
The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders

Abstract
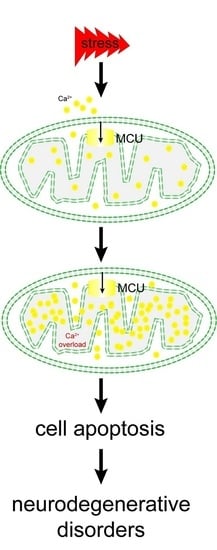
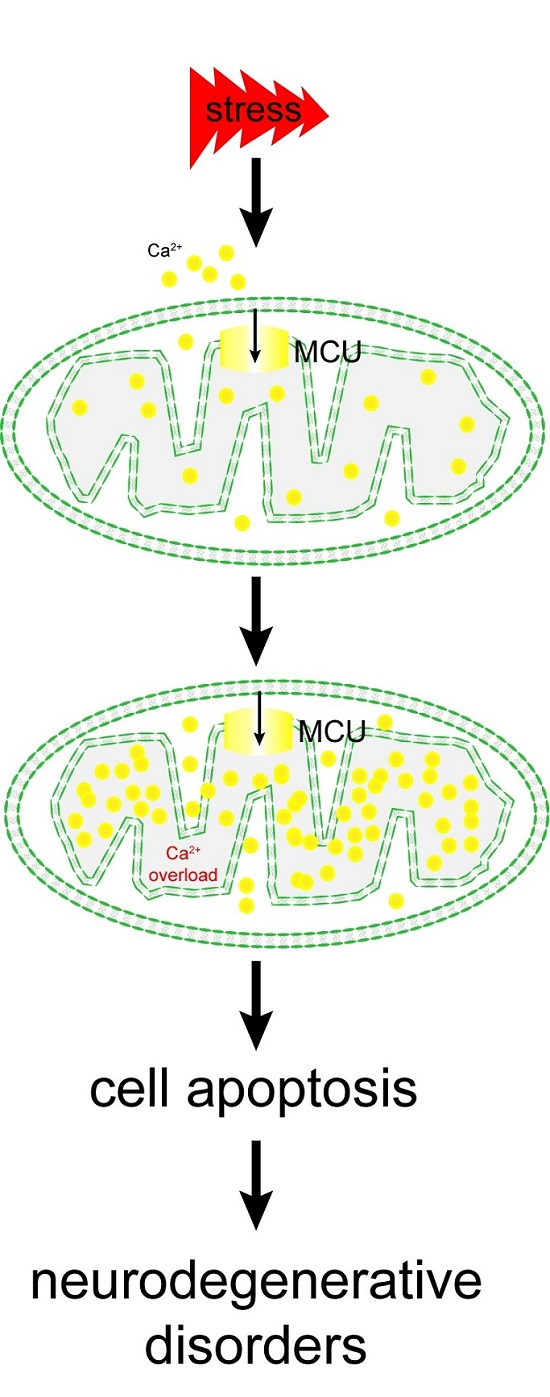
:
1. Introduction
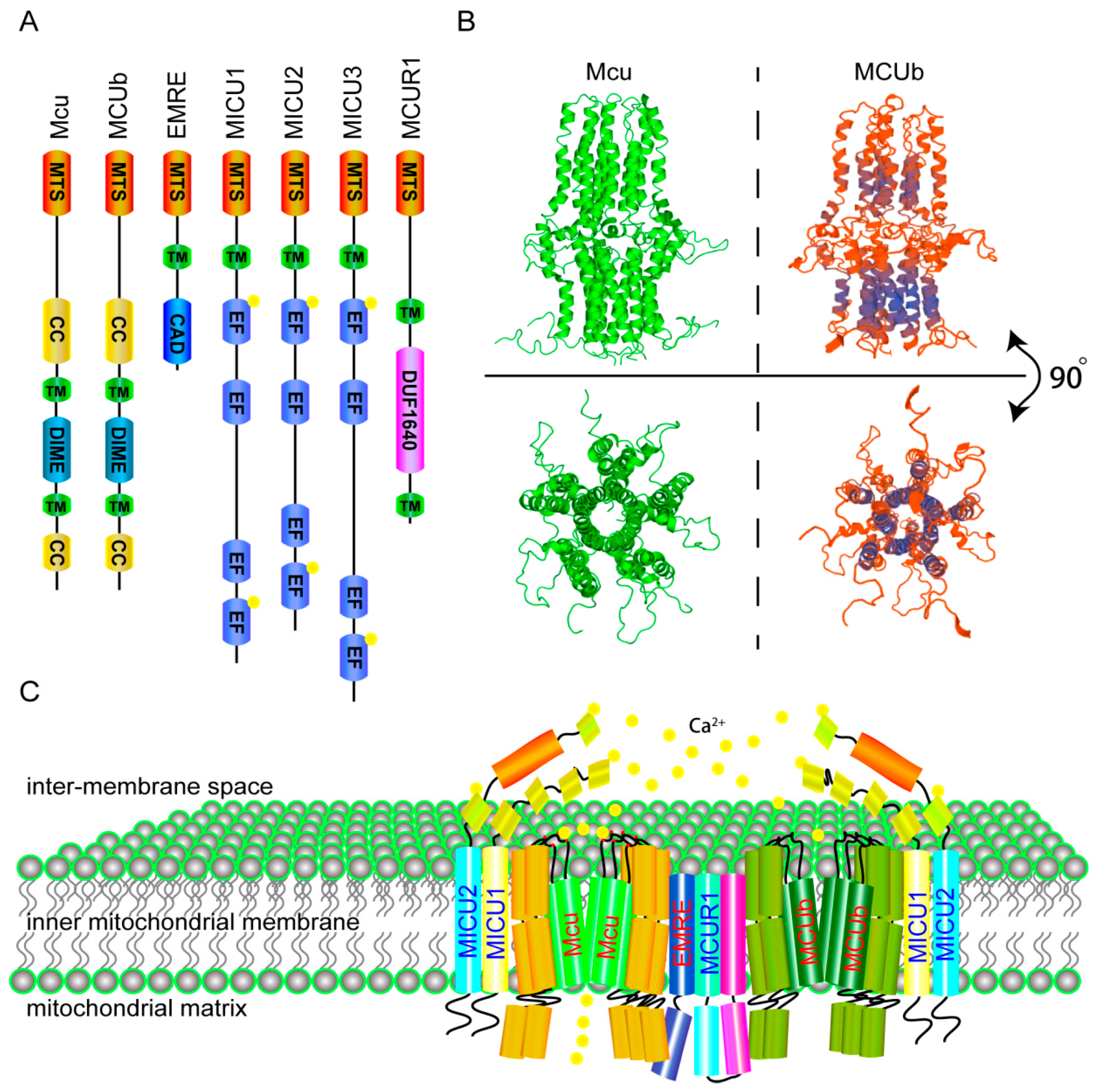
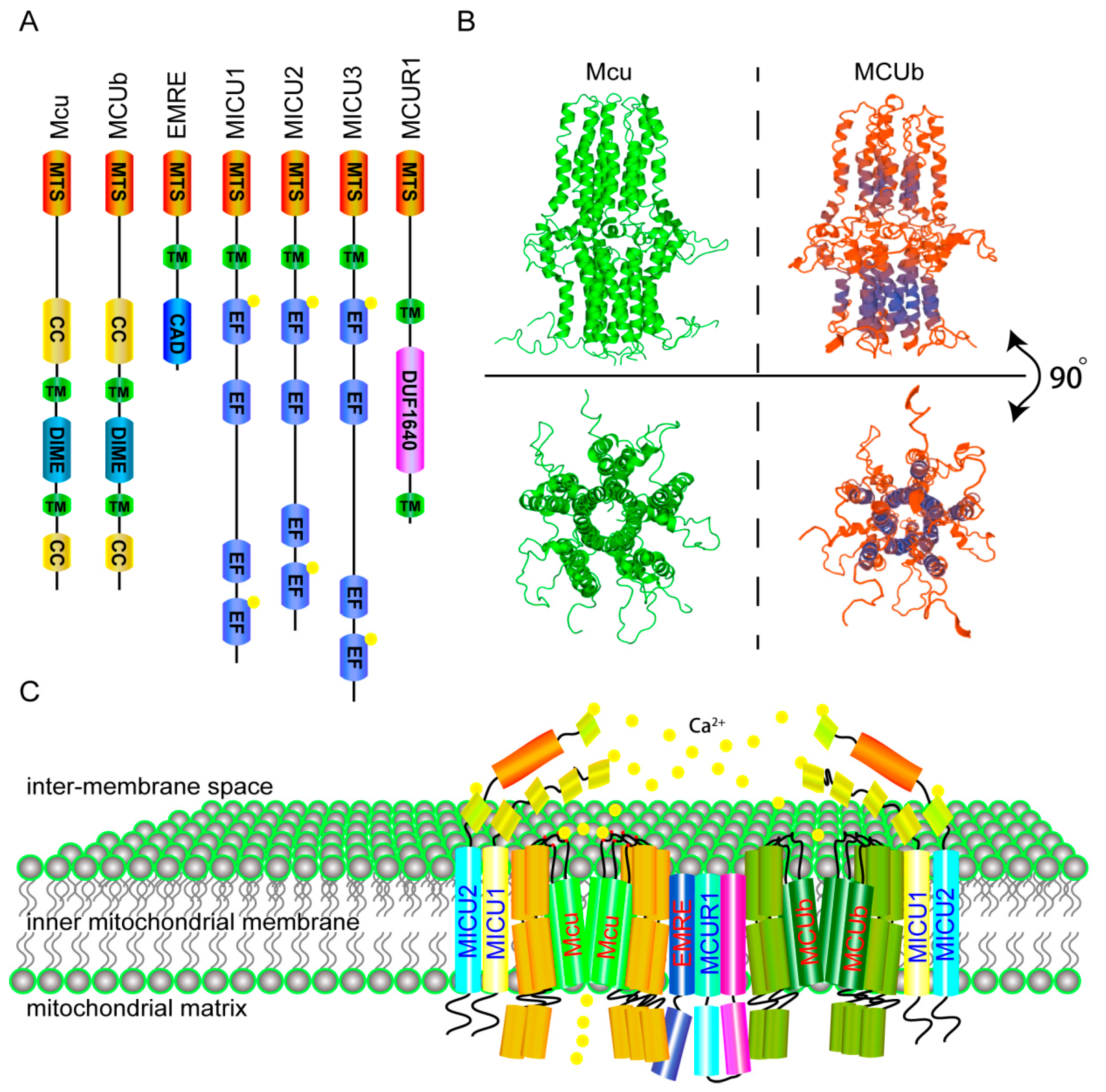
2. The Molecular Components and Calcium Transporting Mechanism of the MCU
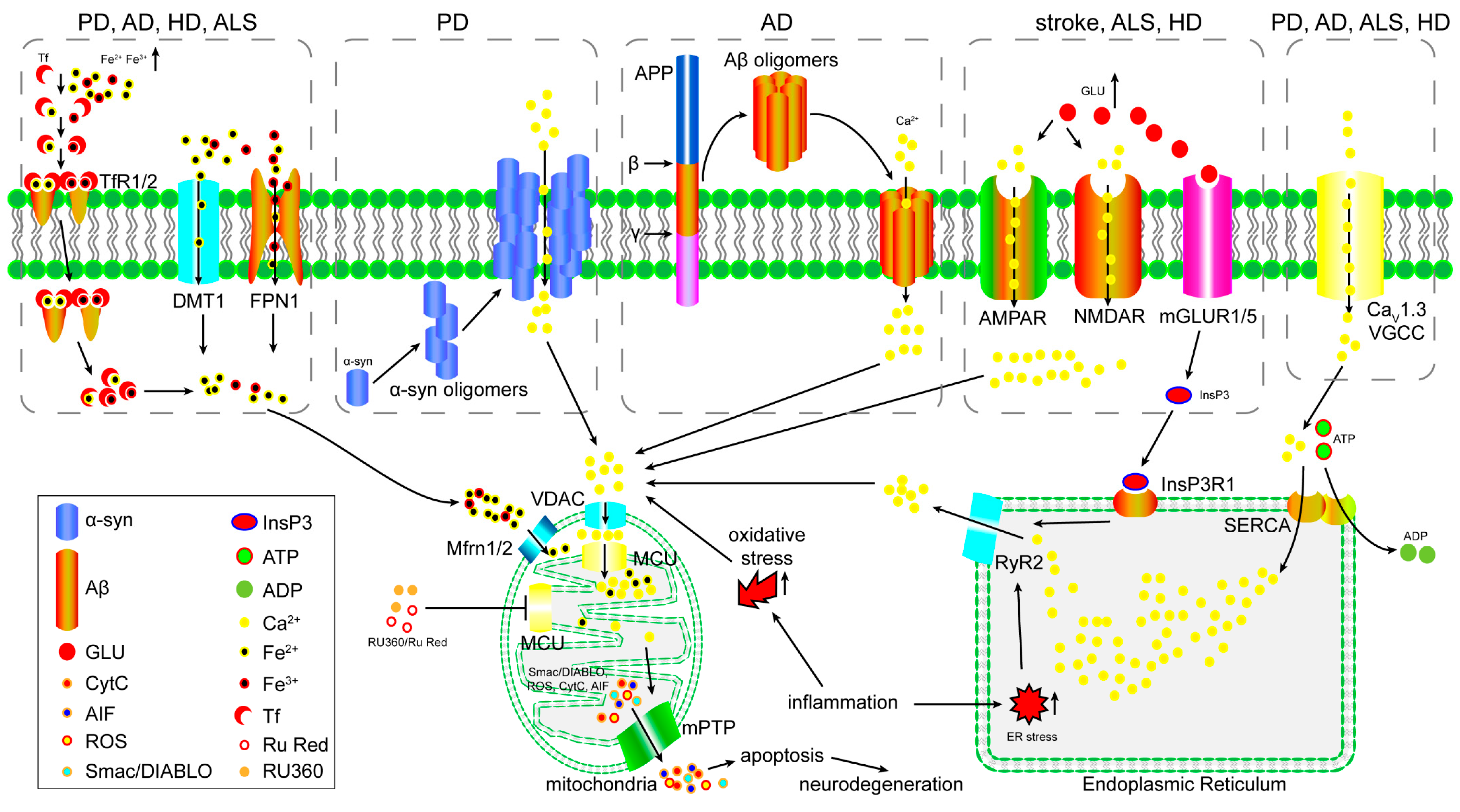
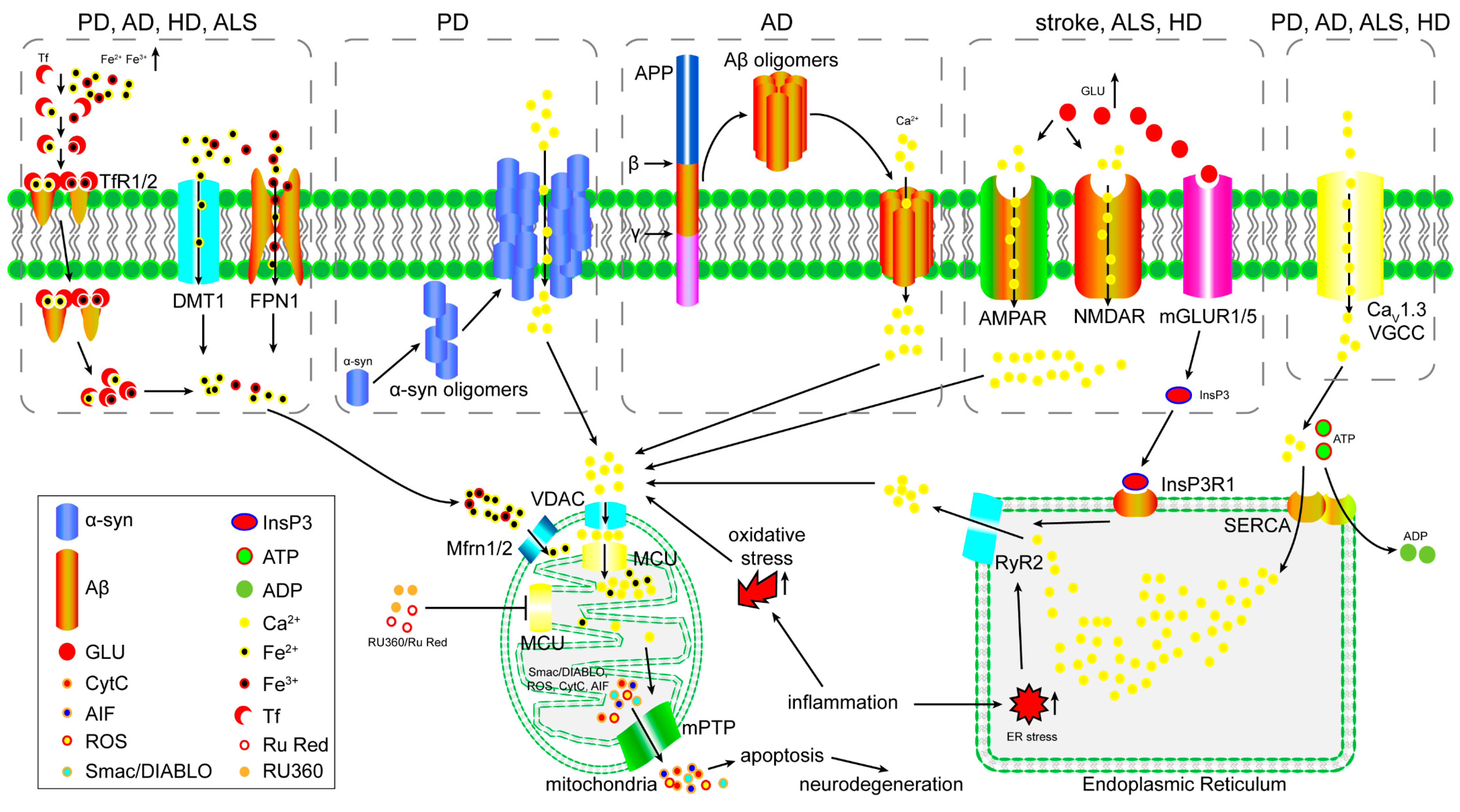
3. The Role of the MCU in Excitotoxicity
4. The Role of the MCU in Iron Overload-Induced Mitochondrial Dysfunction
5. The Role of the MCU in Oxidative Stress-Induced Mitochondrial Dysfunction
6. The Function of the MCU in Inflammation
7. The Potential of the MCU as a Target for Neurodegenerative Disorder Therapy
8. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.; Cowburn, R.F.; Bonkale, W.L.; Ohm, T.G.; Fastbom, J.; Carmody, M.; Kelliher, M. Dysfunctional intracellular calcium homoeostasis: A central cause of neurodegeneration in Alzheimer’s disease. Biochem. Soc. Symp. 2001, 67, 177–194. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Parkinson disease: Calcium channel blockers and Parkinson disease. Nat. Rev. Neurol. 2010, 6, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Cooper, L.A.; Minotti, S.; Durham, H.D. Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: Mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 2011, 42, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Deluca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Mraz, F.R. Strontium and Calcium Uptake by Rat Liver and Kidney Mitochondria. Hw-76000. US At. Energy Comm. 1963, 86, 95–97. [Google Scholar]
- Vasington, F.D. Calcium ion uptake by fragments of rat liver mitochondria and its dependence on electron transport. J. Biol. Chem. 1963, 238, 1841–1847. [Google Scholar] [PubMed]
- Browning, M.; Baudry, M.; Lynch, G. Evidence that high frequency stimulation influences the phosphorylation of pyruvate dehydrogenase and that the activity of this enzyme is linked to mitochondrial calcium sequestration. Prog. Brain Res. 1982, 56, 317–337. [Google Scholar] [PubMed]
- Budde, R.J.; Fang, T.K.; Randall, D.D. Regulation of the phosphorylation of mitochondrial pyruvate dehydrogenase complex in situ: Effects of respiratory substrates and calcium. Plant Physiol. 1988, 88, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Komuniecki, R. Characterization of the α-ketoglutarate dehydrogenase complex from Fasciola hepatica: Potential implications for the role of calcium in the regulation of helminth mitochondrial metabolism. Mol. Biochem. Parasitol. 1996, 81, 243–246. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.J.; Denton, R.M. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell. Biochem. 