Identification of Key Candidate Genes and Pathways in Colorectal Cancer by Integrated Bioinformatical Analysis


Abstract
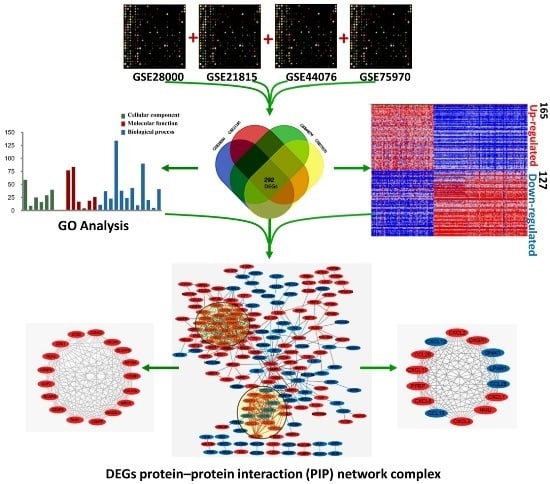
:
1. Introduction
2. Results
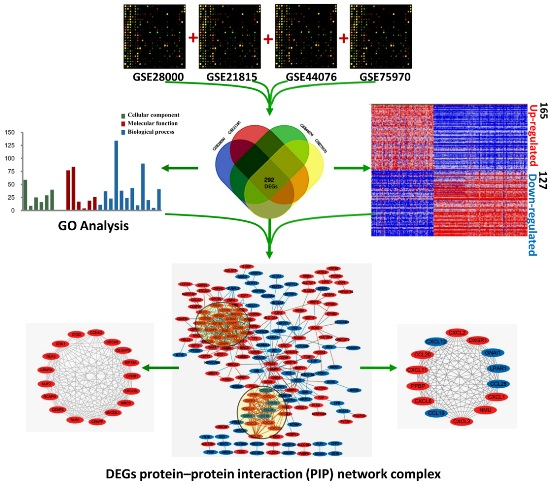
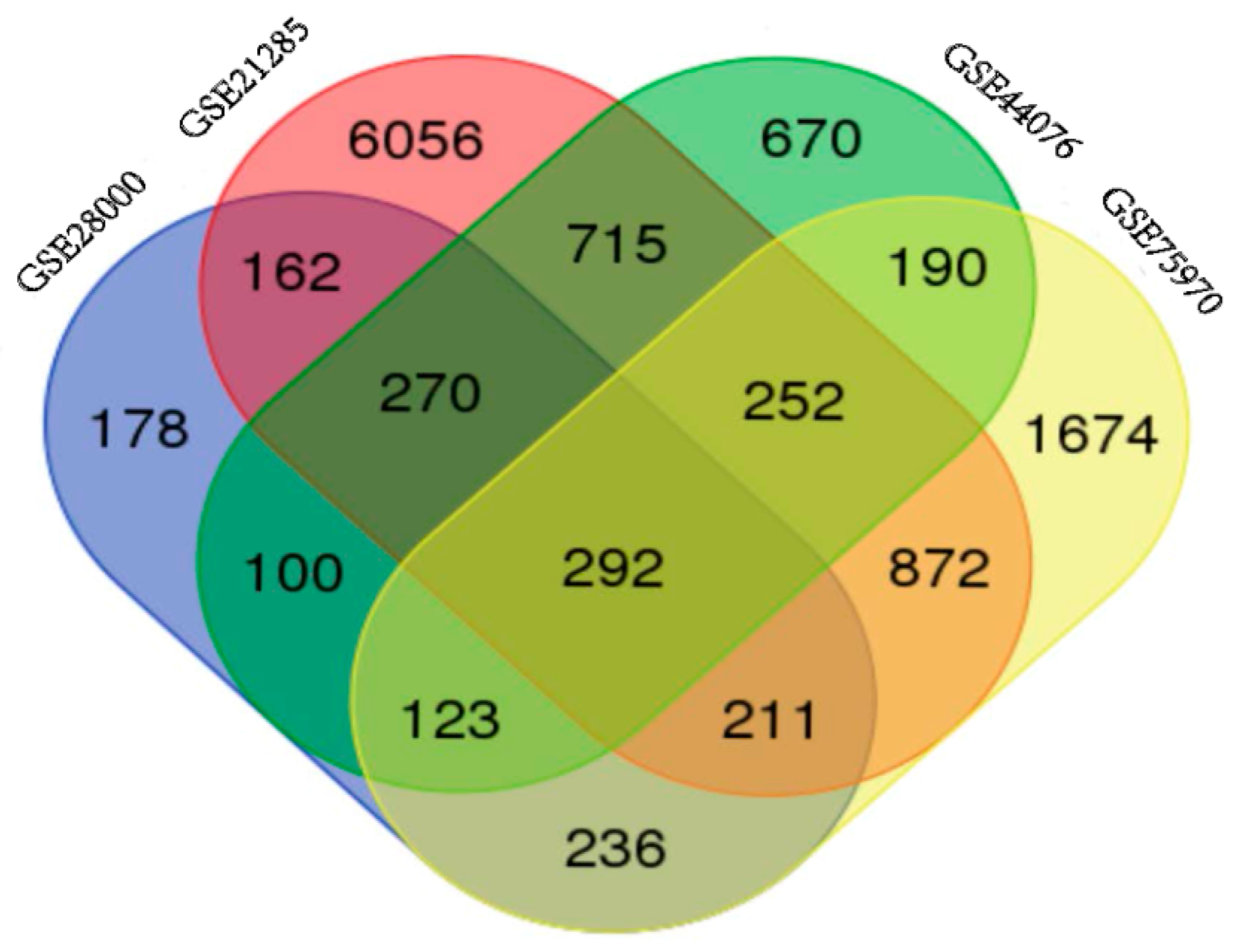
2.1. Identification of DEGs in Colorectal Cancers
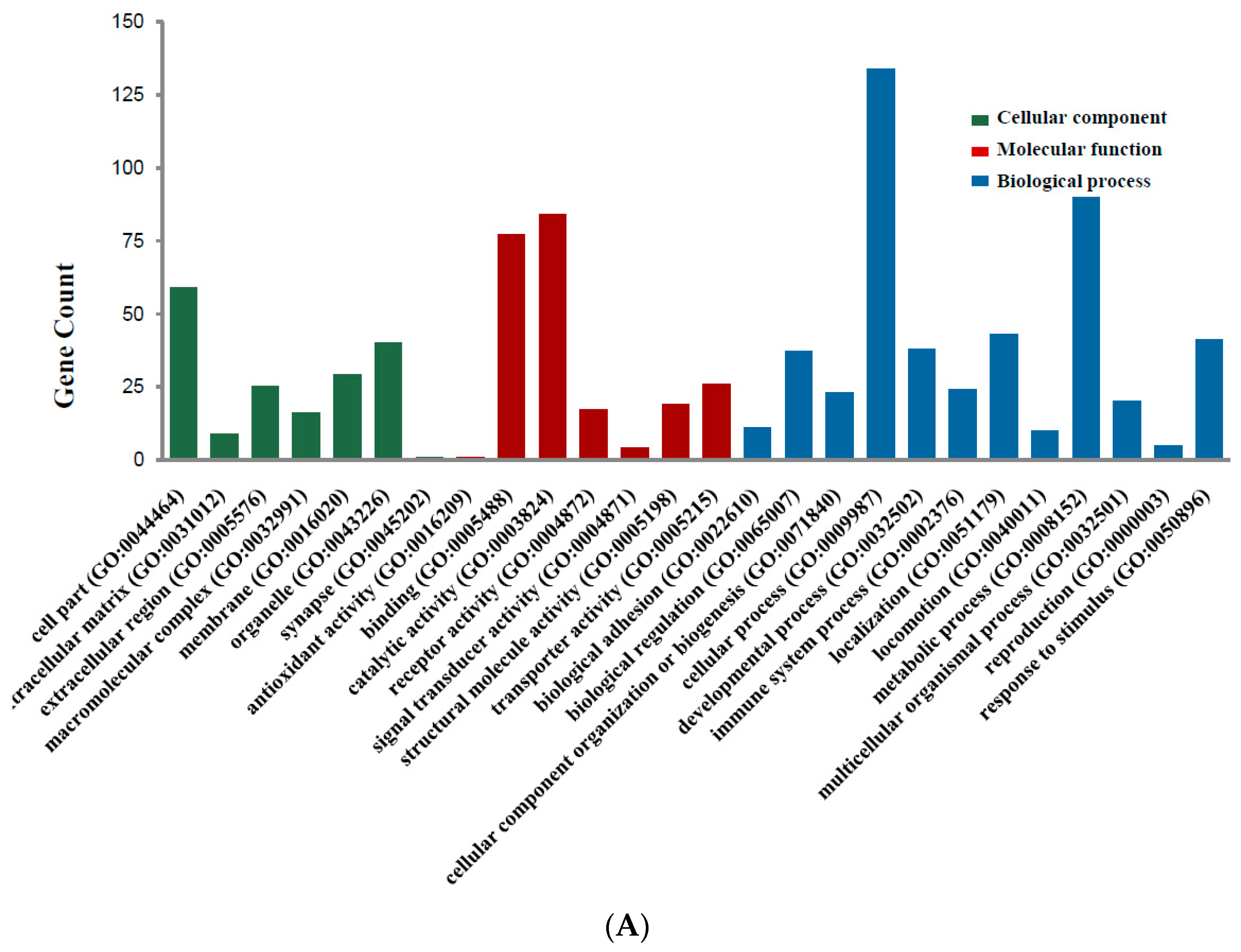
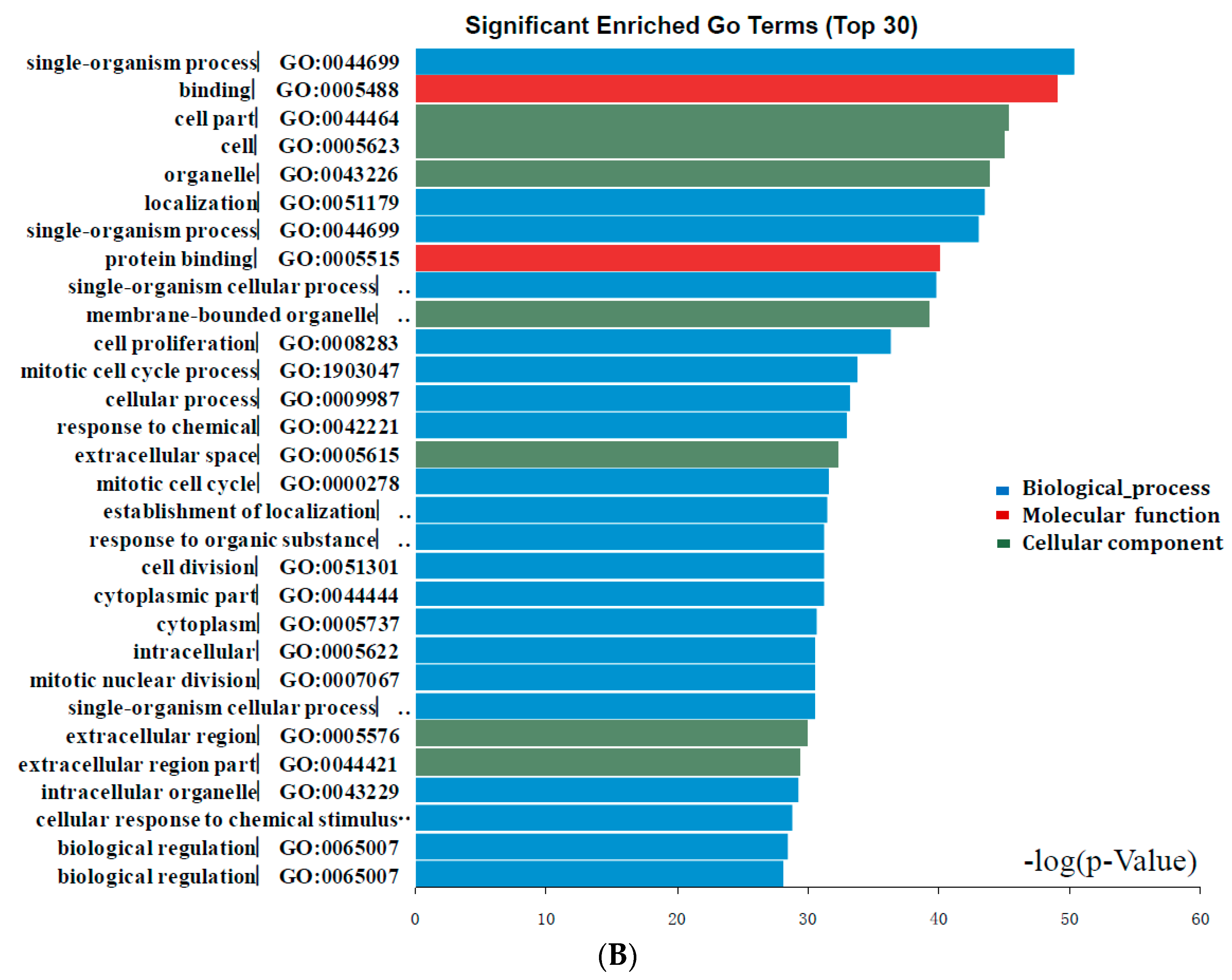
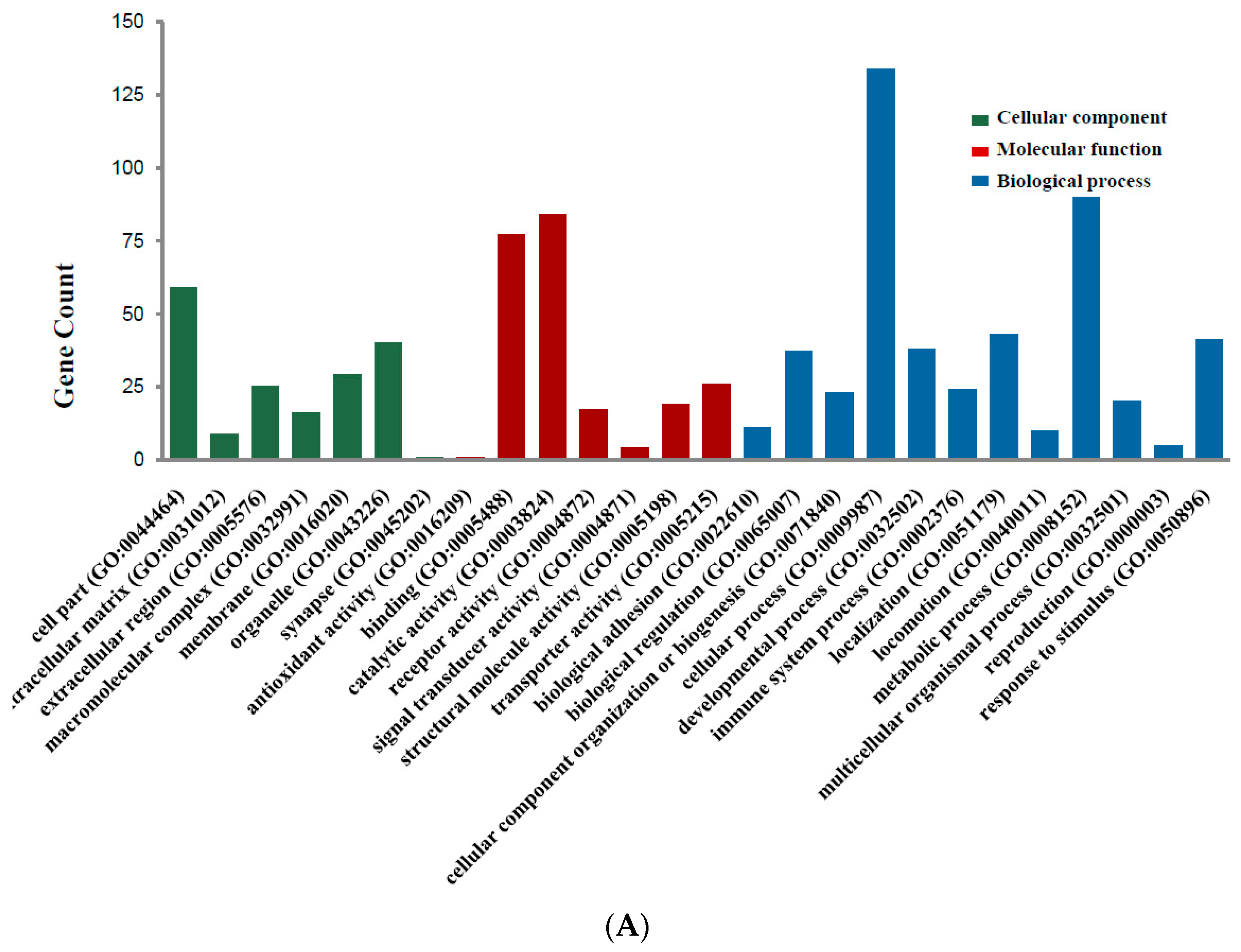
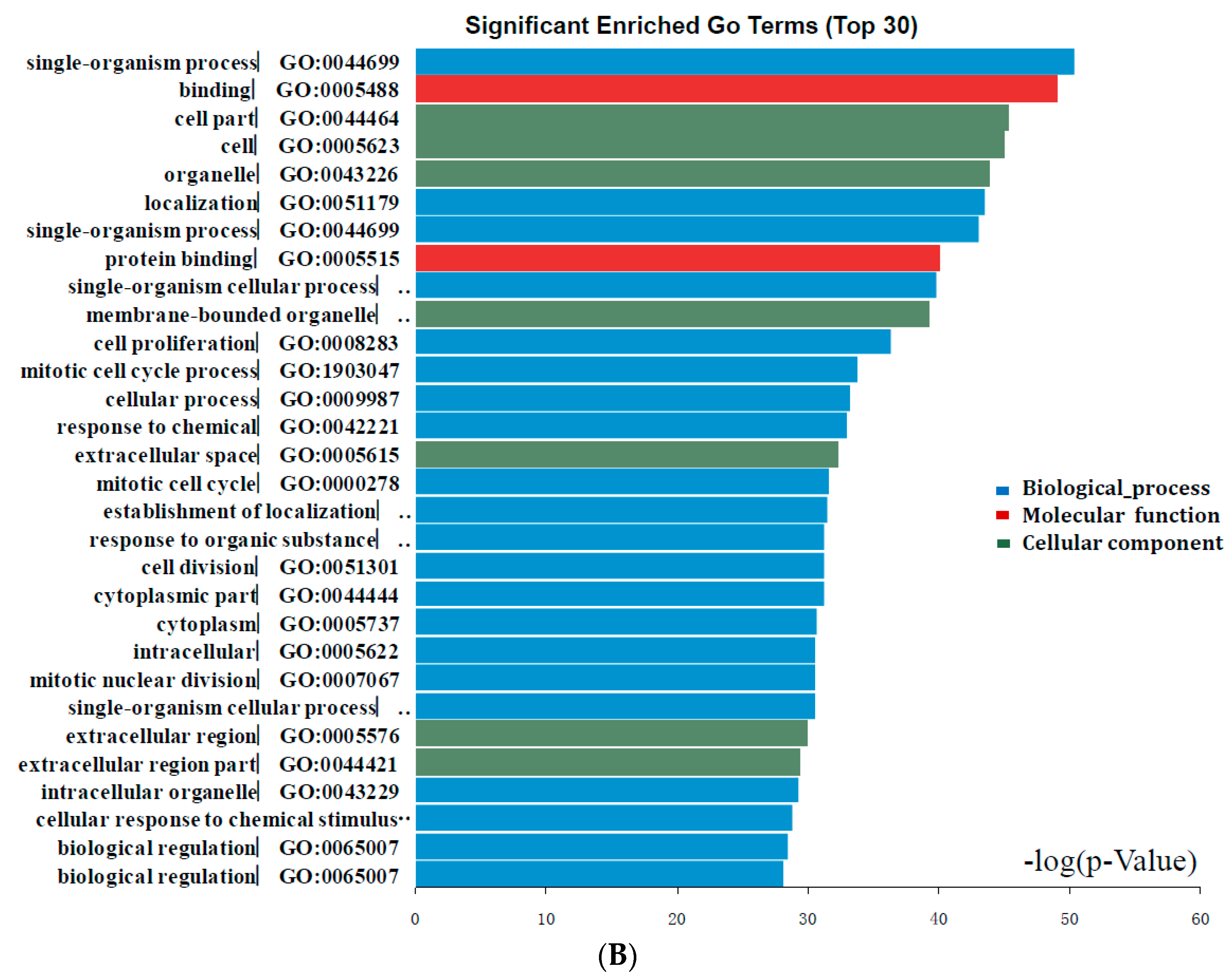
2.2. DEGs Gene Ontology Analysis in Colorectal Cancers
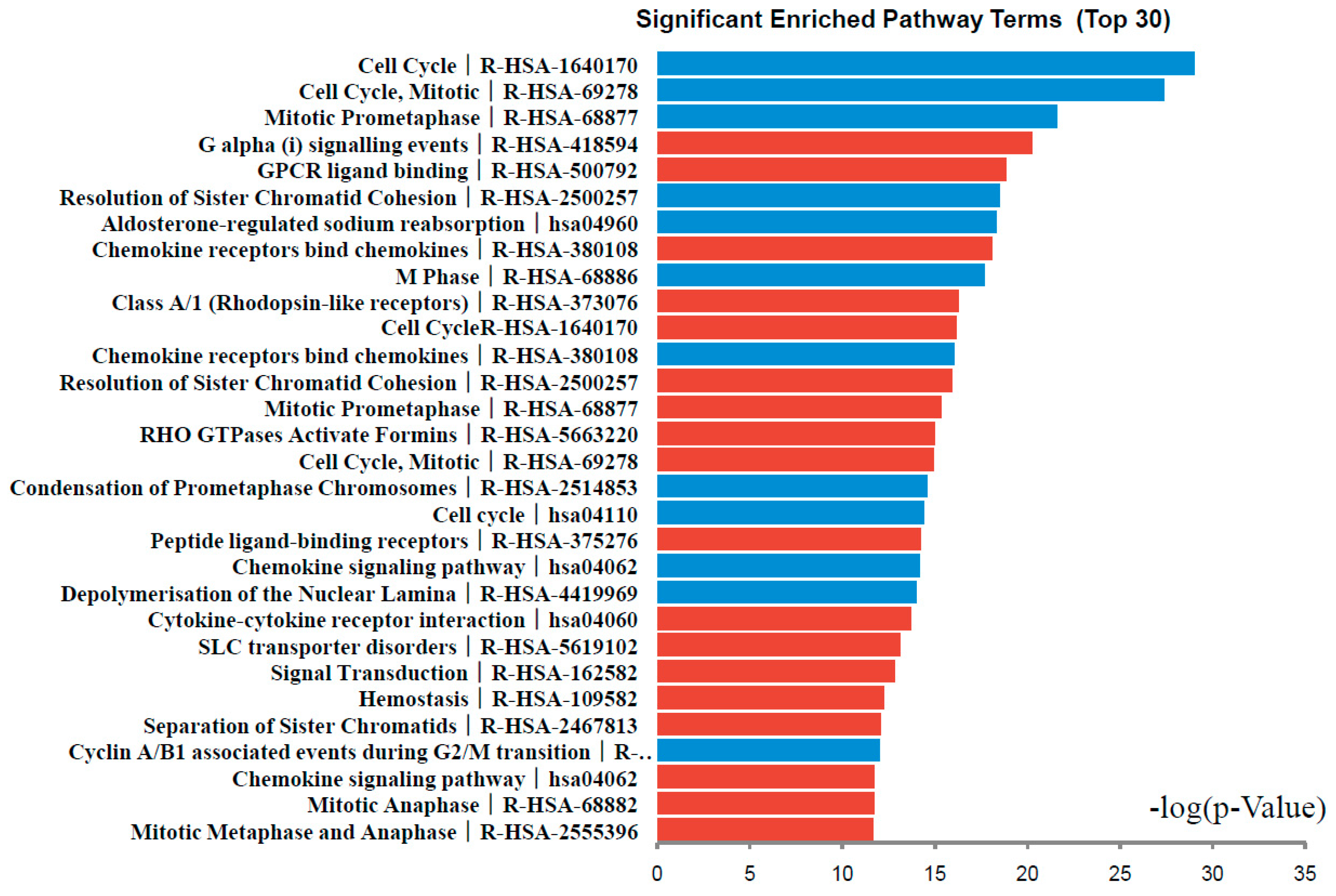
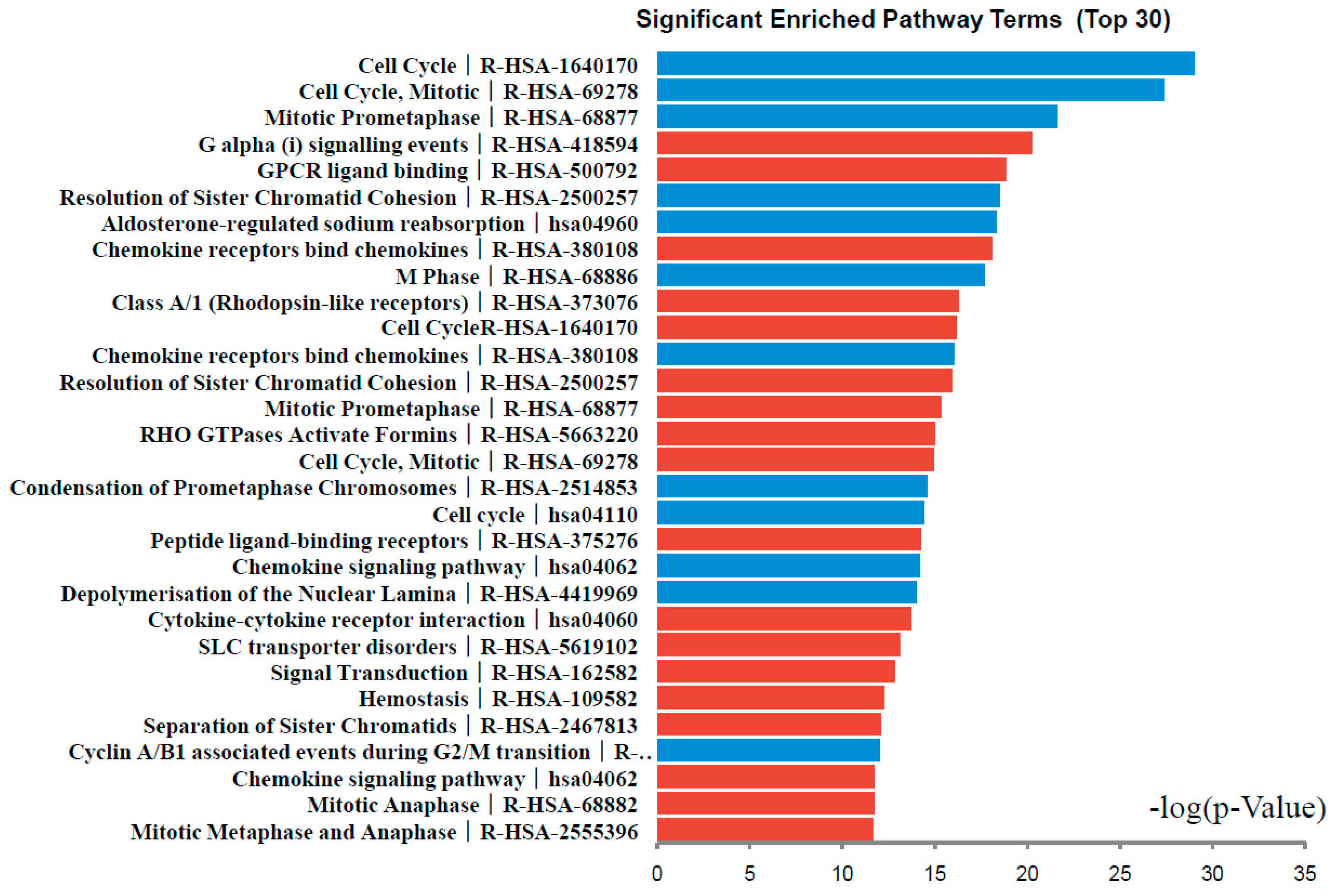
2.3. Signaling Pathway Enrichment Analysis
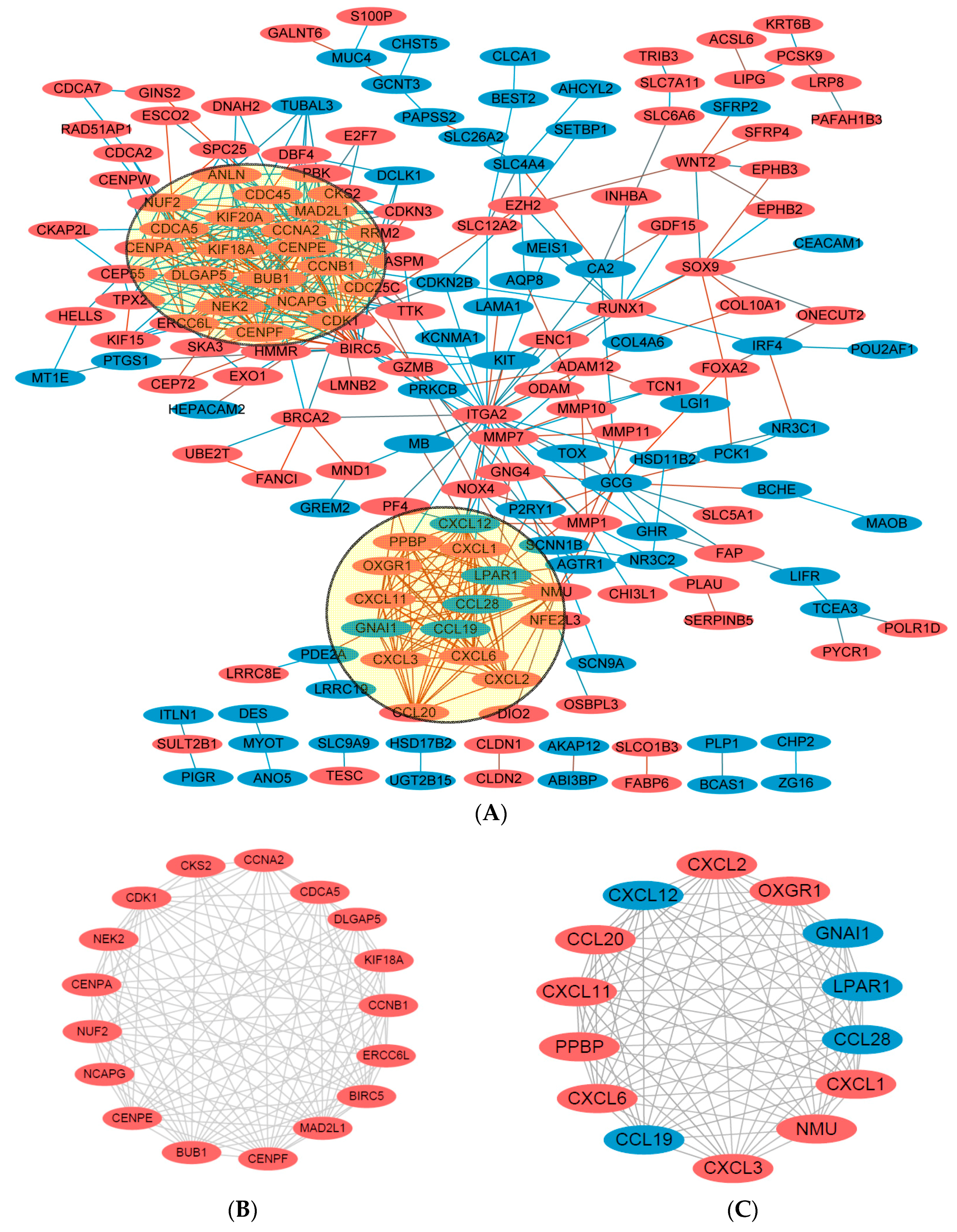
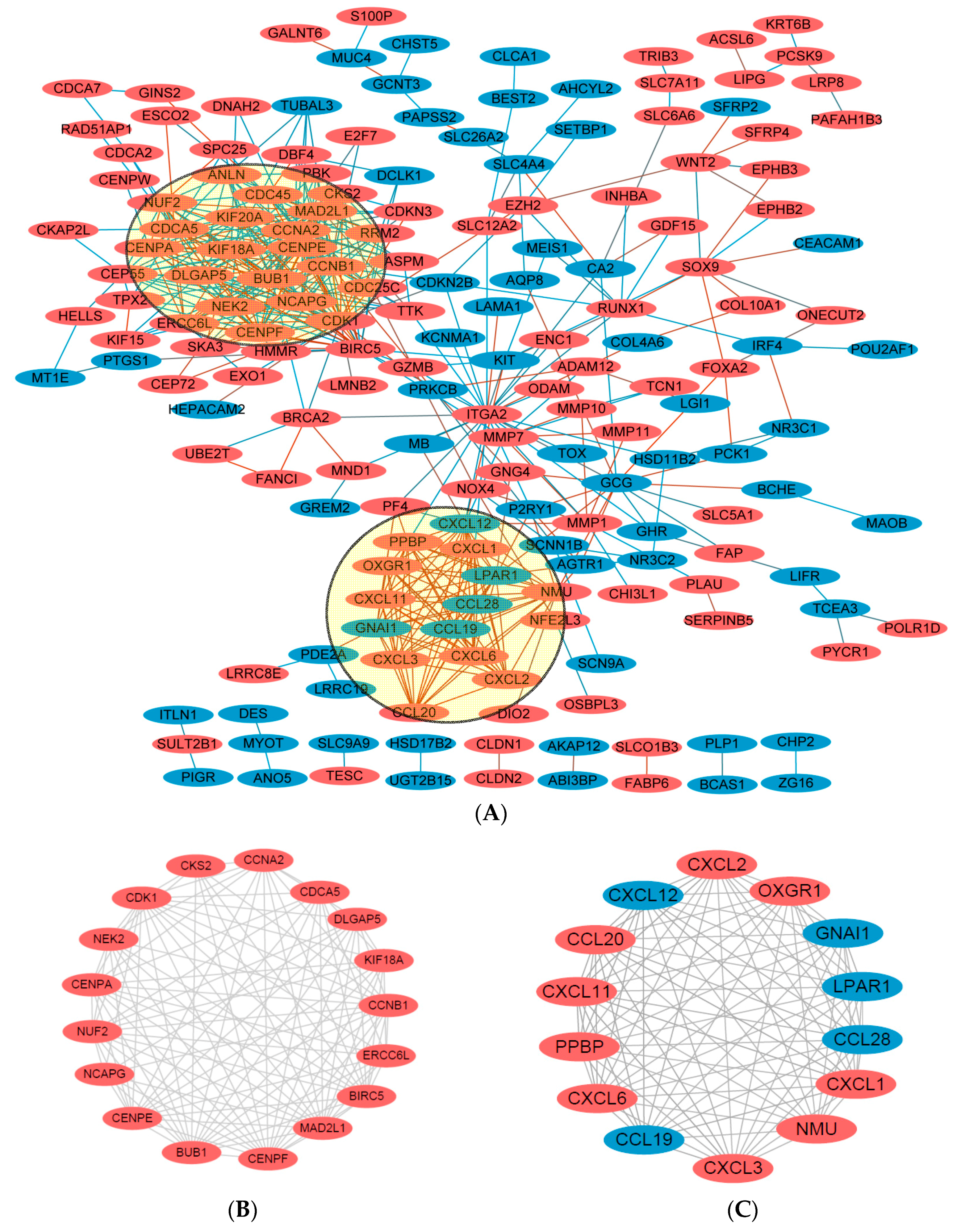
2.4. Key Candidate Genes and Pathways Identification with DEGs Protein–Protein Interaction Network (PPI) and Modular Analysis
2.5. Validation of the DEGs in TCGA Dataset
3. Discussion
4. Materials and Methods
4.1. Microarray Data Information and DEGs Identification
4.2. Gene Ontology and Pathway Enrichment Analysis
4.3. Integration of Protein–Protein Interaction (PPI) Network, Modular Analysis and Significant Candidate Genes and Pathway Identification
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| DEG | Differentially expressed genes |
| PPI | Protein–protein interaction network |
| GPCR | G-protein couple receptor |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Salas, N.; Dominguez, G.; Barderas, R.; Mendiola, M.; Garcia-Albeniz, X.; Maurel, J.; Batlle, J.F. Clinical relevance of colorectal cancer molecular subtypes. Crit. Rev. Oncol. Hematol. 2017, 109, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Jovov, B.; Araujo-Perez, F.; Sigel, C.S.; Stratford, J.K.; McCoy, A.N.; Yeh, J.J.; Keku, T. Differential gene expression between African American and European American colorectal cancer patients. PLoS ONE 2012, 7, e30168. [Google Scholar] [CrossRef] [PubMed]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Mimori, K.; Ishii, H.; Yokobori, T.; Takatsuno, Y.; Sato, T.; Toh, H.; Onoyama, I.; Nakayama, K.I.; Baba, H.; et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: Clinical significance. Int. J. Cancer 2010, 126, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Pamplona, R.; Berenguer, A.; Cordero, D.; Mollevi, D.G.; Crous-Bou, M.; Sole, X.; Pare-Brunet, L.; Guino, E.; Salazar, R.; Santos, C.; et al. Aberrant gene expression in mucosa adjacent to tumor reveals a molecular crosstalk in colon cancer. Mol. Cancer 2014, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Sole, X.; Crous-Bou, M.; Cordero, D.; Olivares, D.; Guino, E.; Sanz-Pamplona, R.; Rodriguez-Moranta, F.; Sanjuan, X.; de Oca, J.; Salazar, R.; et al. Discovery and validation of new potential biomarkers for early detection of colon cancer. PLoS ONE 2014, 9, e106748. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Lebrec, J.J.; Huizinga, T.W.; Toes, R.E.; Houwing-Duistermaat, J.J.; van Houwelingen, H.C. Integration of gene ontology pathways with North American Rheumatoid Arthritis Consortium genome-wide association data via linear modeling. BMC Proc. 2009, 3 (Suppl. S7), S94. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J. Use of Biomarkers in Screening for Cancer. Adv. Exp. Med. Biol. 2015, 867, 27–39. [Google Scholar] [PubMed]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, β-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat.Rev. 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Roy, I.; Veldkamp, C.T.; Volkman, B.F.; Dwinell, M.B. Chemokines in colitis: MicroRNA control. Gut 2014, 63, 1202–1204. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, Y.; Li, Z.; Fang, W.; Yang, Y.; Li, X.; Li, Z.; Xiong, B.; Chen, Z.; Wang, J.; et al. MicroRNA profiling in Muc2 knockout mice of colitis-associated cancer model reveals epigenetic alterations during chronic colitis malignant transformation. PLoS ONE 2014, 9, e99132. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Wu, Z.; Huang, L.; Qiu, H.; Wang, L.; Li, L.; Yao, L.; Kang, K.; Qu, J.; Wu, Y.; et al. Mulberry fruit prevents LPS-induced NF-κB/pERK/MAPK signals in macrophages and suppresses acute colitis and colorectal tumorigenesis in mice. Sci. Rep. 2015, 5, 17348. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Trinchieri, G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 2011, 11, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Grandis, J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front. Biosci. 