1. Introduction
Oceans cover more than 75% of the Earth’s surface and represent one of the most complex ecosystems, being an important resource for the discovery of novel molecules with distinct modes of action [
1,
2]. The interest in marine agents is growing and their biomedical potential has slowly gained prime significance among natural products’ research, attracting scientific and economic interests worldwide [
1,
2,
3,
4]. The chemical diversity of a wide variety of natural products from marine animals, have been providing therapeutic agents to treat various diseases, including those arising from acute or chronic inflammatory processes [
2].
The phylum Cnidaria comprises over 11,000 species, including sea anemones [
2]. In this work two sea anemone species were studied:
Actinia equina (Linnaeus, 1767) and
Anemonia sulcata (Pennant, 1777). These organisms are benthic as adults and lack the physical defense of a mineralized skeleton. Thus, they produce several secondary metabolites that often act as chemical defenses against predation, adverse conditions, fouling, or infection [
5]. Nevertheless, little is described about their biological activity.
With the increasing consumption of novel foods there have been reports on the human consumption of several species of sea anemones as foodstuffs [
6]. However, to the best of our knowledge, no previous work has addressed the composition of sea anemone extracts and their biological effects. In order to mimic the route of ingestion, we have used aqueous extracts of
A. equina and
A. sulcata with the aim of studying their in vitro anti-inflammatory effects in a murine macrophage cell line (RAW 264.7), as well as potential toxicity upon human gastric adenocarcinoma cells (AGS), with emphasis in the mechanism of action.
3. Materials and Methods
3.1. Chemicals and Standards
LPS from Escherichia coli, sulfanilamide, dichlorodihydrofluorescein diacetate (DCDHF-DA), MTT, DPX mountant, sodium pyruvate, β-nicotinamide adenine dinucleotide reduced form (NADH), Triton X-100, N-(1-naphthyl)ethylenediamine dihydrochloride, Giemsa dye, sodium deoxycholate, trizma hydrochloride, dimethyl sulfoxide (DMSO), LOX from Glycine max (L.) Merr. (Type V-S; EC 1.13.11.12), PLA2 (EC.3.1.1.4, from honey bee venom (Apis mellifera), magnesium chloride (MgCl2), 1,2-dilinoleoyl-sn-glycero-3-phosphocholine, Trypan blue, iodomethane, propylene carbonate, picolinic acid, propano-2-ol, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), sucrose, 1,4-dithiothreitol (DTT), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), 2,2′,2″,2′′′-(ethane-1,2-diyldinitrilo)tetraacetic acid (EDTA), palmitic acid, dexamethasone and phorbol 12-myristate 13-acetate (PMA) were from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), Hank’s balanced salt solution (HBSS), 0.05% Trypsin-EDTA (1X) and penicillin-streptomycin solution (penicillin 5000 units/mL and streptomycin 5000 µg/mL) were purchased from GIBCO® by life technologies™, Invitrogen (Grand Island, NY, USA). Caspase-3/-7 luminescent assay kit was from Promega Corporation. Staurosporine and Z-VAD.fmk were obtained by Santa Cruz Biotechnology (Dallas, Texas, USA). Acetonitrile, potassium dihydrogen phosphate and methanol were purchased from Merck (Darmstadt, Germany). Hydrochloric acid was from VWR International, LLC. Ammonia, phosphoric acid and ethyl ether were from Panreac (Barcelona, Spain). Ac-Leu-Glu-His-Asp-7-Amino-4-trifluoromethylcoumarin (caspase-9 substrate) was purchased from CPC Scientific (Sunnyvale, CA, USA). Z-Leu-Glu-Val-Asp-AFC (caspase-4 substrate) was obtained from Innovagen (Lund, Sweeden). Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid was from AMRESCO (Solon, OH, USA).
3.2. Sampling and Extract Preparation
Individuals of A. sulcata and A. equina were collected in Praia da Luz (Porto, Portugal) in July 2014. After collection, samples were placed on ice and immediately transported to the laboratory, washed with a saline solution to remove contaminants, frozen and lyophilized (Lyophilizer Lacone Freezone 4,5—Kansas City, MO, USA). Afterwards, the samples were pulverized and 5 g were sifted (910 µm). After this procedure 100 mL of extractor solvent (water) were added. The extraction occurred in 30 min under stirring (200 rpm) and at room temperature. Both extracts were centrifuged (ROTOFIX 32A, Hettich, Tuttlingen, Germany) at 400 rpm for 10 min and then filtered under reduced pressure. Lastly, aqueous extracts were frozen at −20 °C, lyophilized for seven days, and then stored in a desiccator until analysis.
3.3. Metabolic Profile Analysis
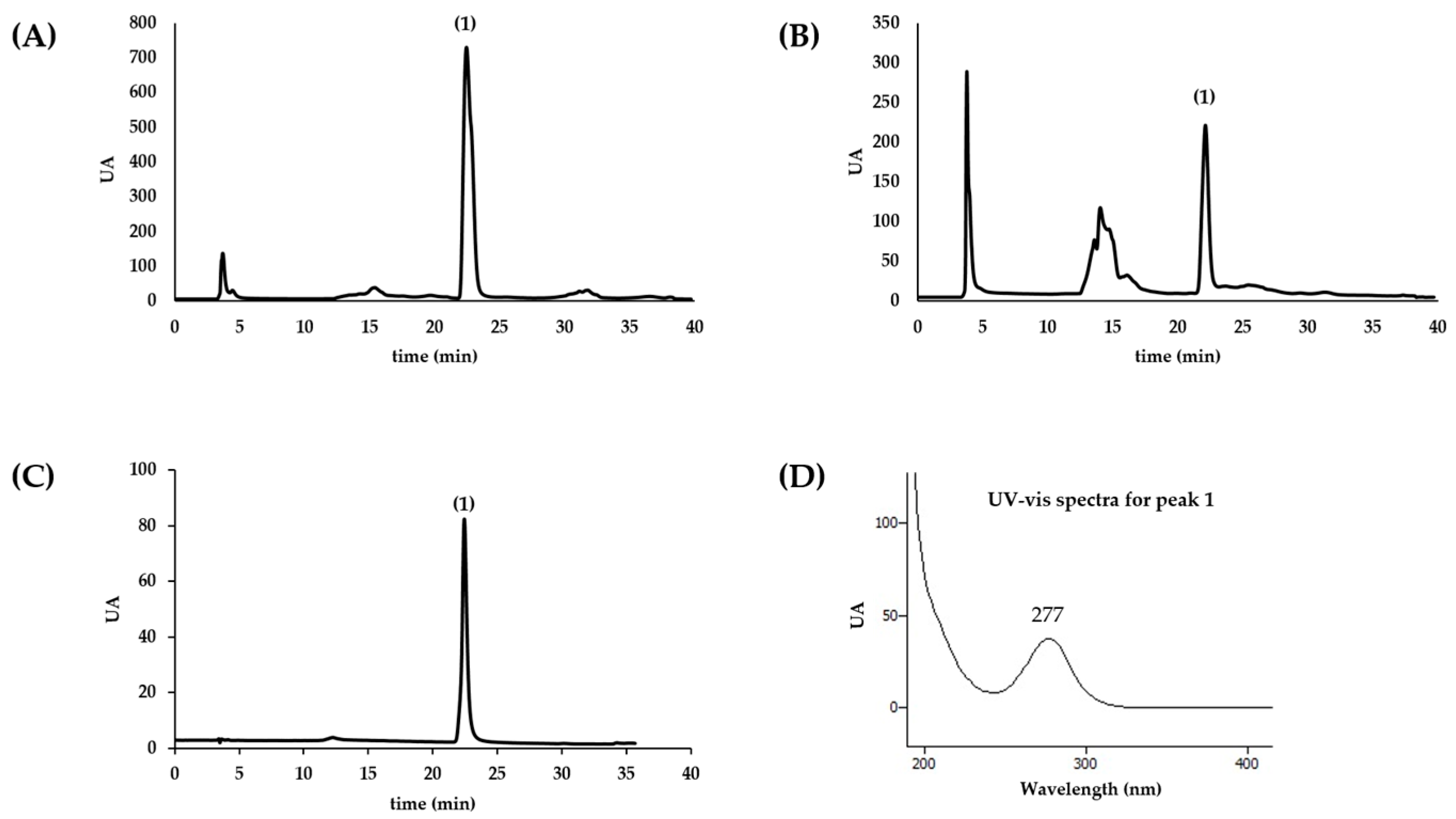
The chromatographic profile of aqueous extract of sea anemones was established by HPLC-DAD (Gilson 811C Dynamic Mixer, Gilson HPLC Dynamic Mixer). Compounds present in the extracts were separated with reverse phase column (Waters Spherisorb, C18, 5 µm ODS2, 250 × 4.6 mm Analytical Cartridge, Part NO. PSS839540, Dublin, Ireland), at room temperature and using a gradient elution method. The mobile phase consisted of two solvents: water (A) and acetonitrile (B). Elution started with 100% B and a gradient was used to obtain 85% A at 15 min, followed by 30% A at 35 min. The injection volume was 20 µL and the flow rate 0.9 mL/min. The spectral data were collected in the range of 190–700 nm and elution was monitored at 280, 320, and 350 nm. The data were processed using the software Clarity (Europa Science Ltd., Cambridge, UK). Homarine quantification was achieved by the interpolation of the absorbance recorded at 280 nm in the chromatograms relative to a calibration curve constructed with the authentic standard analyzed under the same conditions. At least three independent analysis were conducted.