1995, 149–150, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Irigoin, F.; Inada, N.M.; Fernandes, M.P.; Piacenza, L.; Gadelha, F.R.; Vercesi, A.E.; Radi, R. Mitochondrial calcium overload triggers complement-dependent superoxide-mediated programmed cell death in Trypanosoma cruzi. Biochem. J. 2009, 418, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.C.; Bygrave, F.L. The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. Biochem. J. 1974, 140, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Luthra, R.; Olson, M.S. The inhibition of calcium uptake and release by rat liver mitochondria by ruthenium red. FEBS Lett. 1977, 81, 142–146. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; de Groot, J.; Speciner, L.; et al. MICU2, a Paralog of MICU1, Resides within the Mitochondrial Uniporter Complex to Regulate Calcium Handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; de Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.X.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.L.; Liu, Z.J.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.Y.; Sun, J.H.; Teng, Y.J.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Pan, X.; Nguyen, T.; Liu, J.; Holmstrom, K.M.; Finkel, T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 2014, 449, 384–385. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shamugapriya, S.; Zhang, X.Q.; Rajan, S.; Mallilankaraman, K.; Gandhirajan, R.K.; Vagnozzi, R.J.; Ferrer, L.M.; Sreekrishnanilayam, K.; et al. MICU1 Motifs Define Mitochondrial Calcium Uniporter Binding and Activity. Cell Rep. 2013, 5, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, X.; Li, S.; Wang, Z.; Liu, Y.; Feng, J.; Zhu, Y.; Shen, Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014, 33, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Liu, J.; Holmstrom, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yamagoshi, R.; Harada, K.; Kawano, M.; Minami, N.; Ido, Y.; Kuwahara, K.; Fujita, A.; Ozono, M.; Watanabe, A.; et al. Analysis of the structure and function of EMRE in a yeast expression system. Biochim. Biophys. Acta 2016, 1857, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) Is a Cytochrome c Oxidase Assembly Factor and Not a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015, 21, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Budd, S.L.; Nicholls, D.G. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 1996, 67, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mallory, M.; Alford, M.; Tanaka, S.; Masliah, E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J. Neuropathol. Exp. Neurol. 1997, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid β peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M.; Zhang, J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp. Neurol. 2009, 220, 230–233. [Google Scholar] [CrossRef] [PubMed]
- DeLorenzo, R.J.; Sun, D.A.; Blair, R.E.; Sombati, S. An in vitro model of stroke-induced epilepsy: Elucidation of the roles of glutamate and calcium in the induction and maintenance of stroke-induced epileptogenesis. Int. Rev. Neurobiol. 2007, 81, 59–84. [Google Scholar] [PubMed]
