yamada, M.; Matsumoto, S.; Monden, T.; Satoh, T.; Mori, M. Mouse sterol response element binding protein-1c gene expression is negatively regulated by thyroid hormone. Endocrinology 2006, 147, 4292–4302. [Google Scholar] [CrossRef] [PubMed]
- Cable, E.E.; Finn, P.D.; Stebbins, J.W.; Hou, J.; Ito, B.R.; van Poelje, P.D.; Linemeyer, D.L.; Erion, M.D. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology 2009, 49, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Swierczynski, J.; Mitchell, D.A.; Reinhold, D.S.; Salati, L.M.; Stapleton, S.R.; Klautky, S.A.; Struve, A.E.; Goodridge, A.G. Triiodothyronine-induced accumulations of malic enzyme, fatty acid synthase, acetyl-coenzyme A carboxylase, and their mRNAs are blocked by protein kinase inhibitors. Transcription is the affected step. J. Biol. Chem. 1991, 266, 17459–17466. [Google Scholar] [PubMed]
- Hashimoto, K.; Ishida, E.; Matsumoto, S.; Okada, S.; Yamada, M.; Satoh, T.; Monden, T.; Mori, M. Carbohydrate response element binding protein gene expression is positively regulated by thyroid hormone. Endocrinology 2009, 150, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, K.; Billon, C.; Bissler, M.; Beylot, M.; Lobaccaro, J.M.; Vanacker, J.M.; Samarut, J. Thyroid hormone receptor β (TRβ) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J. Biol. Chem. 2010, 285, 28156–28163. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Ruggiero, F.M. Effect of hyperthyroidism on the transport of pyruvate in rat-heart mitochondria. Biochim. Biophys. Acta 1988, 935, 79–86. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M. Enhanced activity of the tricarboxylate carrier and modification of lipids in hepatic mitochondria from hyperthyroid rats. Arch. Biochem. Biophys. 1990, 278, 425–430. [Google Scholar] [CrossRef]
- Vaulont, S.; Munnich, A.; Decaux, J.F.; Kahn, A. Transcriptional and post-transcriptional regulation of L-type pyruvate kinase gene expression in rat liver. J. Biol. Chem. 1986, 261, 7621–7625. [Google Scholar] [PubMed]