2008, 13, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Jin, R.; Qu, G.; Wang, X.; Li, Z.; Yuan, Z.; Zhao, C.; Siwko, S.; Shi, T.; Wang, P.; et al. GPR116, an adhesion G-protein-coupled receptor, promotes breast cancer metastasis via the Gαq-p63RhoGEF-Rho GTPase pathway. Cancer Res. 2013, 73, 6206–6218. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, I.P.; Zohn, I.E.; Der, C.J. Rho GTPase-dependent transformation by G protein-coupled receptors. Oncogene 2001, 20, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.K.; Burgess, A.W.; Gulbis, J.M. Structure and function of LGR5: An enigmatic G-protein coupled receptor marking stem cells. Protein Sci. 2014, 23, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Song, M.; Bai, R.; Cheng, S.; Yan, W. Identification of potential therapeutic targets for colorectal cancer by bioinformatics analysis. Oncol. Lett. 2016, 12, 5092–5098. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Zhang, S.; Chen, X.; Hu, S. Gene expression profile analysis of colorectal cancer to investigate potential mechanisms using bioinformatics. Oncol. Targets Ther. 2015, 8, 745–752. [Google Scholar]
- Watanabe, T.; Itabashi, M.; Shimada, Y.; Tanaka, S.; Ito, Y.; Ajioka, Y.; Hamaguchi, T.; Hyodo, I.; Igarashi, M.; Ishida, H.; et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) Guidelines 2014 for treatment of colorectal cancer. Int. J. Clin. Oncol. 2015, 20, 207–239. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Ho, K.S.; Eu, K.W.; Cheah, P.Y. A susceptibility gene set for early onset colorectal cancer that integrates diverse signaling pathways: Implication for tumorigenesis. Clin. Cancer Res. 2007, 13, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Yamauchi, M.; Nishihara, R.; Lochhead, P.; Qian, Z.R.; Kuchiba, A.; Kim, S.A.; Mima, K.; Sukawa, Y.; Jung, S.; et al. Tumor LINE-1 methylation level and microsatellite instability in relation to colorectal cancer prognosis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Yamauchi, M.; Nishihara, R.; Kim, S.A.; Mima, K.; Sukawa, Y.; Li, T.; Yasunari, M.; Zhang, X.; Wu, K.; et al. Prognostic significance and molecular features of signet-ring cell and mucinous components in colorectal carcinoma. Ann. Surg. Oncol. 2015, 22, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
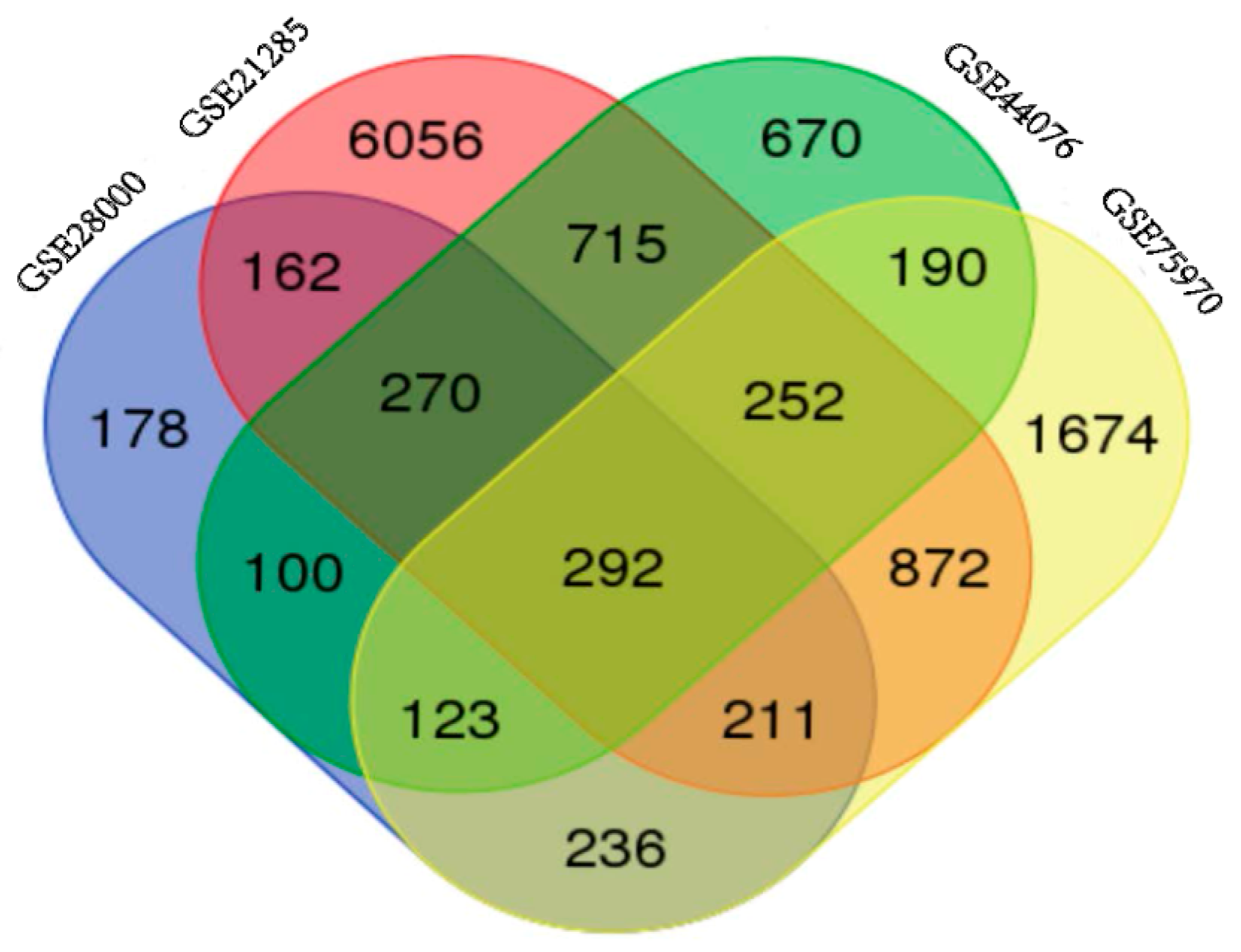