- Riederer, P.; Lange, K.W.; Kornhuber, J.; Jellinger, K. Glutamate receptor antagonism: Neurotoxicity, anti-akinetic effects, and psychosis. J. Neural Transm. Suppl. 1991, 34, 203–210. [Google Scholar] [PubMed]
- Akaike, A.; Katsuki, H.; Kume, T.; Maeda, T. Reactive oxygen species in NMDA receptor-mediated glutamate neurotoxicity. Park. Relat. Disord. 1999, 5, 203–207. [Google Scholar] [CrossRef]
- Iihara, K.; Joo, D.T.; Henderson, J.; Sattler, R.; Taverna, F.A.; Lourensen, S.; Orser, B.A.; Roder, J.C.; Tymianski, M. The influence of glutamate receptor 2 expression on excitotoxicity in Glur2 null mutant mice. J. Neurosci. 2001, 21, 2224–2239. [Google Scholar] [PubMed]
- Castilho, R.F.; Hansson, O.; Ward, M.W.; Budd, S.L.; Nicholls, D.G. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurosci. 1998, 18, 10277–10286. [Google Scholar] [PubMed]
- Attucci, S.; Clodfelter, G.V.; Thibault, O.; Staton, J.; Moroni, F.; Landfield, P.W.; Porter, N.M. Group I metabotropic glutamate receptor inhibition selectively blocks a prolonged Ca2+ elevation associated with age-dependent excitotoxicity. Neuroscience 2002, 112, 183–194. [Google Scholar] [CrossRef]
- Vergun, O.; Keelan, J.; Khodorov, B.I.; Duchen, M.R. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J. Physiol. 1999, 519 Pt 2, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Spaethling, J.M.; Klein, D.M.; Singh, P.; Meaney, D.F. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 2008, 25, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, F.; Iqbal, Z. Regulation of N-methyl-d-aspartate receptor-mediated calcium transport and norepinephrine release in rat hippocampus synaptosomes by polyamines. Neurochem. Res. 1994, 19, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.K.; Kassai, H.; Watabe, A.M.; Aiba, A.; Manabe, T. Functional coupling of the metabotropic glutamate receptor, InsP3 receptor and L-type Ca2+ channel in mouse CA1 pyramidal cells. J. Physiol. 2012, 590, 3019–3034. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Grillner, S.; Wallen, P. Calcium dynamics during NMDA-induced membrane potential oscillations in lamprey spinal neurons—Contribution of L-type calcium channels (Cav1.3). J. Physiol. 2013, 591, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Bornman, M.S. Calcium-mediated aponecrosis plays a central role in the pathogenesis of estrogenic chemical-induced neurotoxicity. Med. Hypotheses 2005, 65, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.W.; Rego, A.C.; Frenguelli, B.G.; Nicholls, D.G. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurosci. 2000, 20, 7208–7219. [Google Scholar] [PubMed]