- Spence, J.T.; Pitot, H.C.; Zalitis, G. Regulation of ATP-citrate lyase in primary cultures of adult rat hepatocytes. J. Biol. Chem. 1979, 254, 12169–12173. [Google Scholar] [PubMed]
- González-Manchón, C.; Butta, N.; Ferrer, M.; Ayuso, M.S.; Parrilla, R. Molecular cloning and functional characterization of the human cytosolic malic enzyme promoter: Thyroid hormone responsiveness. DNA Cell Biol. 1997, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- González-Manchón, C.; Ayuso, M.S.; Parrilla, R. AP-1 and T3RE cis elements operate as a functional unit in the transcriptional control of the human malic enzyme gene. Gene 1999, 226, 111–119. [Google Scholar] [CrossRef]
- Fritz, S.R.; Kletzien, R.F. Regulation of glucose-6-phosphate dehydrogenase by diet and thyroid hormones. Mol. Cell Endocrinol. 1987, 51, 13–17. [Google Scholar] [CrossRef]
- Thompson, K.S.; Towle, H.C. Localization of the carbohydrate response element of the rat L-type pyruvate kinase gene. J. Biol. Chem. 1991, 266, 8679–8682. [Google Scholar] [PubMed]
- Davis, P.J.; Davis, F.B. Nongenomic actions of thyroid hormone. Thyroid 1996, 6, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.B.; Maloney, M.; Kinlaw, W.B. “Spot 14” protein functions at the pretranslational level in the regulation of hepatic metabolism by thyroid hormone and glucose. J. Biol. Chem. 1997, 272, 2163–2166. [Google Scholar] [PubMed]
- Gharbi-Chihi, J.; Facchinetti, T.; Bergé-Lefranc, J.L.; Bonne, J.; Torresani, J. Triiodothyronine control of ATP-citrate lyase and malic enzyme during differentiation of a murine preadipocyte cell line. Horm. Metab. Res. 1991, 23, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.A.; Lee, J.J.; Park, S.W.; Ahn, Y.H.; Kim, K.S. The roles of sterol regulatory element-binding proteins in the transactivation of the rat ATP citrate-lyase promoter. J. Biol. Chem. 2000, 275, 30280–30286. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hillgartner, F.B. Alterations in retinoid X receptor-α expression contribute to cell-type dependent differences in thyroid hormone regulation of malic enzyme transcription. Mol. Cell. Endocrinol. 2000, 164, 41–52. [Google Scholar] [CrossRef]