3.4. Screening for Alkaloids
Screening for alkaloids was performed in order to assess the possible presence of these in both extracts [
20]. For alkaloid extraction, extracts (1 g) were weighed, heated with 10 mL of 10% hydrochloric acid and then filtered. Ammonia solution (1:1) was added to alkalinize the solution and alkaloids were extracted with ethyl ether. The ether phase was separated and 10 mL of 10% HCl solution were added. Finally, the solution was divided in 4 test tubes, three of them receiving either the Dragendorff, Mayer, or Bertrand’s reagent, the fourth one serving as control. Formation of precipitates with all reagents were assessed.
3.5. Synthesis of Homarine
As there was no homarine commercially available at the time this study was conducted; as such, we have synthesized this molecule: 1.5 g iodomethane was added to a well-stirred suspension of 1.0 g picolinic acid (0.81 mmol) in 20 mL propylene carbonate. After two days, 100 mL ether were added and the resulting yellow solid was recrystallized in methanol/ether. Identity of the molecule was confirmed by 1H-NMR (400 MHz, MeOD) δ ppm: 4.55 (3H, s), 8.11 (1H, td, J = 7 Hz, J = 1 Hz), 8.36 (1H, dd, J = 8 Hz, J = 1 Hz), 8.65 (1H, td, J = 8 Hz, J = 1 Hz), 8.96 (1H, dd, J = 7 Hz, J = 1 Hz); 13C-NMR (101 MHz, MeOD) δ ppm: 49.00, 129.53, 129.59, 147.69, 148.58, 150.78, 163.73.
3.6. Cell Culture