- Parihar, M.S.; Brewer, G.J. Simultaneous age-related depolarization of mitochondrial membrane potential and increased mitochondrial reactive oxygen species production correlate with age-related glutamate excitotoxicity in rat hippocampal neurons. J. Neurosci. Res. 2007, 85, 1018–1032. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Duchen, M.R. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta 2008, 1777, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Tan, Y.W.; Hagenston, A.M.; Martel, M.A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.A.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Casella, L.; Albertini, A.; Bellei, C.; Zucca, F.A.; Engelen, M.; Zadlo, A.; Szewczyk, G.; Zareba, M.; Sarna, T. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson’s disease. J. Neurochem. 2008, 106, 1866–1875. [Google Scholar] [PubMed]
- Liu, Y.; Connor, J.R. Iron and ER stress in neurodegenerative disease. Biometals 2012, 25, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.J.; Shoham, S.; Connor, J.R. Iron and neurodegenerative disorders. Brain Res. Bull. 2001, 55, 155–164. [Google Scholar] [CrossRef]
- Shoham, S.; Youdim, M.B. Iron involvement in neural damage and microgliosis in models of neurodegenerative diseases. Cell. Mol. Biol. 2000, 46, 743–760. [Google Scholar] [PubMed]
- Hagemeier, J.; Geurts, J.J.; Zivadinov, R. Brain iron accumulation in aging and neurodegenerative disorders. Expert Rev. Neurother. 2012, 12, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K. [Iron accumulation and neurodegenerative diseases]. Nihon Rinsho. Jpn. J. Clin. Med. 2016, 74, 1161–1167. [Google Scholar]
- Urrutia, P.J.; Mena, N.P.; Nunez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Chew, K.C.; Ang, E.T.; Tai, Y.K.; Tsang, F.; Lo, S.Q.; Ong, E.; Ong, W.Y.; Shen, H.M.; Lim, K.L.; Dawson, V.L.; et al. Enhanced autophagy from chronic toxicity of iron and mutant A53T α-synuclein: Implications for neuronal cell death in Parkinson disease. J. Biol. Chem. 2011, 286, 33380–33389. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-H.; Gao, W.-J.; Shao, T.-M.; Xie, H.-L.; Bai, J.-T.; Zhao, J.-Y.; Chai, X.-Q. Age-related changes of brain iron load changes in the frontal cortex in APPswe/PS1ΔE9 transgenic mouse model of Alzheimer’s disease. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. 2015, 30, 118–123. [Google Scholar]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS ONE 2016, 11, e0161341. [Google Scholar] [CrossRef] [PubMed]
- Harley, A.; Cooper, J.M.; Schapira, A.H. Iron induced oxidative stress and mitochondrial dysfunction: Relevance to Parkinson’s disease. Brain Res. 1993, 627, 349–353. [Google Scholar] [CrossRef]
- Vatassery, G.T.; DeMaster, E.G.; Lai, J.C.; Smith, W.E.; Quach, H.T. Iron uncouples oxidative phosphorylation in brain mitochondria isolated from vitamin E-deficient rats. Biochim. Biophys. Acta 2004, 1688, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.Y.; Hao, S.Y.; Sun, X.Y.; Zhang, D.D.; Gao, X.; Yu, Z.; Li, K.Y.; Hang, C.H. Blockage of mitochondrial calcium uniporter prevents iron accumulation in a model of experimental subarachnoid hemorrhage. Biochem. Biophys. Res. Commun. 2015, 456, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Sripetchwandee, J.; KenKnight, S.B.