- Towle, H.C.; Mariash, C.N.; Oppenheimer, J.H. Changes in the hepatic levels of messenger ribonucleic acid for malic enzyme during induction by thyroid hormone or diet. Biochemistry 1980, 19, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Shimano, H.; Korn, B.S.; Bashmakov, Y.; Horton, J.D. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J. Biol. Chem. 1998, 273, 35299–35306. [Google Scholar] [CrossRef] [PubMed]
- Czyzewska, U.; Tylicki, A.; Siemieniuk, M.; Strumilo, S. Changes of activity and kinetics of certain liver and heart enzymes of hypothyroid and T3-treated rats. J. Physiol. Biochem. 2012, 68, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Miksicek, R.J.; Towle, H.C. Changes in the rates of synthesis and messenger RNA levels of hepatic glucose-6-phosphate and 6-phosphogluconate dehydrogenases following induction by diet or thyroid hormone. J. Biol. Chem. 1982, 257, 11829–11835. [Google Scholar] [PubMed]
- Mariash, C.N.; Seelig, S.; Schwartz, H.L.; Oppenheimer, J.H. Rapid synergistic interaction between thyroid hormone and carbohydrate on mRNA S14 induction. J. Biol. Chem. 1986, 261, 9583–9586. [Google Scholar] [PubMed]
- Zhu, Q.; Anderson, G.W.; Mucha, G.T.; Parks, E.J.; Metkowski, J.K.; Mariash, C.N. The Spot 14 protein is required for de novo lipid synthesis in the lactating mammary gland. Endocrinology 2005, 146, 3343–3350. [Google Scholar] [CrossRef] [PubMed]
- Mater, M.K.; Thelen, A.P.; Pan, D.A.; Jump, D.B. Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J. Biol. Chem. 1999, 274, 32725–32732. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tsatsos, N.G.; Towle, H.C. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J. Biol. Chem. 2005, 280, 12019–12027. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.G.; Wilcox, H.G.; Heimberg, M. Formation of the very low density lipoprotein and metabolism of [1-14C]-oleate by perfused livers from rats treated with triiodothyronine or propylthiouracil. Metabolism 1987, 30, 135–146. [Google Scholar] [CrossRef]