Murine macrophage-like cell line, RAW 264.7, was from the American Type Culture Collection (LGC Standards S.L.U., Barcelona, Spain) and AGS cells were from Sigma-Aldrich. Cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin, and grown in an incubator at 37 °C, in a humidified atmosphere of 5% CO2. AGS cells were trypsinized and subjected to centrifugation at 1300 rpm for 5 min and then suspended in flasks. RAW cells were harvested by scraping and then suspended in flasks for growth.
3.7. MTT
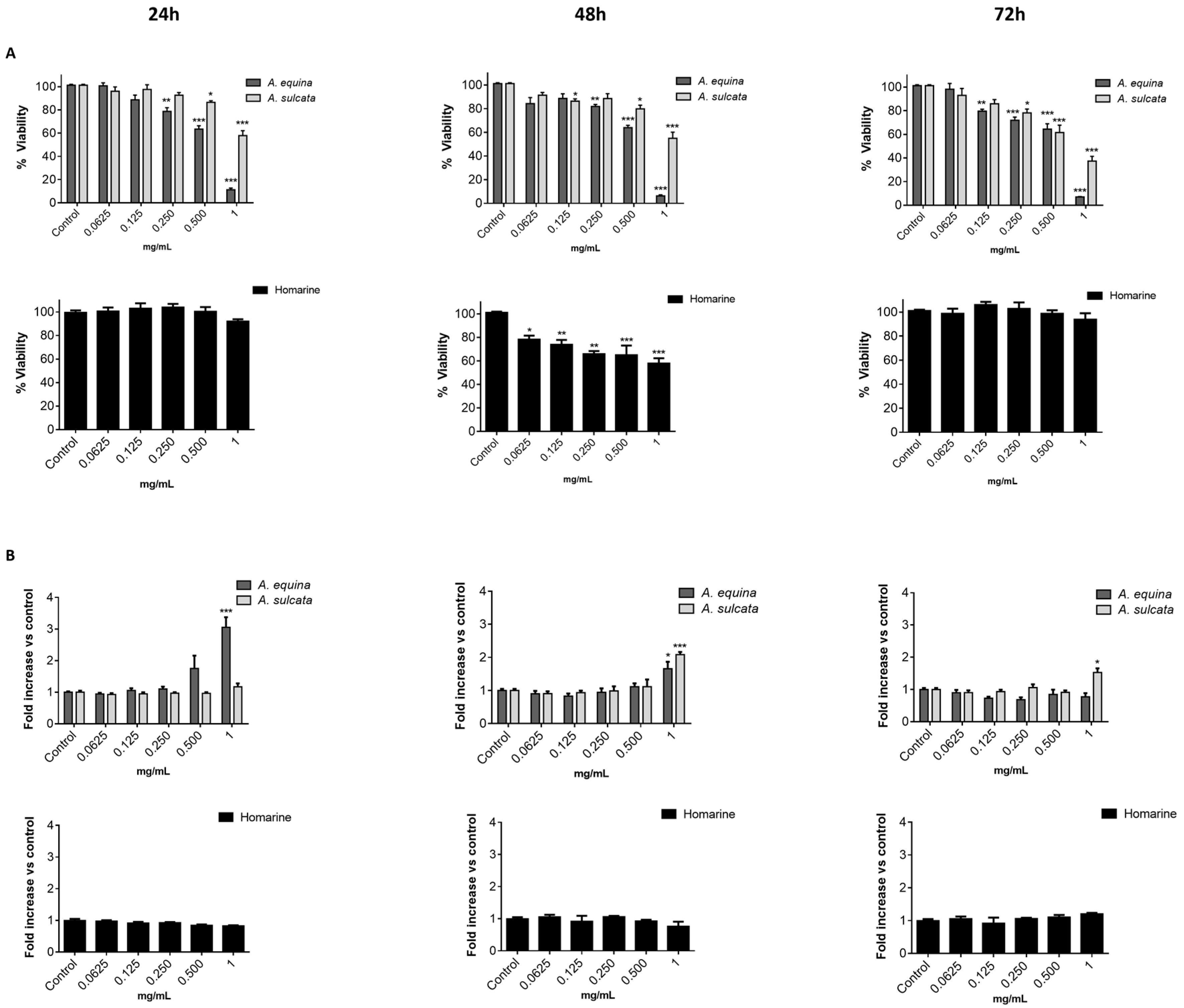
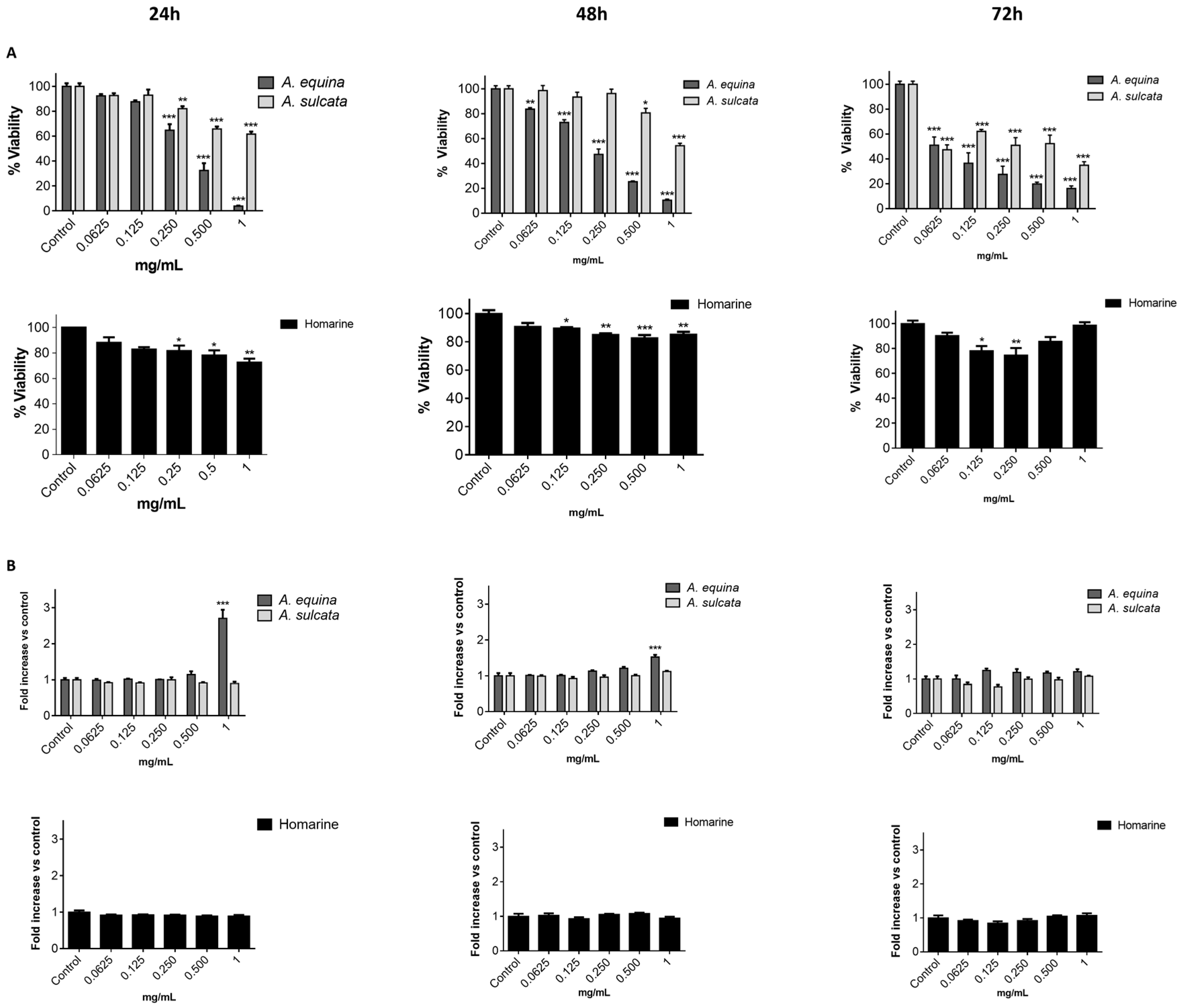
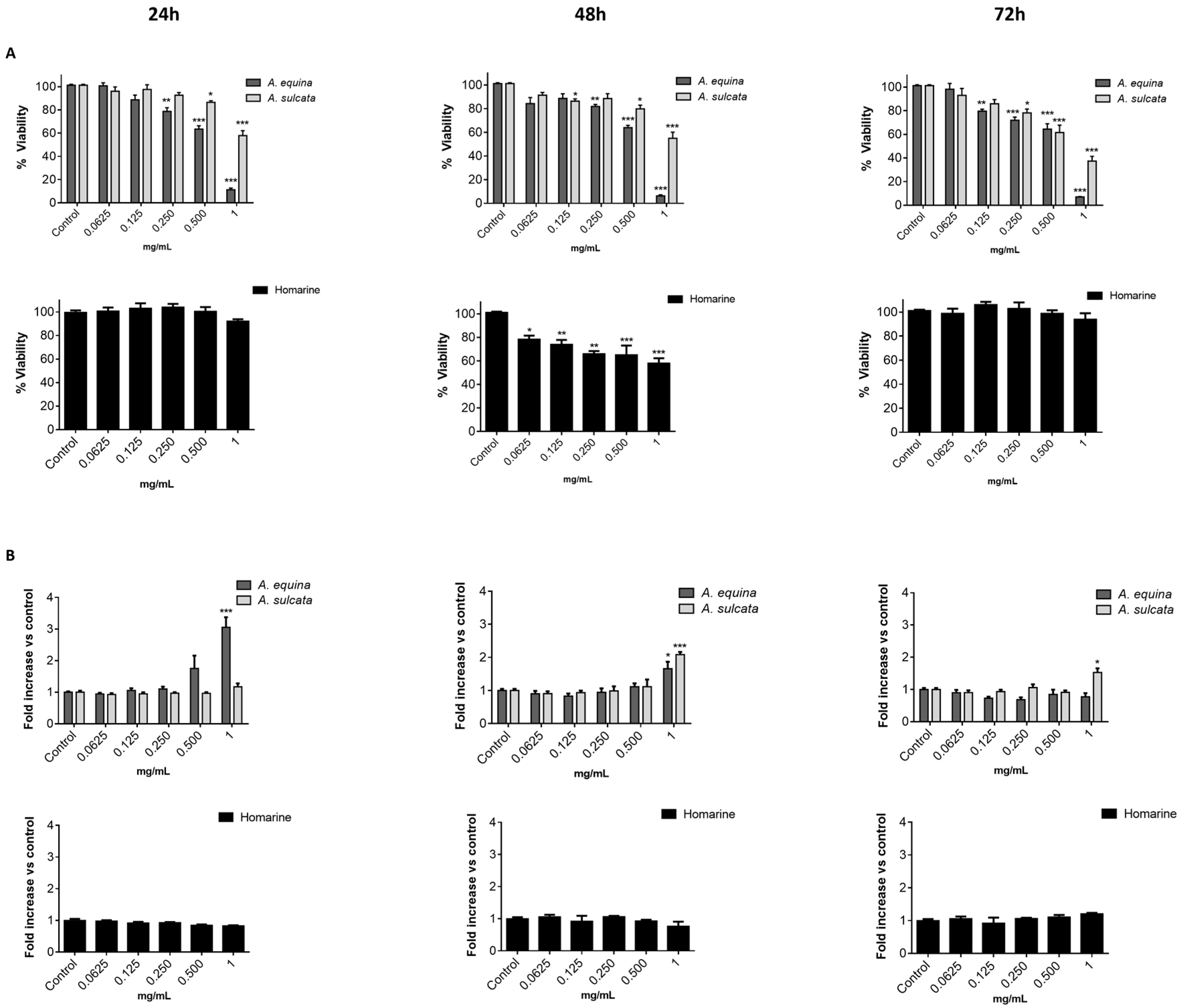
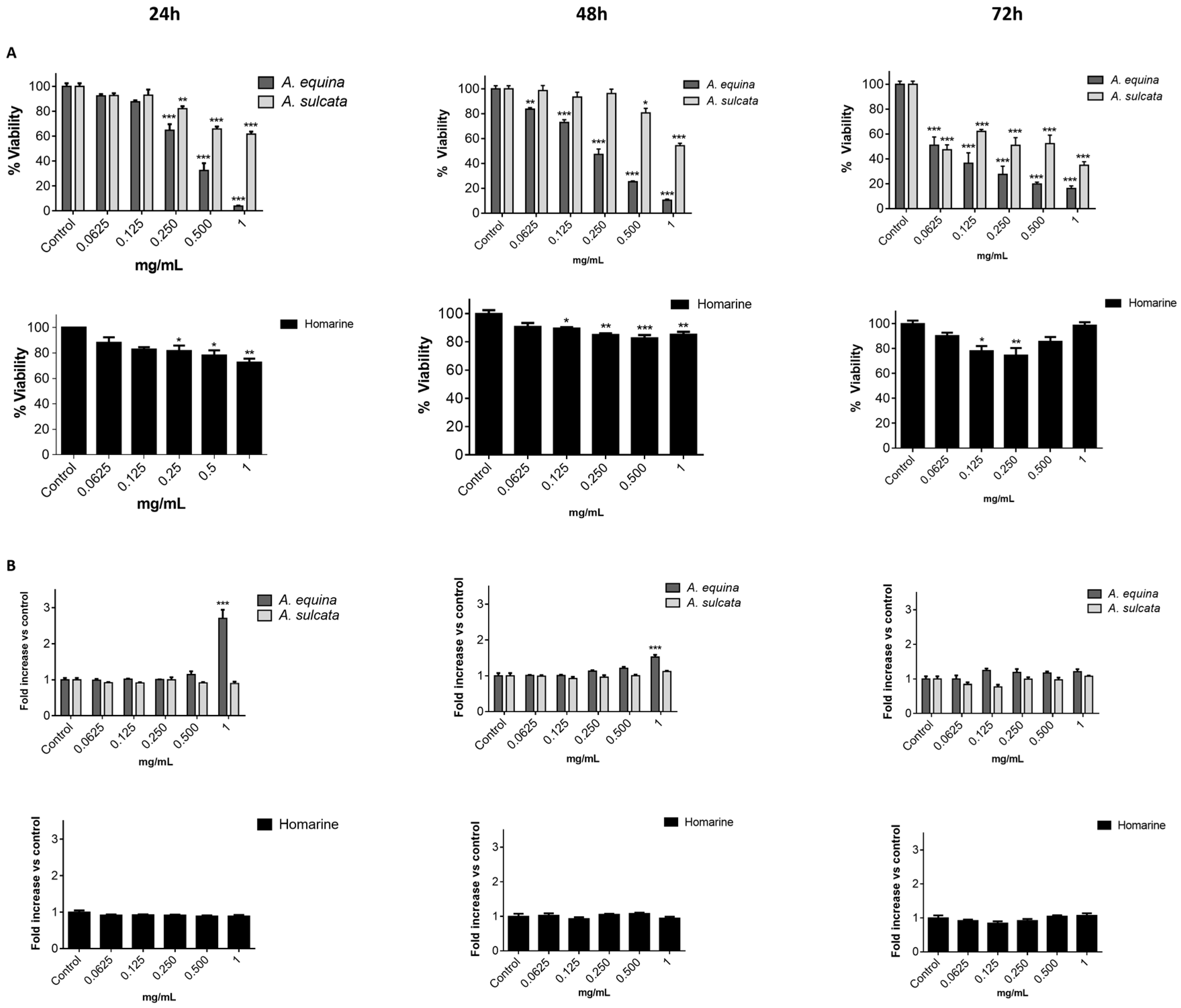
Cell viability was evaluated by the MTT reduction assay [
21]. Cells were cultured in 96-well plates (15,000 cells/well for AGS and 25,000 cells/well for RAW) and allowed to attach for 24 h. After incubation with extracts or molecules (0.0625–1 mg/mL) for 24, 48 and 72 h, MTT (0.5 mg/mL final concentration) was added to each well and the plate was incubated for 90 min at 37 °C. Formazan crystals were dissolved by addition of a DMSO:isopropanol mixture (3:1) and then quantified spectrophotometrically at 560 nm in microplate reader (Multiskan ASCENT, Haverhill, MA, USA). The results of cell viability correspond to the mean ± standard error of at least three independent experiments performed in triplicate and are expressed as the percentage of the untreated control cells.