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Blockade of mitochondrial calcium uniporter prevents cardiac mitochondrial dysfunction caused by iron overload. Acta Physiol. 2014, 210, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Sripetchwandee, J.; Sanit, J.; Chattipakorn, N.; Chattipakorn, S.C. Mitochondrial calcium uniporter blocker effectively prevents brain mitochondrial dysfunction caused by iron overload. Life Sci. 2013, 92, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Neurochem. Res. 2008, 33, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Secretases, oxidative stress, and perturbed calcium homeostasis in AD and stroke. J. Gen. Physiol. 2008, 132, 14a. [Google Scholar]
- Klonowski-Stumpe, H.; Schreiber, R.; Grolik, M.; Schulz, H.U.; Haussinger, D.; Niederau, C. Effect of oxidative stress on cellular functions and cytosolic free calcium of rat pancreatic acinar cells. Am. J. Physiol. 1997, 272, G1489–G1498. [Google Scholar] [PubMed]
- Gutierrez-Martin, Y.; Martin-Romero, F.J.; Henao, F.; Gutierrez-Merino, C. Alteration of cytosolic free calcium homeostasis by SIN-1: High sensitivity of L-type Ca2+ channels to extracellular oxidative/nitrosative stress in cerebellar granule cells. J. Neurochem. 2005, 92, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.A.; Molshatzki, N.; Schwammenthal, Y.; Merzeliak, O.; Toashi, M.; Sela, B.; Tanne, D. Serum Calcium Levels and Long-Term Mortality in Patients with Acute Stroke. Cerebrovasc. Dis. 2011, 31, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Hao, Y.M.; Chen, H.; He, Q.; Yuan, Z.Q.; Cheng, J.B. Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 2015, 6, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.M.; Liang, D.D.; et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Zheng, S.F.; Leng, J.; Wang, S.L.; Zhao, T.; Liu, J. Inhibition of mitochondrial calcium uniporter protects neurocytes from ischemia/reperfusion injury via the inhibition of excessive mitophagy. Neurosci. Lett. 2016, 628, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Wang, S.L.; Luan, H.H. Effects of the mitochondrial calcium uniporter on cerebral edema in a rat model of cerebral ischemia reperfusion injury. Neural Regen. Res. 2011, 6, 1720–1724. [Google Scholar]
- Barsukova, A.G.; Bourdette, D.; Forte, M. Mitochondrial calcium and its regulation in neurodegeneration induced by oxidative stress. Eur. J. Neurosci. 2011, 34, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Baune, B.T. Inflammation and neurodegenerative disorders: Is there still hope for therapeutic intervention? Curr. Opin. Psychiatry 2015, 28, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Fenoglio, C.; Scarpini, E. Inflammation in neurodegenerative disorders: Friend or foe? Curr. Aging Sci. 2008, 1, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Whitney, N.P.; Eidem, T.M.; Peng, H.; Huang, Y.; Zheng, J.C. Inflammation mediates varying effects in neurogenesis: Relevance to the pathogenesis of brain injury and neurodegenerative disorders. J. Neurochem. 