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEGs | Genes Name |
|---|---|
| Up-regulated | MMP7, FOXQ1, CLDN1, KRT23, TESC, MMP1, GDF15, ASCL2, CXCL3, CXCL1, CTHRC1, TRIB3, SLC6A6, INHBA, CHI3L1, MMP11, CLDN2, SLCO4A1, ACSL6, NFE2L3, FABP6, NEBL, CXCL2, COL10A1, AZGP1, MACC1, CGREF1, KLK10, GZMB, KRT6B, TPX2, FAP, CELSR1, PLAU, ANLN, MMP12, CXCL11, LRP8, ENC1, GALNT6, SOX9, NEK2, PMAIP1, SLCO1B3, GNG4, TMEM206, EP55, CST1, SQLE, OSBPL3, CADPS, SERPINB5, BIRC5, BUB1, MMP10, TCN1, ADAM12, S100P, NUFIP1, SKA3, SORD, SFRP4, SULT2B1, CDCA5, SIM2, ASB9, ERCC6L, DEFA6, MAD2L1, RRM2, ZNRF3, E2F7, FAM3B, CCNB1, EPHB3, CDK1, SPC25, WNT2, SLC7A11, TNS4, NMU, CENPF, ITGA2, PAFAH1B3, KIF20A, CXCL6, MND1, CCNA2, CCL20, FOXA2, ODAM, DIO2, RNF43, FERMT1, PYCR1, RNF183, PAQR4, GPR143, UBE2T, BRCA2, UNC5CL, CKS2, RUNX1, DBF4, CEP72, XKRX, CDC45, KIF18A, CENPW, FAM150A, PPBP, RAB15, CMTM8, FANCI, ECE2, NOX4, ASPM, SLC5A1, PCSK9, TTK, FAM64A, CKAP2L, KDELR3, LIPG, CDCA2, NCAPG, KIF15, ESCO2, PBK, NUF2, EZH2, NUP62CL, CDKN3, EPHB2, KCTD14, OXGR1, CNPY3, GINS2, LMNB2, DLGAP5, PSAT1, POLR1D, HPDL, REG1B, CDC25C, EXO1, ASPHD1, LRRC8E, SFTA2, HMMR, GPX2, DNAH2, HELLS, CLCN5, SLC12A2, RAD51AP1, CENPA, TRIM29, ONECUT2, SYNCRIP, CENPE, PF4, CDCA7, BACE2, CKAP2L, KDELR3, LIPG, CDCA2, NCAPG, KIF15, ESCO2, PBK |
| Down-regulated | CLCA4, AQP8, GUCA2B, ZG16, CA2, CLCA1, ITLN1, HSD17B2, CHP2, AKR1B10, CEACAM7, GCG, SLC4A4, BEST2, LAMA1, HRASLS2, FCGBP, SCARA5, HEPACAM2, SCNN1B, MT1H, CDKN2B, LDHD, SLC26A2, UGT2B15, HHLA2, VSIG2, SCIN, MUC4, GCNT3, INSL5, SDCBP2, MAMDC2, BTNL8, LRRC19, AHCYL2, NR3C2, CXCL12, MT1G, PIGR, TNFRSF17, TMEM100, MYOT, HSD11B2, STMN2, SLC17A4, NAP1L2, TUBAL3, MT1E, CEACAM1, FAM150B, BCAS1, KIF16B, PTPRH, GREM2, CA12, BTNL3, FABP1, LIFR, SYNPO2, MB, ATP1A2, CDH19, DENND2A, TSPAN7, PAPSS2, SCUBE2, ATP2A3, CILP, DPT, MAOB, KIT, GFRA2, CCL28, TCEA3, CCL19, GHR, P2RY1, CNTN3, PCOLCE2, DES, PADI2, POU2AF1, IRF4, SIAE, MFAP4, ANGPTL1, CHST5, ABI3BP, ANO5, ABCA8, HSPB3, SCGN, FAM132A, CR2, STOX2, PTGS1, PRKCB, LPAR1, SLC25A23, NR3C1, NDN, SLC17A8, PDE2A, OLFM1, KCNMA1, SERTAD4, LGI1, KIAA2022, COL4A6, BCHE, TOX, PRKAA2, SETBP1, AGTR1, MEIS1, ASPA, ZSCAN18, CHRNA3, SCN9A, SLC9A9, SFRP2, GNAI1, PLP1, AKAP12, DCLK1, PCK1 |
| Term | Description | Count | p-Value |