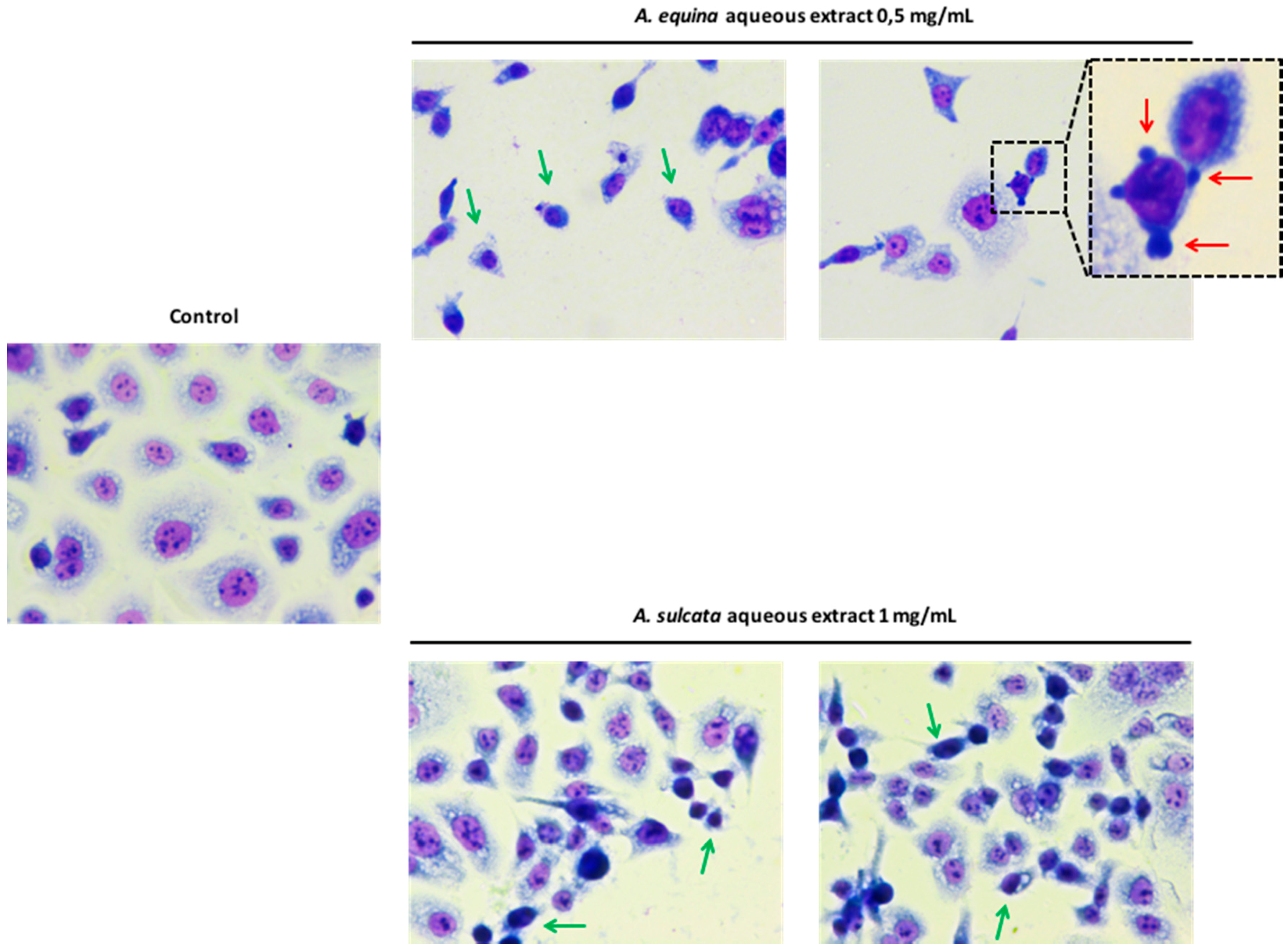
3.8. Morphological Studies
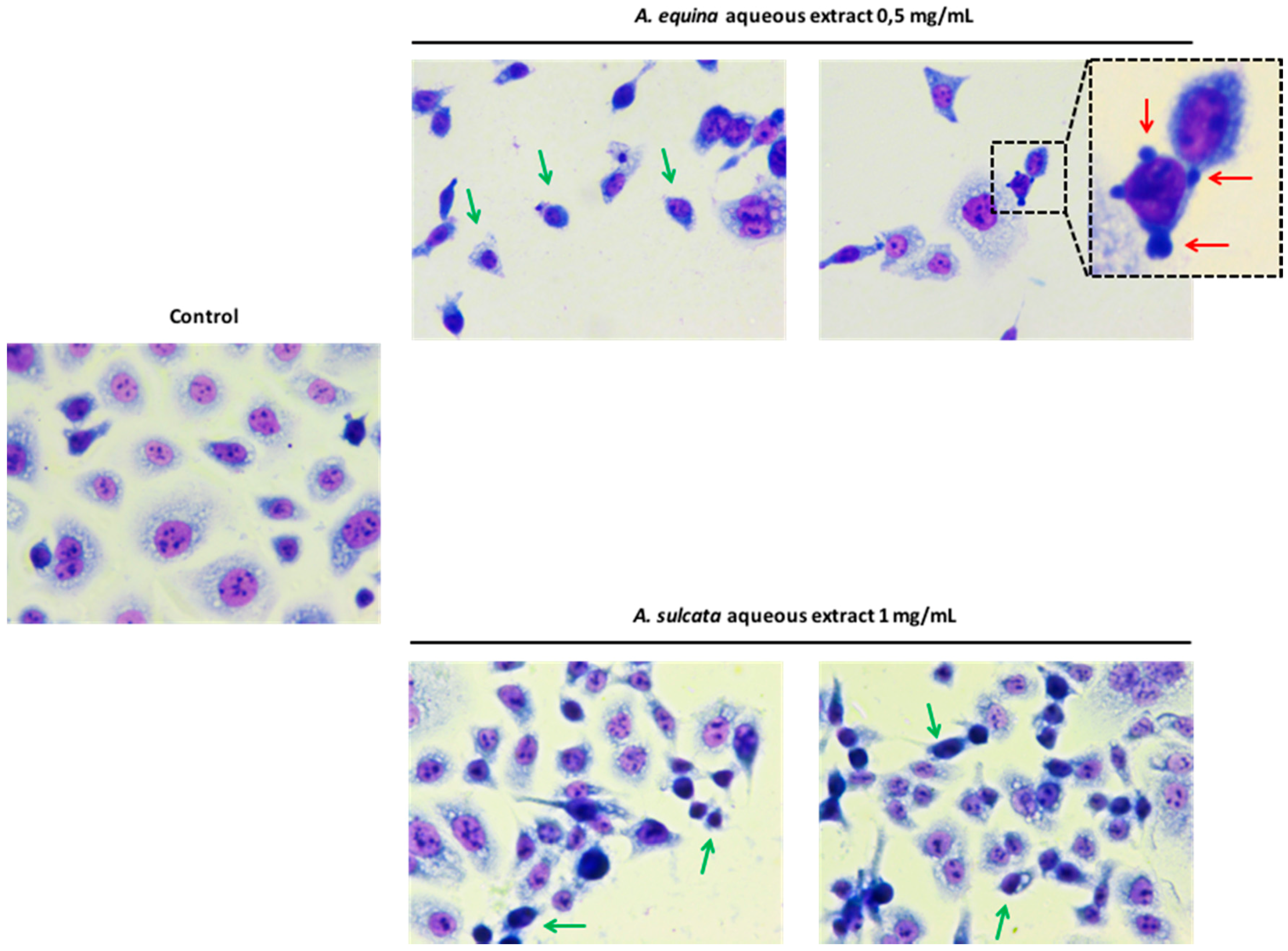
AGS cells were cultured at a density of 50,000 cells/well in coverslips placed in 24-well plates, as previously described [
22], with some modifications. Briefly, after incubation with different concentrations of the aqueous extracts (0.5 mg/mL of
A. equina and 1 mg/mL of
A. sulcata) for 24 h, cells were washed twice with HBSS and fixed in coverslips with cold methanol, at 4 °C for 30 min. Giemsa dye (1:10) was then added and kept for 30 min at room temperature, after which cells were repeatedly washed with water and then mounted in DPX.
3.9. Membrane Integrity
In order to evaluate the membrane integrity, the activity extracellular LDH was measured as previously described [
12]. Briefly, cells were cultured in 96-well plates as described for the MTT assay and allowed to attach for 24 h at 37 °C. After this, cells were incubated with extracts or the molecule (0.0625–1 mg/mL) for 24, 48, and 72 h. LDH released into the culture media after 24, 48, and 72 h in culture media supernatant was evaluated by monitoring the decrease of NADH (252.84 µM) during the conversion of pyruvate (14.993 mM) to lactate, at 340 nm in microplate reader (Multiskan ASCENT, Haverhill, MA, USA). Triton X-100 1% was used as positive control for cell lysis (30 min). All of the results correspond to the fold-increase of absorbance in treated versus untreated cells.