2009, 108, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.E.; Murphy, S.P. Inflammatory events at the blood brain barrier: Regulation of adhesion molecules, cytokines, and chemokines by reactive nitrogen and oxygen species. Brain Behav. Immun. 1997, 11, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Kielland, A.; Blom, T.; Nandakumar, K.S.; Holmdahl, R.; Blomhoff, R.; Carlsen, H. In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radic. Biol. Med. 2009, 47, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Lien, J.C.; Chen, C.J.; Liu, Y.C.; Wang, C.Y.; Ping, C.F.; Lin, Y.F.; Huang, A.C.; Lin, C.W. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hui, B.; Li, J.; Geng, M.Y. Sulfated polymannuroguluronate, a novel anti-acquired immune deficiency syndrome drug candidate, decreased vulnerability of PC12 cells to human immunodeficiency virus tat protein through attenuating calcium overload. J. Neurosci. Res. 2008, 86, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Heath, M.C. Role of calcium in signal transduction during the hypersensitive response caused by basidiospore-derived infection of the cowpea rust fungus. Plant Cell 1998, 10, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.A.; Olsen, R.G. In vitro infection of cell lines with HTLV-I and SIVmac results in altered intracellular free calcium concentration and increased membrane polarization. Int. J. Cancer 1989, 44, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.B.; Liao, Y.J.; Zhou, L.J.; Peng, S.Y.; Chen, H.; Yuan, Z.Q. Amplified RLR signaling activation through an interferon-stimulated gene-endoplasmic reticulum stress-mitochondrial calcium uniporter protein loop. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liao, W.; Gao, W.; Huang, J.; Gao, Y. Intermittent hypoxia protects cerebral mitochondrial function from calcium overload. Acta Neurol. Belg. 2013, 113, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Hirota, Y.; Genka, C.; Nakazawa, H.; Nakaya, H.; Sato, T. Opening of mitochondrial K(ATP) channels attenuates the ouabain-induced calcium overload in mitochondria. Circ. Res. 2001, 89, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kuroda, Y. Molecular mechanism of neurodegeneration induced by Alzheimer’s β-amyloid protein: Channel formation and disruption of calcium homeostasis. Brain Res. Bull. 2000, 53, 389–397. [Google Scholar] [CrossRef]
- Marongiu, R.; Spencer, B.; Crews, L.; Adame, A.; Patrick, C.; Trejo, M.; Dallapiccola, B.; Valente, E.M.; Masliah, E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 2009, 108, 1561–1574. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Chen, Q.; Wang, X.; Wang, Q.C.; Wang, Y.; Cheng, H.P.; Guo, C.; Sun, Q.; Chen, Q.; Tang, T.S. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 2013, 288, 3070–3084. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Magrane, J.; Starkov, A.A.; Manfredi, G. The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain J. Neurol. 