|---|---|---|---|
| Up-regulated | |||
| GO:0044699 | single-organism process | 129 | 1.10 × 10−13 |
| GO:0008283 | cell proliferation | 41 | 1.16 × 10−11 |
| GO:0005615 | extracellular space | 32 | 1.82 × 10−10 |
| GO:0044763 | single-organism cellular process | 114 | 6.45 × 10−10 |
| GO:0065007 | biological regulation | 110 | 3.35 × 10−9 |
| GO:0005576 | extracellular region | 60 | 5.56 × 10−9 |
| GO:0000278 | mitotic cell cycle | 24 | 1.51 × 10−8 |
| GO:1903047 | mitotic cell cycle process | 23 | 1.68 × 10−8 |
| GO:0044421 | extracellular region part | 53 | 1.74 × 10−8 |
| GO:0009987 | cellular process | 129 | 2.59 × 10−8 |
| Down-regulated | |||
| GO:0044699 | single-organism process | 106 | 6.94 × 10−16 |
| GO:0005488 | binding | 110 | 1.68 × 10−15 |
| GO:0044464 | cell part | 115 | 2.24 × 10−14 |
| GO:0005623 | cell | 115 | 2.65 × 10−14 |
| GO:0043226 | organelle | 103 | 6.36 × 10−14 |
| GO:0051179 | localization | 66 | 7.96 × 10−14 |
| GO:0005515 | protein binding | 92 | 8.65 × 10−13 |
| GO:0044763 | single-organism cellular process | 96 | 1.03 × 10−12 |
| GO:0043227 | membrane-bounded organelle | 97 | 1.58 × 10−12 |
| GO:1903047 | mitotic cell cycle process | 23 | 6.85 × 10−11 |
| Pathway | Name | Gene Count | p-Value | Genes |
|---|---|---|---|---|
| Up-regulated DEG | ||||
| Reactome:R-HSA-418594 | G α (i) signalling events | 11 | 8.21 × 10−7 | CXCL2 NMU CXCL6 CCL28 CXCL3 PPBP OXGR1 GNG4 CXCL11 INSL5 LPAR1 |
| Reactome:R-HSA-500792 | GPCR ligand binding | 14 | 2.10 × 10−6 | CXCL2 WNT2 NMU GCG CXCL6 CCL28 CXCL3 PPBP OXGR1 GNG4 CXCL11 P2RY1 INSL5 LPAR1 |
| Reactome:R-HSA-380108 | Chemokine receptors bind chemokines | 6 | 3.62 × 10−6 | CXCL2 CXCL11 CXCL3 CXCL6 CCL28 PPBP |
| Reactome:R-HSA-373076 | Class A/1 (Rhodopsin-ike receptors) | 11 | 1.24 × 10−5 | CXCL2 NMU CXCL6 CCL28 CXCL3 PPBP OXGR1 CXCL11 P2RY1 INSL5 LPAR1 |
| Reactome:R-HSA-1640170 | Cell Cycle | 15 | 1.36 × 10−5 | DBF4 TPX2 BIRC5 CEP72 CENPA CENPF MND1 GINS2 CENPE BRCA2 CDC25C MAD2L1 BUB1 RRM2 TUBAL3 |
| Reactome:R-HSA-2500257 | Resolution of Sister Chromatid Cohesion | 7 | 1.64 × 10−5 | BIRC5 CENPA CENPF CENPE MAD2L1 BUB1 TUBAL3 |
| Reactome:R-HSA-68877 | Mitotic Prometaphase | 7 | 2.45 × 10−5 | BIRC5 CENPA CENPF CENPE MAD2L1 BUB1 TUBAL3 |