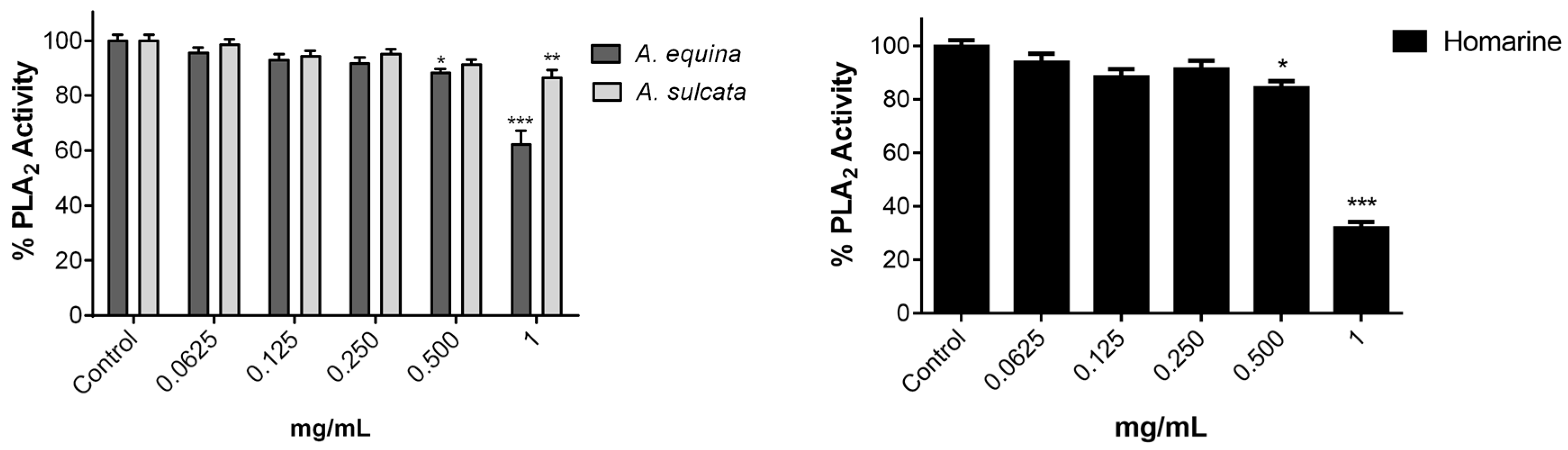
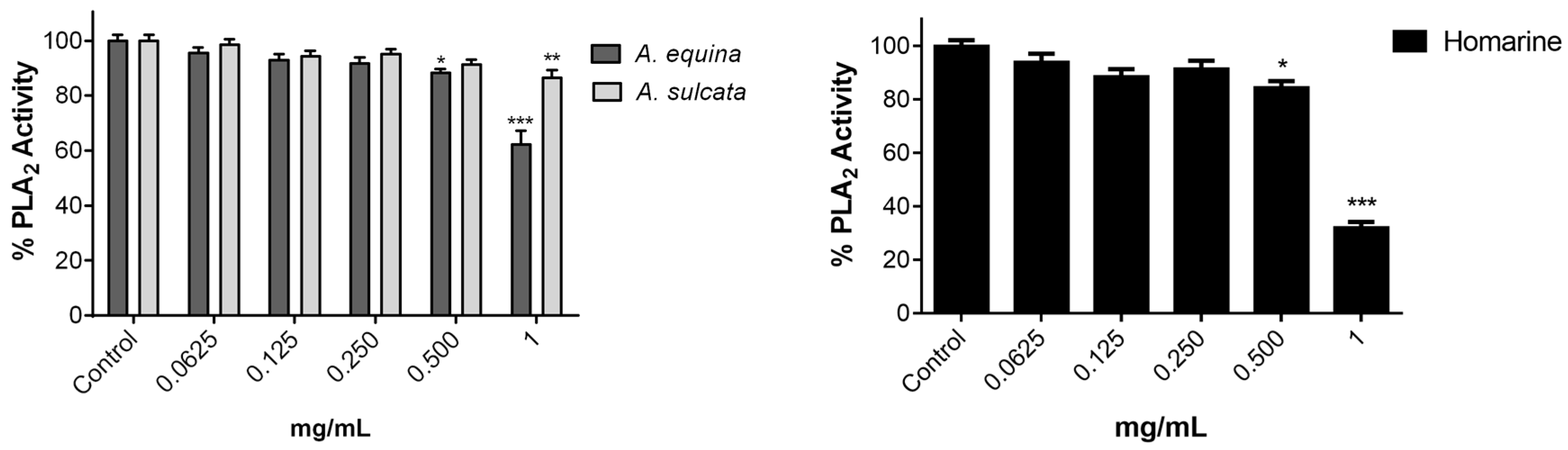
3.10. Inhibition of PLA2
Inhibition of PLA
2 assay was measured as previously described [
15] with some modifications. Phospholipase A
2 was solubilized in water and used at a final concentration of 0.25 µg/mL in 3 mM deoxycholate dissolved in 50 mM Tris-HCl buffer (pH 8.5). The solution of phosphatidylcholine substrate was prepared by drying aliquots of a dilinoleoyl phosphatidylcholine stock solution in chloroform under a stream of N
2; the film obtained was rapidly dispersed, up to a final concentration of 1.3 mM, in 10 mM deoxycholate dissolved in 50 mM Tris buffer, pH 8.5. In the assay, the substrate was used in a final concentration of 65 μM in 50 mM Tris-HCl buffer (pH 8.5). Type V lipoxygenase from
Glycine max (soybean) LOX was used as a co-enzyme in a final concentration of 0.23 µg/mL in 3 mM deoxycholate dissolved in 50 mM Tris-HCl buffer, pH 8.5. The extracts were dissolved in the same buffer. Both enzymes were mixed with 50 µL of the extracts or molecule (0.0625–1 mg/mL) and the reaction started by the adding of PLA
2 substrate. The linoleic acid released by PLA
2 is oxidized by 5-LOX and then phospholipase activity was assessed by monitoring the formation of conjugated dienes of the hydroperoxide product at 234 nm in a microplate reader (Multiskan ASCENT, Haverhill, MA, USA).
Controls without either PLA2, 5-LOX or substrate were always carried out. All assays were performed at 37 °C and each measurement was repeated three times. Values represent the means ± standard error of the mean and are expressed as percentage of the control with enzymes and substrate.