- Prieur, X.; Huby, T.; Coste, H.; Schaap, F.G.; Chapman, M.J.; Rodríguez, J.C. Thyroid hormone regulates the hypotriglyceridemic gene APOA5. J. Biol. Chem. 2005, 280, 27533–27543. [Google Scholar] [CrossRef] [PubMed]
- Laker, M.E.; Mayes, P.A. Effect of hyperthyroidism and hypothyroidism on lipid and carbohydrate metabolism of the perfused rat liver. Biochem. J. 1981, 196, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Al-Tonsi, A.A.; Abdel-Gayoum, A.A.; Saadm, M. The secondary dyslipidemia and deranged serum phosphate concentration in thyroid disorders. Exp. Mol. Pathol. 2004, 76, 182–187. [Google Scholar] [CrossRef] [PubMed]
- De Lange, P.; Cioffi, F.; Senese, R.; Moreno, M.; Lombardi, A.; Silvestri, E.; de Matteis, R.; Lionetti, L.; Mollica, M.P.; Goglia, F.; Lanni, A. Nonthyrotoxic prevention of diet-induced insulin resistance by 3,5-diiodo-l-thyronine in rats. Diabetes 2011, 60, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Demori, I.; Canesi, L.; de Matteis, R.; Goglia, F.; Lanni, A.; Gallo, G.; Vergani, L. 3,5-Diiodo-l-thyronine modulates the expression of genes of lipid metabolism in a rat model of fatty liver. J. Endocrinol. 2012, 212, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Canesi, L.; Salis, A.; Damonte, G.; Compalati, A.D.; Goglia, F.; Gallo, G.; Vergani, L. 3,5-Diiodo-l-thyronine modifies the lipid droplet composition in a model of hepatosteatosis. Cell. Physiol. Biochem. 2014, 33, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, A.; Navarrete-Ramírez, P.; Hernández-Puga, G.; Villalobos, P.; Holzer, G.; Renaud, J. P.; Laudet, V.; Orozco, A. 3,5-T2 is an alternative ligand for the thyroid hormone receptor β1. Endocrinology 2013, 154, 2948–2958. [Google Scholar] [CrossRef] [PubMed]
- Senese, R.; Lasala, P.; Leanza, C.; de Lange, P. New avenues for regulation of lipid metabolism by thyroid hormones and analogs. Front. Physiol. 2014, 5, 475. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Saggerson, D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 2008, 28, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Xu, X.J.; Nelsonm, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Jonas, W.; Lietzow, J.; Wohlgemuth, F.; Hoefig, C.S.; Wiedmer, P.; Schweizer, U.; Köhrle, J.; Schürmann, A. 3,5-Diiodo-l-thyronine (3,5-t2) exerts thyromimetic effects on hypothalamus-pituitary-thyroid axis, body composition, and energy metabolism in male diet-induced obese mice. Endocrinology 2015, 156, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Rochira, A.; Damiano, F.; Marsigliante, S.; Gnoni, G.V.; Siculella, L. 3,5-Diiodo-l-thyronine induces SREBP-1 proteolytic cleavage block and apoptosis in human hepatoma (HepG2) cells. Biochim. Biophys. Acta 2013, 1831, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Fallahi, P.; Ferrari, S.M.; di Domenicantonio, A.; Moreno, M.; Lanni, A.; Goglia, F. 3,5-Diiodo-l-thyronine increases resting metabolic rate and reduces body weight without undesirable side effects. J. Biol. Regul. Homeost. Agents 2011, 25, 655–660. [Google Scholar] [PubMed]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
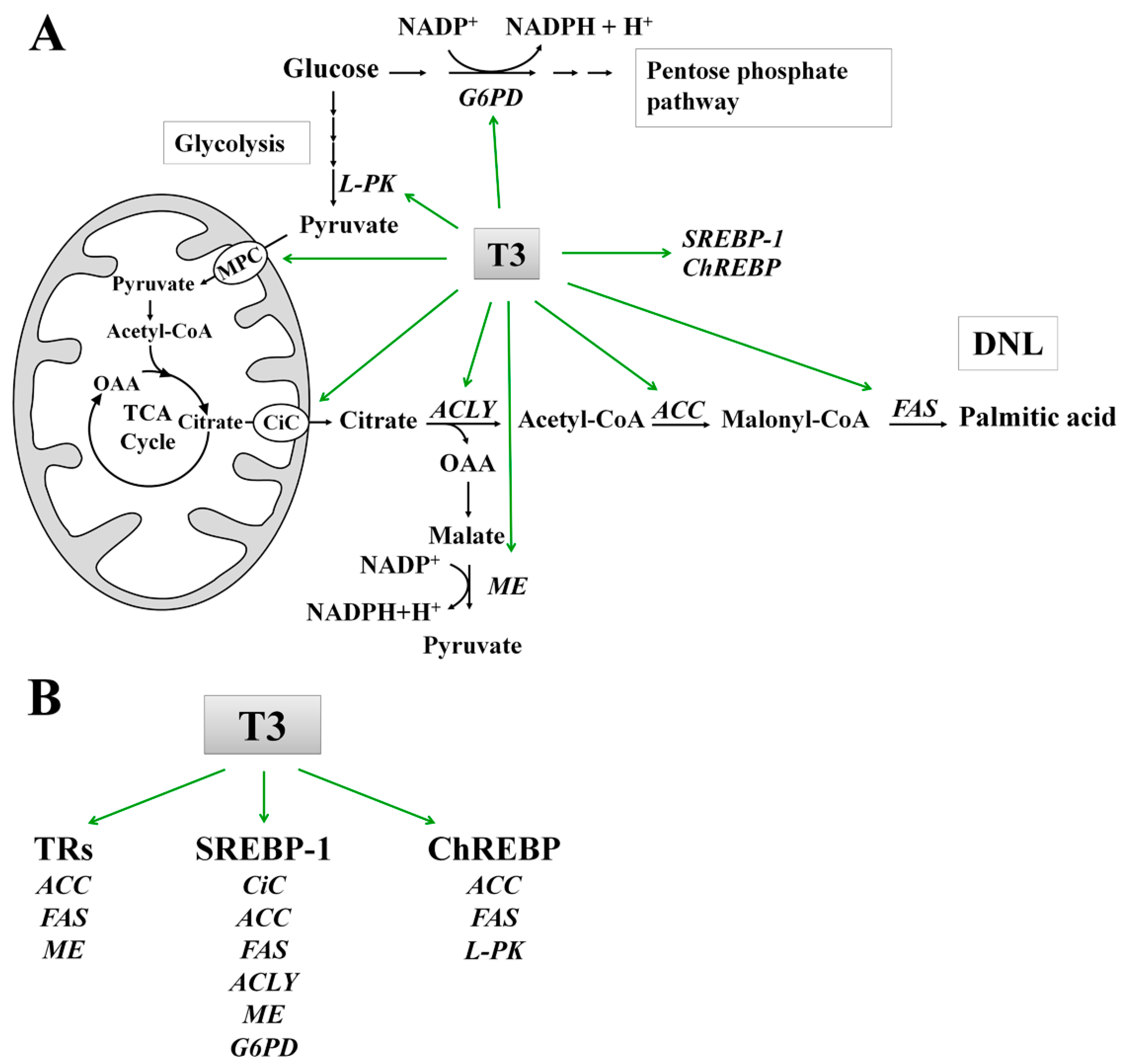

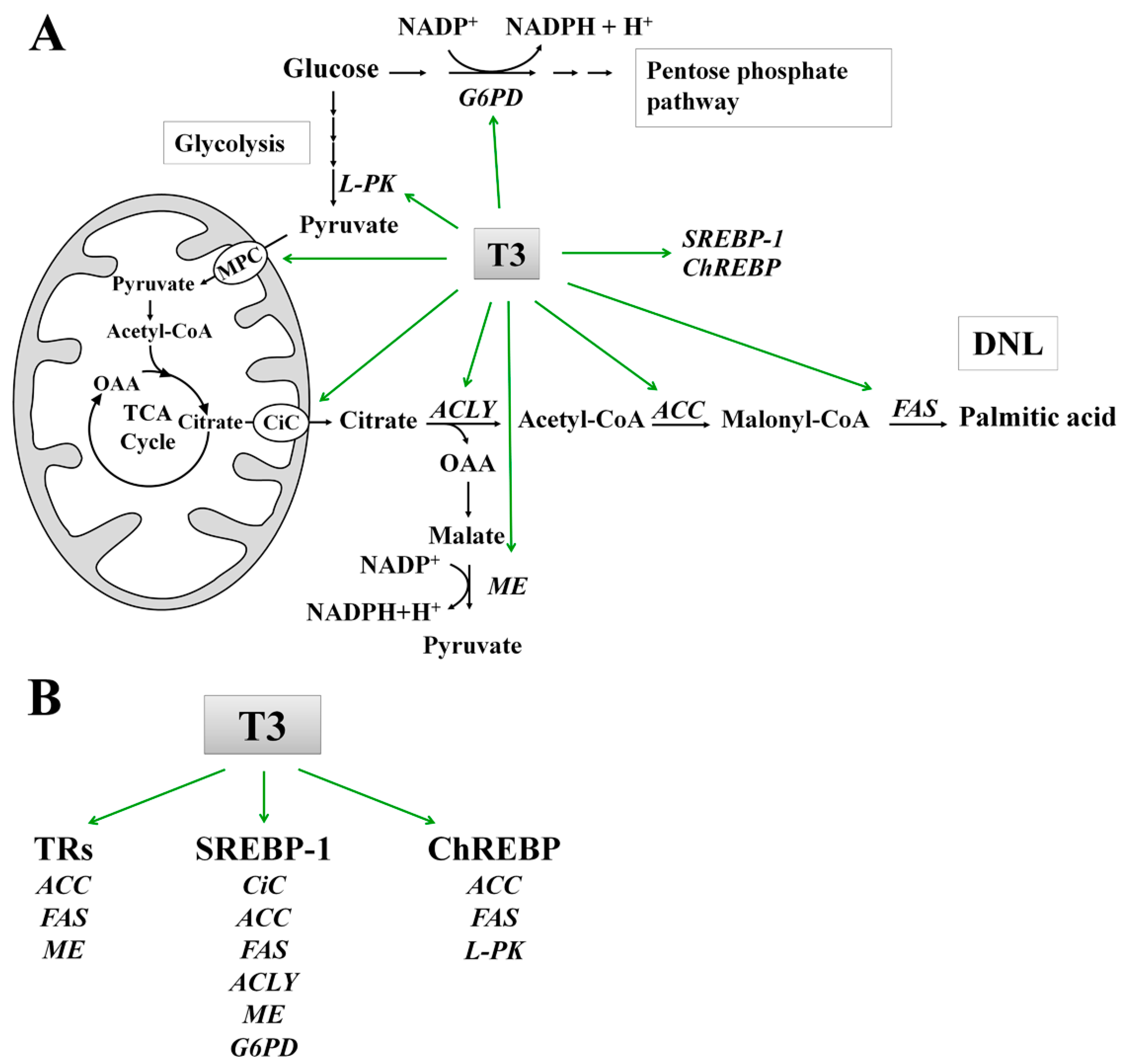{kind=link}
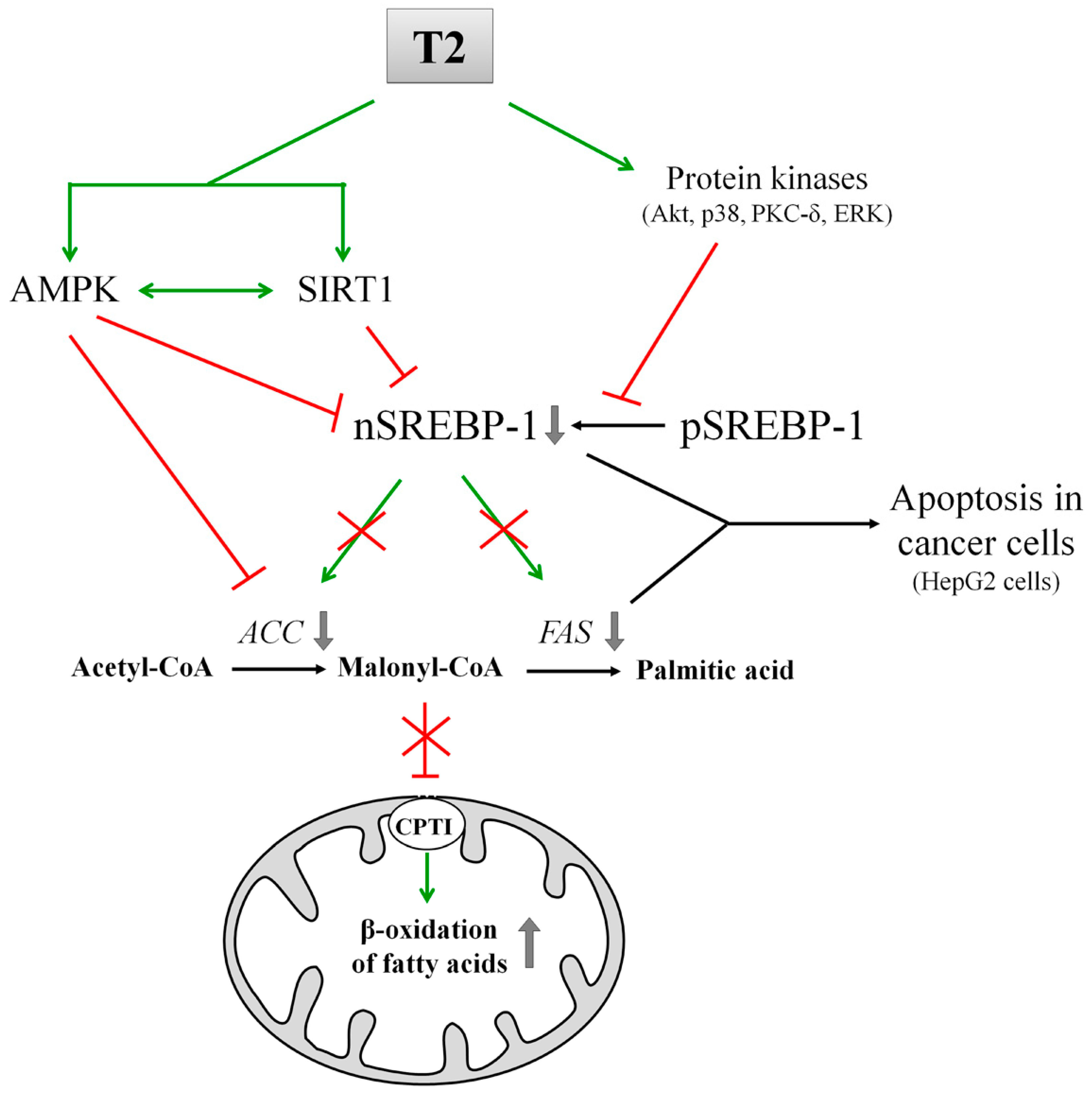{kind=link}
| Experimental Model | Time of Treatment | Effect of T3 on Lipid Metabolism | Mechanism of Action | Ref. | |
|---|---|---|---|---|---|
| In vitro studies | |||||
| HepG2 cells | 24 h | ↑ | FAS mRNA | Genomic | [28] |
| HepG2 cells | Up to 24 h | ↑ | SREBP-1 protein synthesis | Non genomic | [30] |
| Hepatocytes from eu- and hypothyroid rats fed chow diet | 4 h | ↑ | Synthesis of fatty acids and their incorporation into lipid fractions | Not reported | [62] |
| Cultured hepatocytes from chick embryo | Up to 49 h | ↑ | ACC promoter activity and mRNA abundance | Genomic | [66,67] |
| Cultured hepatocytes from chick embryo | Up to 48 h | ↑ | ME, FAS, ACC enzyme activity and mRNA abundance | Genomic | [76,71] |
| Hepatocytes from hypothyroid rats | 24 h | ↑ | ACLY protein level and activity | Not reported | [82] |
| HepG2 cells | 24 h | ↑ | ME promoter activity | Genomic | [83] |
| In vivo studies | |||||