| Reactome:R-HSA-5663220 | RHO GTPases Activate Formins | 7 | 3.12 × 10−5 | BIRC5 CENPA CENPF CENPE MAD2L1 BUB1 TUBAL3 |
| Reactome:R-HSA-69278 | Cell Cycle, Mitotic | 13 | 3.23 × 10−5 | DBF4 TPX2 BIRC5 CEP72 CENPA CENPF GINS2 CENPE CDC25C MAD2L1 BUB1 RRM2 TUBAL3 |
| Reactome:R-HSA-375276 | Peptide ligand-binding receptors | 8 | 5.24 × 10−5 | CXCL2 NMUCXCL6 CCL28 CXCL3 PPBP CXCL11 INSL5 |
| KEGG Pathway:hsa04060 | Cytokine-cytokine receptor interaction | 9 | 7.61 × 10−5 | TNFRSF17 CXCL2 LIFR INHBA CXCL6 CCL28 CXCL3 PPBP CXCL11 |
| Down-regulated DEGs | ||||
| Reactome:R-HSA-1640170 | Cell Cycle | 18 | 1.83 × 10−9 | CDC45 NCAPG KIF20A NEK2 KIF18A CCNB1 NUF2 CCNA2 CDCA5 CDKN2B SPC25 PRKCB CDK1 ESCO2 EXO1 ERCC6L CENPW |
| Reactome:R-HSA-69278 | Cell Cycle, Mitotic | 16 | 5.78 × 10−9 | HMMR CDC45 NCAPG KIF20A NEK2 KIF18A CCNB1 NUF2 CCNA2 CDCA5 CDKN2B SPC25 PRKCB CDK1 ESCO2 ERCC6L |
| Reactome:R-HSA-68877 | Mitotic Prometaphase | 8 | 3.19 × 10−7 | NCAPG KIF18A CCNB1 NUF2 CDCA5 SPC25 CDK1 ERCC6L |
| Reactome:R-HSA-2500257 | Resolution of Sister Chromatid Cohesion | 7 | 2.74 × 10−6 | KIF18A CCNB1 NUF2 CDCA5 SPC25 CDK1 ERCC6L |
| KEGG PathwaY:hsa04960 | Aldosterone-regulated sodium reabsorption | 5 | 3.09 × 10−6 | ATP1A2 NR3C2 PRKCB SCNN1B HSD11B2 |
| Reactome:R-HSA-68886 | M Phase | 10 | 4.77 × 10−6 | NCAPG KIF20A KIF18A CCNB1 NUF2 CDCA5 SPC25 PRKCB CDK1 ERCC6L |
| Reactome:R-HSA-380108 | Chemokine receptors bind chemokines | 5 | 1.48 × 10−5 | CXCL12 CCL20 PF4 CXCL1 CCL19 |
| Reactome:R-HSA-2514853 | Condensation of Prometaphase Chromosomes | 3 | 4.06 × 10−5 | NCAPG CCNB1 CDK1 |
| KEGG Pathway:hsa04110 | Cell cycle | 6 | 4.53 × 10−5 | CDC45 TTK CCNB1 CCNA2 CDKN2B CDK1 |
| KEGG Pathway:hsa04062 | Chemokine signaling pathway | 7 | 5.37 × 10−5 | CXCL12 CCL20 GNAI1 PF4 CXCL1 PRKCB CCL19 |
| Reactome:R-HSA-4419969 | Depolymerisation of the Nuclear Lamina | 3 | 6.20 × 10−5 | CCNB1 PRKCB CDK1 |
| Term | Description | Count | p-Value |
|---|---|---|---|
| R-HSA-68877 | Mitotic Prometaphase | 12 | 2.56 × 10−24 |
| GO:0000819 | sister chromatid segregation | 13 | 5.52 × 10−24 |
| GO:0000280 | nuclear division | 15 | 2.13 × 10−23 |
| GO:0048285 | organelle fission | 15 | 5.03 × 10−23 |
| GO:1903047 | mitotic cell cycle process | 16 | 8.44 × 10−23 |
| GO:0098813 | nuclear chromosome segregation | 13 | 8.98 × 10−23 |