2012, 135, 2865–2874. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.A.; Blazey, T.; Su, Y.; Fagan, A.M.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L. Longitudinal β-Amyloid Deposition and Hippocampal Volume in Preclinical Alzheimer Disease and Suspected Non-Alzheimer Disease Pathophysiology. JAMA Neurol. 2016, 73, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Acuna-Castroviejo, D.; Tan, D.X.; Lopez-Armas, G.; Cruz-Ramos, J.; Munoz, R.; Melnikov, V.G.; Manchester, L.C.; Reiter, R.J. Accumulation of exogenous amyloid-β peptide in hippocampal mitochondria causes their dysfunction: A protective role for melatonin. Oxidative Med. Cell. Longev. 2012, 2012, 843649. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.; Li, X.; Homayouni, R. Interaction of a novel mitochondrial protein, 4-nitrophenylphosphatase domain and non-neuronal SNAP25-like protein homolog 1 (NIPSNAP1), with the amyloid precursor protein family. Eur. J. Neurosci. 2010, 31, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Tillement, L.; Lecanu, L.; Papadopoulos, V. Further evidence on mitochondrial targeting of β-amyloid and specificity of β-amyloid-induced mitotoxicity in neurons. Neuro-Degener. Dis. 2011, 8, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Behera, P.; Bhowmick, P.; Banerjee, K.; Basu, S.; Chakrabarti, S. Aging promotes amyloid-β peptide induced mitochondrial dysfunctions in rat brain: A molecular link between aging and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2011, 27, 753–765. [Google Scholar] [PubMed]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, F.J.; Malik, K.U.; Shea, T.B. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to β-amyloid—Map kinase mediates β-amyloid-induced neurodegeneration. J. Biol. Chem. 1999, 274, 30322–30327. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, F.J.; Linsley, M.D.; Shea, T.B. β-Amyloid-induced calcium influx induces neurodegeneration in culture by oxidative stress rather than tau phosphorylation. Mol. Biol. Cell 1999, 10, 61A. [Google Scholar]
- Durell, S.R.; Guy, H.R.; Arispe, N.; Rojas, E.; Pollard, H.B. Theoretical models of the ion channel structure of amyloid β-protein. Biophys. J. 1994, 67, 2137–2145. [Google Scholar] [CrossRef]
- Lal, R.; Lin, H.; Quist, A.P. Amyloid β ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta 2007, 1768, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Zheng, J.; Nussinov, R. Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007, 93, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Ho, Y.S.; Chang, R.C. Modulation of mitochondrial calcium as a pharmacological target for Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front. Neurol. Neurosci. 2009, 24, 114–125. [Google Scholar] [PubMed]