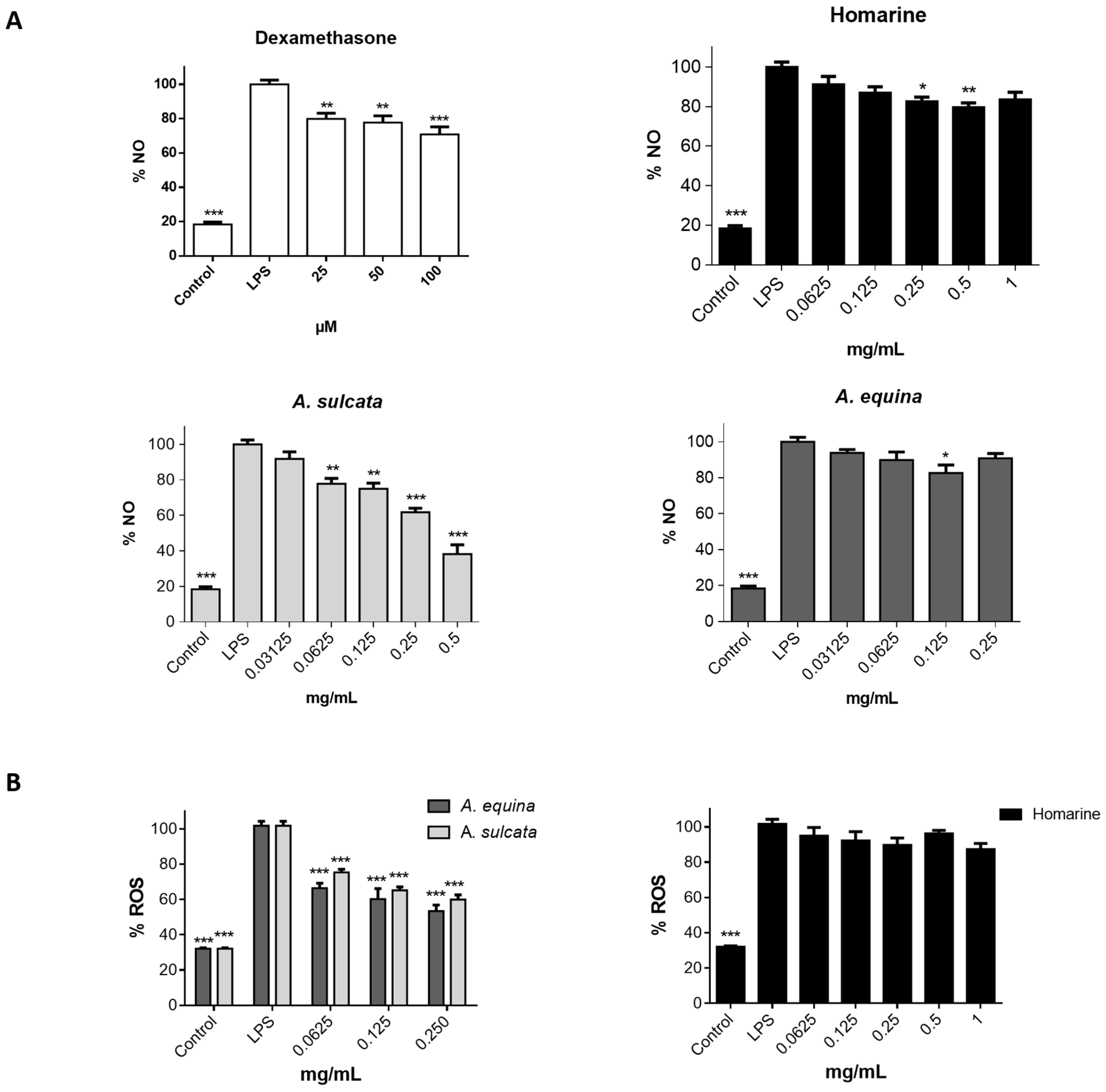
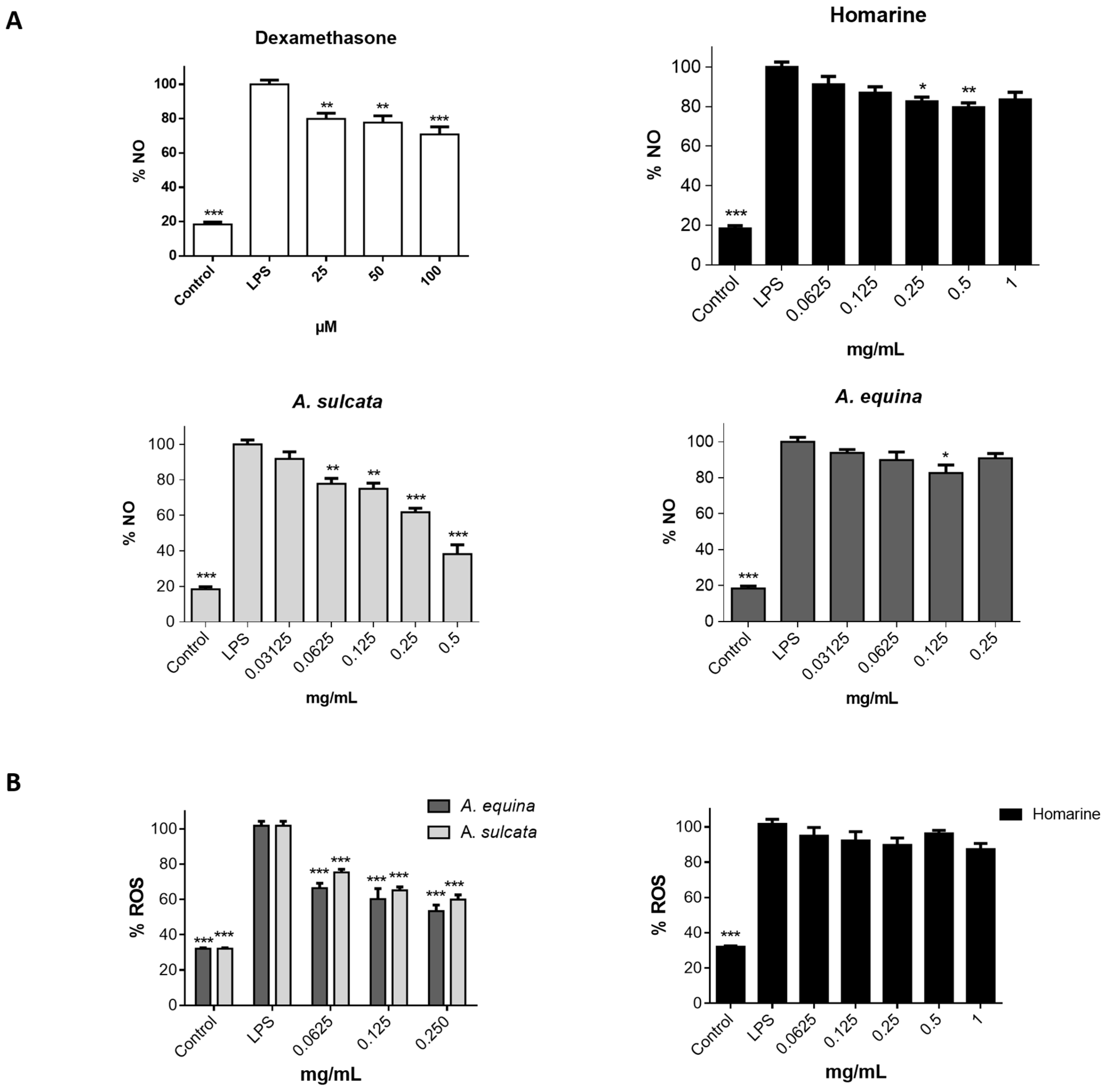
3.11. Evaluation of NO Levels
RAW 264.7 cells were cultured in 96-well plates (35,000 cells/well) for 24 h and then pre-treated with different concentrations of aqueous extract of each sea anemone, homarine, and dexamethasone, for 2 h. Afterwards, LPS was added (final concentration 1 µg/mL) and the plates were incubated for 22 h at 37 °C, in a humidified atmosphere of 5% CO2. The nitrite resulting from the conversion of NO in the culture medium was quantified by mixing 75 µL of culture media with an equal volume of Griess reagent (1% sulfanilamide and 0.1% naphthylethylenediamine dihydrochloride in 2% H3PO4) and incubated for 10 min in the dark, after which absorbance was read at 540 nm in a microplate reader (Multiskan ASCENT, Haverhill, MA, USA). The results correspond to the mean ± standard error of the mean of five independent experiments performed in triplicate and are expressed as percentage of NO in cells exposed to LPS (positive control for NO production).
3.12. Intracellular ROS Levels
Cells were seeded in 96-well black plates (15,000 cells/well for AGS and 25,000 cells/well for RAW 264.7) according to the above-mentioned conditions for the MTT assay. After this, cells were incubated with extracts (0.0625–0.25 mg/mL) or homarine (0.0625–1 mg/mL) for 24 h. Cells were washed with HBSS for 30 min before the end of the incubation period, followed by incubation with a solution of DCDHF-DA (25 µM in HBSS) for 45 min at 37 °C. The quantification of intracellular ROS was performed using fluorescence microplate reader (Cytation™ 3, BioTek, Winooski, VT, USA) (Excitation: 490 nm excitation; Emission: 520 nm).