| GO:0007067 | mitotic nuclear division | 14 | 1.40 × 10−22 |
| GO:0022402 | cell cycle process | 17 | 1.70 × 10−22 |
| GO:0000278 | mitotic cell cycle | 16 | 2.87 × 10−22 |
| R-HSA-2500257 | Resolution of Sister Chromatid Cohesion | 11 | 4.68 × 10−22 |
| GO:0007059 | chromosome segregation | 13 | 6.23 × 10−22 |
| R-HSA-69278 | Cell Cycle, Mitotic | 14 | 1.32 × 10−21 |
| GO:0007049 | cell cycle | 17 | 1.11 × 10−20 |
| R-HSA-1640170 | Cell Cycle | 14 | 1.84 × 10−20 |
| GO:0000775 | chromosome, centromeric region | 11 | 4.37 × 10−20 |
| R-HSA-68886 | M Phase | 12 | 1.09 × 10−19 |
| GO:0000793 | condensed chromosome | 11 | 1.41 × 10−19 |
| GO:0098687 | chromosomal region | 12 | 2.49 × 10−19 |
| GO:0000776 | kinetochore | 10 | 2.70 × 10−19 |
| GO:0000070 | mitotic sister chromatid segregation | 10 | 3.14 × 10−19 |
| GO:0051301 | cell division | 13 | 6.90 × 10−19 |
| GO:0000777 | condensed chromosome kinetochore | 9 | 7.74 × 10−18 |
| GO:0000779 | condensed chromosome, centromeric region | 9 | 1.64 × 10−17 |
| GO:0044427 | chromosomal part | 13 | 6.36 × 10−17 |
| GO:0007062 | sister chromatid cohesion | 9 | 8.20 × 10−17 |
| GO:0051276 | chromosome organization | 14 | 1.02 × 10−16 |
| GO:0051983 | regulation of chromosome segregation | 8 | 2.39 × 10−16 |
| GO:0005694 | chromosome | 13 | 3.03 × 10−16 |
| GO:1902589 | single-organism organelle organization | 15 | 4.31 × 10−16 |
| GO:0007346 | regulation of mitotic cell cycle | 11 | 1.03 × 10−15 |
| Term | Description | Count | p-Value |
|---|---|---|---|
| R-HSA-418594 | G α (i) signaling events | 14 | 1.36 × 10−27 |
| GO:0008009 | chemokine activity | 10 | 6.81 × 10−24 |
| GO:0042379 | chemokine receptor binding | 10 | 3.15 × 10−23 |
| R-HSA-380108 | Chemokine receptors bind chemokines | 10 | 3.15 × 10−23 |
| R-HSA-373076 | Class A/1 (Rhodopsin-like receptors) | 13 | 3.87 × 10−23 |
| GO:0001664 | G-protein coupled receptor binding | 12 | 8.63 × 10−22 |
| R-HSA-500792 | GPCR ligand binding | 13 | 2.51 × 10−21 |
| hsa04062 | Chemokine signaling pathway | 11 | 8.22 × 10−21 |
| R-HSA-375276 | Peptide ligand-binding receptors | 11 | 1.08 × 10−20 |
| GO:0060326 | cell chemotaxis | 11 | 6.67 × 10−20 |
| GO:0002685 | regulation of leukocyte migration | 10 | 2.42 × 10−19 |