| Liver from eu- and hyperthyroid rats fed chow diet. | 4 weeks | ↑ | SREBP-1 protein level | Not reported | [30] |
| Liver from eu- and hypothyroid rats fed chow diet | 4 weeks | ↑ | Mitochondrial citrate carrier expression, nuclear transcription rate and splicing efficiency | Genomic | [53,54] |
| Liver from eu- and hyperthyroid rats fed fat-enriched chow diet. | 7 days | ↑ | Fatty acid synthesis | Not reported | [63] |
| Liver from hypo- and hyperthyroid rats fed chow diet. | 7 days | ↑ | ACC mRNA abundance | Not reported | [69] |
| Liver from eu-, hypo- and hyperthyroid mice fed chow diet. | 5 days | ↓ | SREBP-1 mRNA abundance and SREBP-1 promoter activity | Genomic | [74] |
| Liver from eu-, hypo- and hyperthyroid mice fed chow diet. | 5 days | ↑ | ChREBP mRNA abundance and protein level; ChREBP promoter activity | Genomic | [77] |
| Liver from eu- and hyperthyroid mice fed chow diet or high carbohydrate diet. | Not reported | ↑ | ChREBP mRNA abundance; ChREBP promoter activity | Genomic | [78] |
| Liver from eu-, hypo-, and hyperthyroid rats, starved and refed on carbohydrate-rich diet | 7 days | ↑ | G6PD enzyme activity | Non genomic | [85] |
| Liver from eu- and hyperthyroid rats fed chow diet or high carbohydrate, fat-free diet. | 7 days | ↑ | ME mRNA abundance and enzyme activity | Not reported | [92] |
| Liver from eu- and hyperthyroid rats fed chow diet or high carbohydrate, fat-free diet. | 7 days | ↑ | ME, G6PD and 6PGD enzyme activity, mRNA abundance and relative rate of enzyme synthesis | Genomic and non-genomic | [95] |
| Liver from hypo- and hyperthyroid rats fed chow diet | Up to 4 h | ↑ | Spot 14 protein (S14) mRNA abundance | Not reported | [96] |
| Liver from eu- and hyperthyroid rats fed chow diet | 1 day | ↑ | Induction of citrate carrier activity | Not reported | [80] |
| Experimental Model | Time of Treatment | Effect of T2 on Lipid Metabolism | Ref. |
|---|---|---|---|
| In vitro studies | |||
| FaO cells rendered steatotic by incubation of free fatty acids | 24 h | Reduction in the number and size of lipid droplets in steatotic cells as consequence of triacylglycerols mobilization from lipid droplets. Stimulation of mitochondrial oxidative metabolism of fatty acids. | [106] |