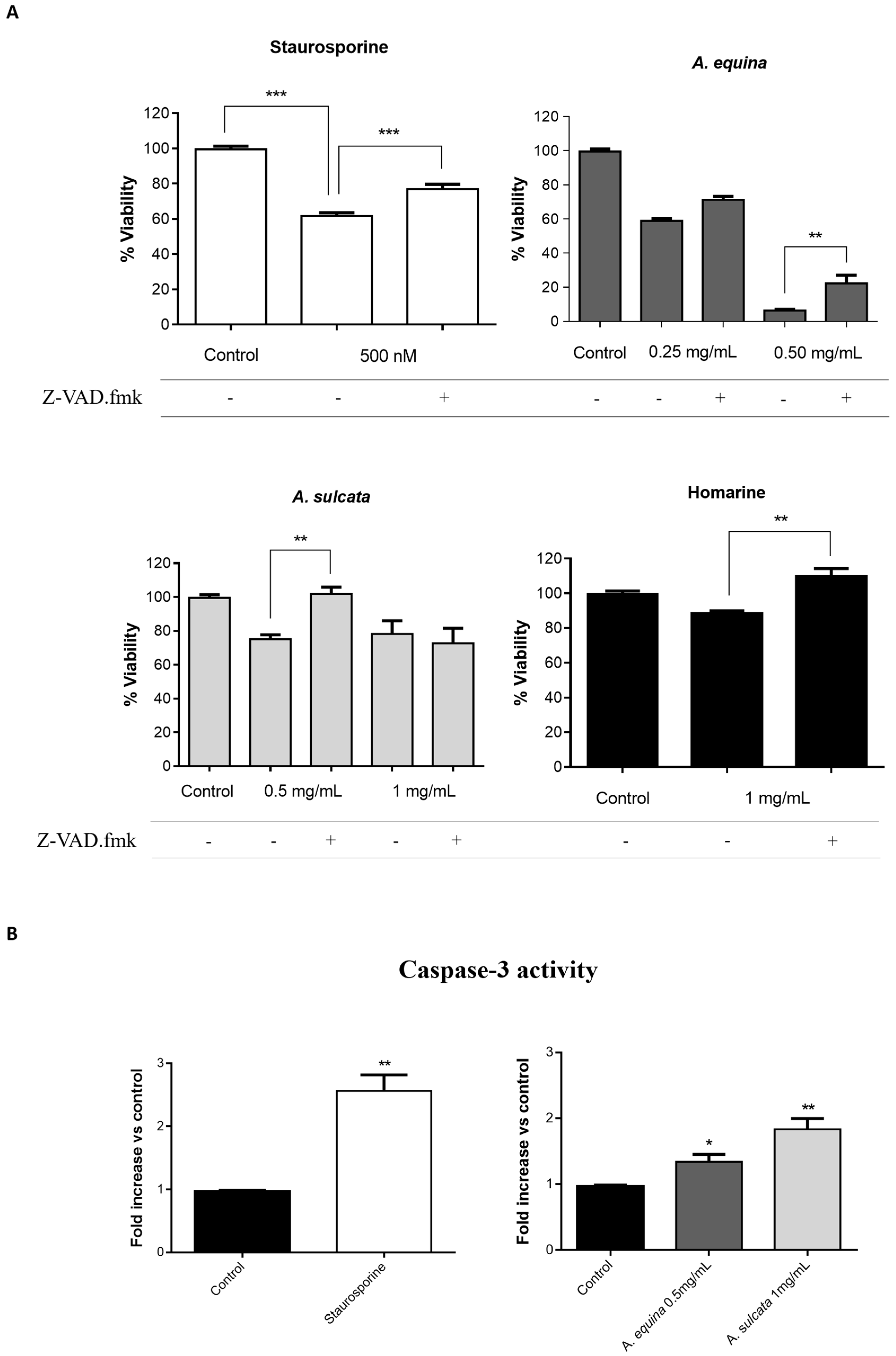
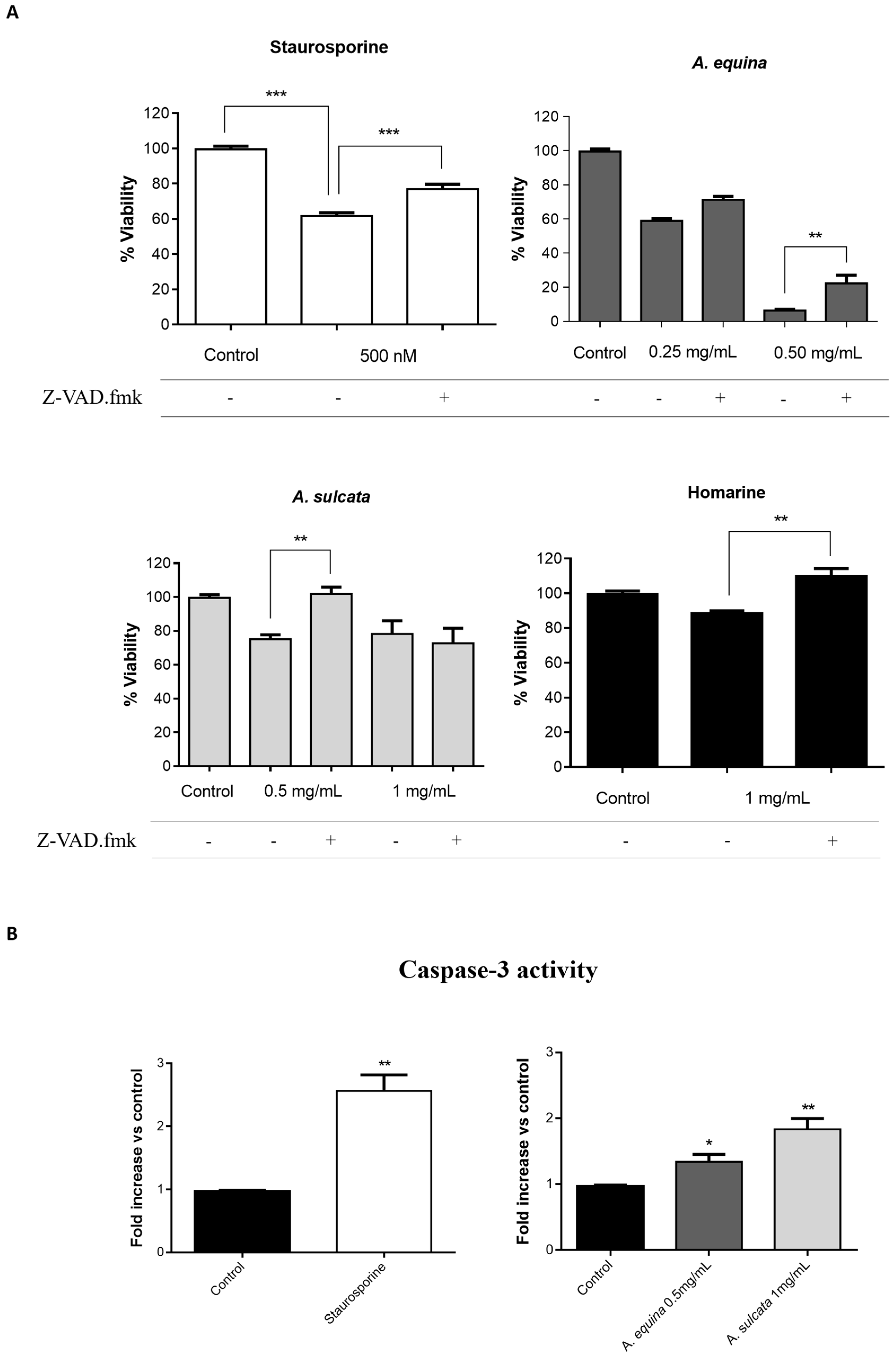
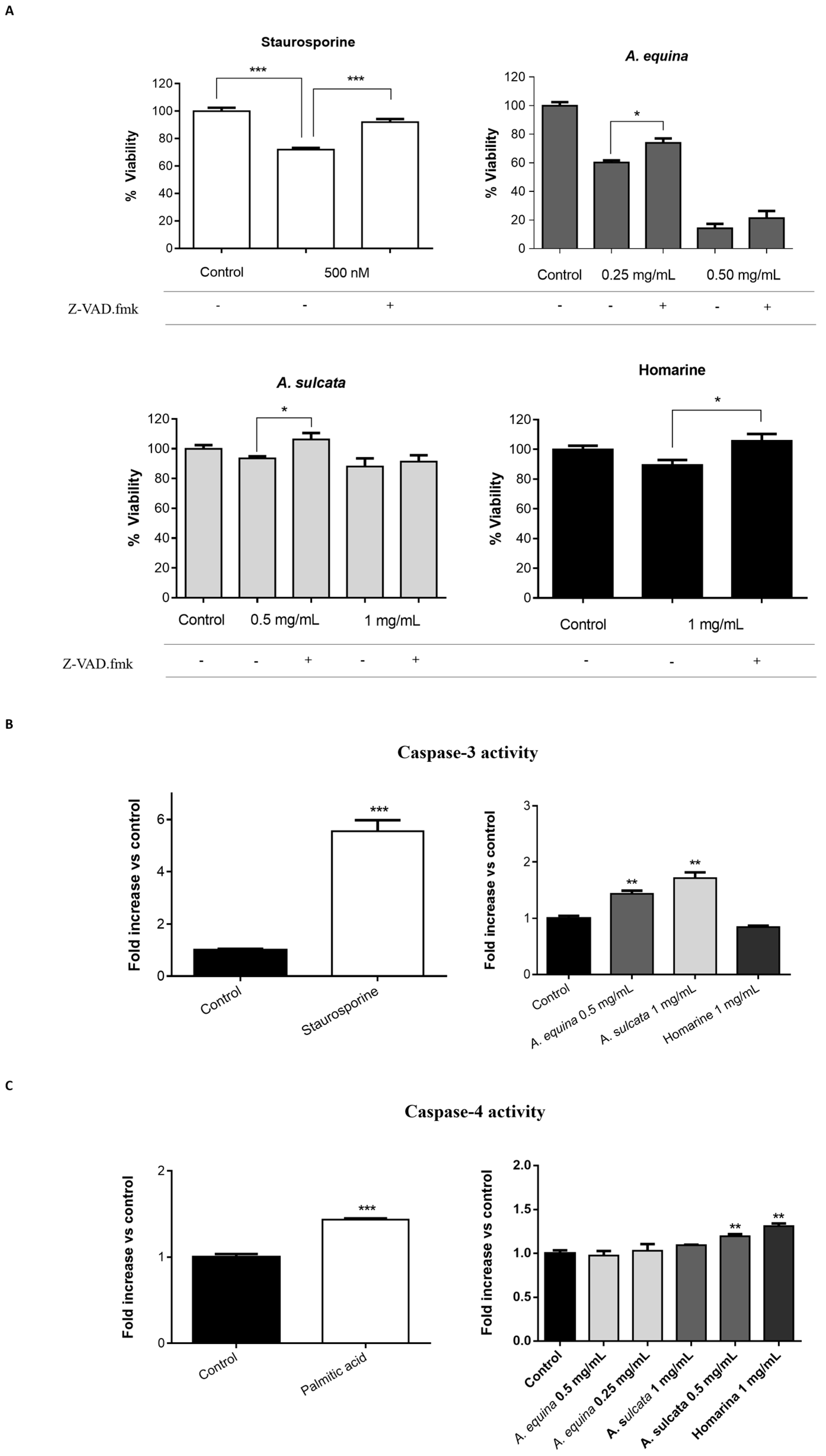
3.13. Caspase Inhibition Assays
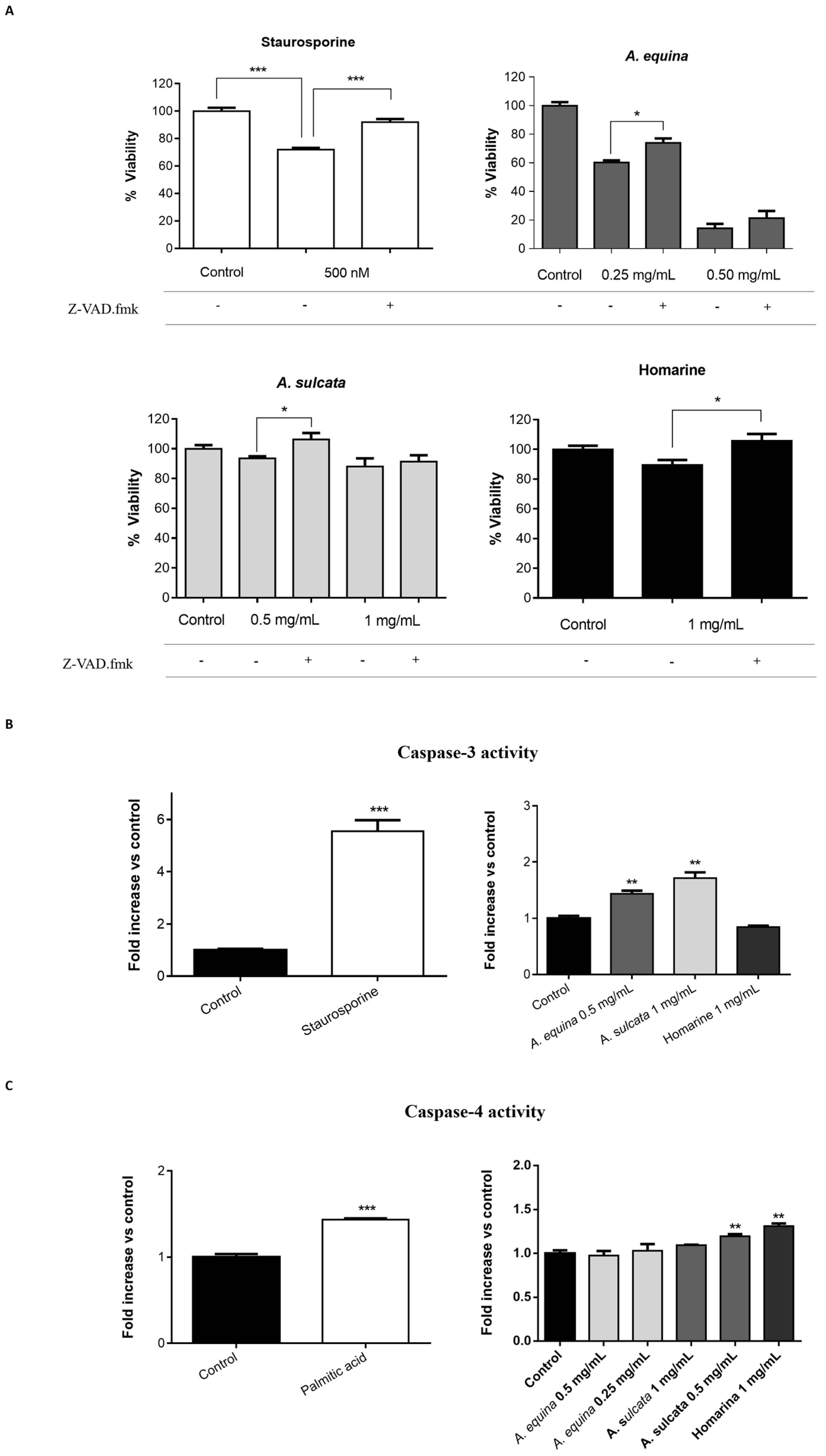
AGS and RAW 264.7 cells were cultured in 96-well plates as described for the MTT assay and allowed to attach for 24 h. After incubation, cells were pre-incubated with Z-VAD.fmk (AGS and RAW 264.7 cells) (50 µM) for 1 h and then extracts (0.25 and 0.5 mg/mL of A. equina and 0.5 and 1 mg/mL of A. sulcata) or homarine (1 mg/mL) were added and incubated for 8 h at 37 °C. After this, MTT assay was conducted.
3.14. Caspases Activity Assays
3.14.1. Caspase-3
Activity of caspase-3 was monitored using the Caspase-Glo® 3/7 kit Assay. After incubation in 96-well white plates of RAW 264.7 and AGS cells, as previously described for the MTT assay, cells were treated with the extracts (0.5 mg/mL of A. equina and 1 mg/mL of A. sulcata) or homarine (1 mg/mL) and incubated for 8 h at 37 °C. After this Caspase-Glo® 3/7 buffer and Caspase-Glo® 3/7 substrate were added to supernatant of the cells in the same quantity during 35 min at 22 °C and the luminescent signal was measured using a microplate reader (Cytation™ 3, BioTek, Winooski, VT, USA). In both cell lines, caspase-3 is the effector caspase and, thus, results from the Caspase-Glo® 3/7 kit Assay can be attributed to caspase-3. Staurosporine was used as a positive control.
3.14.2. Caspase-4
For the assessment of caspase-4 activity, AGS cells were cultured in 96-well black plates, as previously described for the MTT assay, and allowed to attach for 24 h. After this, cells were incubated with extracts (0.25 and 0.5 mg/mL for A. equina and 0.5 and 1 mg/mL for A. sulcata) or homarine (1 mg/mL) for 8 h at 37 °C and then the supernatant was removed and substrate of caspase-4 (50 µM) was incubated for 150 min at 37 °C. Fluorescence was determined in a microplate reader (Cytation™ 3, BioTek, Winooski, VT, USA) (Excitation: 400 nm; Emission: 505 nm). Palmitic acid (1 mM) was used as a positive control for caspase-4 activation.
3.14.3. Caspase-9
AGS and RAW 264.7 cells were cultured in 12-wells plates (75,000 cells/well and 200,000 cells/well, respectively), as described for the MTT assay, and allowed to attach for 24 h. After this, cells were pre-incubated with extracts (0.25 and 0.5 mg/mL for
A. equina and 0.5 and 1 mg/mL for
A. sulcata) or homarine (1 mg/mL) for 8 h at 37 °C, after which the culture medium was removed and cells were washed with HBSS. RAW 264.7 cells were harvested by scraping and AGS cells were trypsinized and subjected to centrifugation at 1300 rpm for 3 min. Afterwards, lysis buffer (25 mM HEPES, 5 mM EDTA, 1 mM EGTA, 5 mM MgCl