| R-HSA-388396 | GPCR downstream signaling | 14 | 2.58 × 10−19 |
| GO:0070098 | chemokine-mediated signaling pathway | 9 | 2.86 × 10−19 |
| GO:0045236 | CXCR chemokine receptor binding | 7 | 1.26 × 10−18 |
| GO:0002687 | positive regulation of leukocyte migration | 9 | 4.32 × 10−18 |
| GO:0007186 | G-protein coupled receptor signaling pathway | 14 | 7.31 × 10−18 |
| GO:0050921 | positive regulation of chemotaxis | 9 | 9.82 × 10−18 |
| GO:0005125 | cytokine activity | 10 | 1.07 × 10−17 |
| R-HSA-372790 | Signaling by GPCR | 14 | 1.20 × 10−17 |
| hsa04060 | Cytokine-cytokine receptor interaction | 10 | 6.02 × 10−17 |
| GO:0005126 | cytokine receptor binding | 10 | 8.95 × 10−17 |
| GO:0002690 | positive regulation of leukocyte chemotaxis | 8 | 1.25 × 10−16 |
| GO:0030595 | leukocyte chemotaxis | 9 | 2.93 × 10−16 |
| GO:0050920 | regulation of chemotaxis | 9 | 3.23 × 10−16 |
| GO:0002688 | regulation of leukocyte chemotaxis | 8 | 4.83 × 10−16 |
| GO:0006935 | chemotaxis | 11 | 5.74 × 10−16 |
| GO:0042330 | taxis | 11 | 5.86 × 10−16 |
| GO:0050900 | leukocyte migration | 10 | 1.08 × 10−15 |
| GO:0030335 | positive regulation of cell migration | 10 | 2.69 × 10−15 |
| GO:2000147 | positive regulation of cell motility | 10 | 3.62 × 10−15 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Bao, Y.; Ma, M.; Yang, W. Identification of Key Candidate Genes and Pathways in Colorectal Cancer by Integrated Bioinformatical Analysis. Int. J. Mol. Sci. 2017, 18, 722. https://doi.org/10.3390/ijms18040722
Guo Y, Bao Y, Ma M, Yang W. Identification of Key Candidate Genes and Pathways in Colorectal Cancer by Integrated Bioinformatical Analysis. International Journal of Molecular Sciences. 2017; 18(4):722. https://doi.org/10.3390/ijms18040722
Chicago/Turabian StyleGuo, Yongchen, Yonghua Bao, Ming Ma, and Wancai Yang. 2017. "Identification of Key Candidate Genes and Pathways in Colorectal Cancer by Integrated Bioinformatical Analysis" International Journal of Molecular Sciences 18, no. 4: 722. https://doi.org/10.3390/ijms18040722
APA StyleGuo, Y., Bao, Y., Ma, M., & Yang, W. (2017). Identification of Key Candidate Genes and Pathways in Colorectal Cancer by Integrated Bioinformatical Analysis. International Journal of Molecular Sciences, 18(4), 722. https://doi.org/10.3390/ijms18040722