| HepG2 cells | Up to 48 h | Induction of SREBP-1 proteolytic cleavage block and apoptosis in human hepatoma. | [114] |
| In vivo studies | |||
| Liver from eu- and T2-treated rats fed chow diet or high fat diet | Up to 4 weeks | Reduction of hepatic fatty accumulation induced by a high-fat diet. Induction of fatty acid oxidation rate and of CPT I activity. | [20] |
| Liver from hypo-, eu- and T2-treated hypothyroid rats fed chow diet | 1h | Increment of CPT-I activity and of total rate of fatty acid oxidation. | [15] |
| Liver from eu- and T2-treated rats fed chow diet or high fat diet | Up to 4 weeks | Deacetylation of peroxisome proliferator–activated receptor (PPAR)-γ and of SREBP-1 through the activation of SIRT1. Up-regulation of genes involved in the mitochondrial biogenesis and down-regulation of lipogenic genes. | [104] |
| Liver from eu- and T2-treated rats fed chow diet or high fat diet | 30 days | Prevention of pathways leading to lipid storage in lipid droplets. Mobilization of lipids from lipid droplets and secretion as VLDL. | [105] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damiano, F.; Rochira, A.; Gnoni, A.; Siculella, L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. Int. J. Mol. Sci. 2017, 18, 744. https://doi.org/10.3390/ijms18040744
Damiano F, Rochira A, Gnoni A, Siculella L. Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. International Journal of Molecular Sciences. 2017; 18(4):744. https://doi.org/10.3390/ijms18040744
Chicago/Turabian StyleDamiano, Fabrizio, Alessio Rochira, Antonio Gnoni, and Luisa Siculella. 2017. "Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms" International Journal of Molecular Sciences 18, no. 4: 744. https://doi.org/10.3390/ijms18040744
APA StyleDamiano, F., Rochira, A., Gnoni, A., & Siculella, L. (2017). Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. International Journal of Molecular Sciences, 18(4), 744. https://doi.org/10.3390/ijms18040744