2, and 5 mM DTT, pH 7.4) was added for 5 min, with subsequent centrifugation at 14,500 rpm for 15 min, at 4 °C. The supernatants were removed and assayed for protein content by the Bradford method [
23]. In order to measure caspase activity, aliquots of cell extracts containing 50 µg of protein were added to a reaction buffer (25 mM HEPES, 0.1% CHAPS, 10% sucrose, and 5 mM DTT pH 7.4). The reaction was initiated after addition of the substrate for caspase-9 (50 µM) and fluorescence was determined after 90 min for RAW 264.7 cells and 120 min for AGS cells at 37 °C, in a microplate reader (Cytation™ 3, BioTek, Winooski, VT, USA) (Excitation: 405 nm; Emission: 535 nm).
3.15. Statistical Analysis
Statistical analysis was performed using KaleidaGraph 4.0 Synergy Software Inc (Reading, PA, USA). One-way ANOVA, followed by a Scheffe multiple range test, was used to determine the statistical significance in comparison to cells treated and untreated.
F values of all statistical analysis are shown in the
Tables S1–S3 of Supplemental Information. Data are expressed as the mean ± standard error of the mean. In all cases, values of
p ≤ 0.05 were considered statistically significant.
4. Conclusions
This work constitutes the first study addressing the chemical composition and biological effects of aqueous extracts from the sea anemones A. equina and A. sulcata. Metabolite profiling of these species showed that, in both cases, the alkaloid homarine was the major metabolite.
A. equina was more toxic that A. sulcata for both gastric and macrophage cells. Both extracts revealed to exert their toxic effects by activation of caspase-3. Furthermore, both extracts were effective in preventing LPS-induced increase of NO, in addition to being able to significantly lower the ROS levels produced by macrophages when displaying an inflammatory phenotype. In addition, A. equina showed mild ability to inhibit PLA2. In several of the parameters studied, such as NO production and PLA2 inhibition, the major metabolite homarine displayed significant activity that could be responsible for the activity of the extracts, however we cannot rule of the possible contribution of other molecules present.
This is the first report on the toxicity of aqueous extracts of this species. In light of the recent interest of sea anemones as gourmet ingredients, our results on the toxicity of these organisms justify further studies to further assess the safety of sea anemones for human consumption. In addition, we show that at sub-toxic concentrations the aqueous extracts display mild anti-inflammatory activity, thus, raising the interest in the exploitation of this material as a source of bioactive molecules.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}