- Ying, Z.; Lin, F.; Gu, W.; Su, Y.; Arshad, A.; Qing, H.; Deng, Y. α-Synuclein increases U251 cells vulnerability to hydrogen peroxide by disrupting calcium homeostasis. J. Neural Transm. 2011, 118, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Melachroinou, K.; Xilouri, M.; Emmanouilidou, E.; Masgrau, R.; Papazafiri, P.; Stefanis, L.; Vekrellis, K. Deregulation of calcium homeostasis mediates secreted α-synuclein-induced neurotoxicity. Neurobiol. Aging 2013, 34, 2853–2865. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Levin, J.; Kamp, F.; Kretzschmar, H.; Giese, A.; Botzel, K. Single-channel electrophysiology reveals a distinct and uniform pore complex formed by α-synuclein oligomers in lipid membranes. PLoS ONE 2012, 7, e42545. [Google Scholar] [CrossRef] [PubMed]
- Clarimon, J.; Eerola, J.; Hellstrom, O.; Peuralinna, T.; Tienari, P.J.; Singleton, A.B. Assessment of PINK1 (PARK6) polymorphisms in Finnish PD. Neurobiol. Aging 2006, 27, 906–907. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Abou-Sleiman, P.M.; Jain, S.; Ahmadi, K.R.; Wood, N.W. Assessment of a DJ-1 (PARK7) polymorphism in Finnish PD. Neurology 2004, 62, 2335. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Apel, A.; Schulte, C.; Schneiderhan-Marra, N.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.C.; Cormier, F.; et al. Inflammatory profile in LRRK2-associated prodromal and clinical PD. J. Neuroinflamm. 2016, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.W.; Guo, M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr. Opin. Neurobiol. 2007, 17, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Pilsl, A.; Winklhofer, K.F. Parkin, PINK1 and mitochondrial integrity: Emerging concepts of mitochondrial dysfunction in Parkinson’s disease. Acta Neuropathol. 2012, 123, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Vives-Bauza, C.; Acin-Perez, R.; Yamamoto, A.; Tan, Y.; Li, Y.; Magrane, J.; Stavarache, M.A.; Shaffer, S.; Chang, S.; et al. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and α-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS ONE 2009, 4, e4597. [Google Scholar] [CrossRef] [PubMed]
- Di Nottia, M.; Masciullo, M.; Verrigni, D.; Petrillo, S.; Modoni, A.; Rizzo, V.; Di Giuda, D.; Rizza, T.; Niceta, M.; Torraco, A.; et al. DJ-1 modulates mitochondrial response to oxidative stress: Clues from a novel diagnosis of PARK7. Clin. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Miguel Martins, L.; Saraiva, L. LRRK2, but not pathogenic mutants, protects against H2O2 stress depending on mitochondrial function and endocytosis in a yeast model. Biochim. Biophys. Acta 2014, 1840, 2025–2031. [Google Scholar] [CrossRef] [PubMed]
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1−/− zebrafish. Eur. J. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606. [Google Scholar] [PubMed]
- Shirotani, T.; Shima, K.; Iwata, M.; Kita, H.; Chigasaki, H. Calcium Accumulation Following Middle Cerebral-Artery Occlusion in Stroke-Prone Spontaneously Hypertensive Rats. J. Cereb. Blood Flow Metab. 1994, 14, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Vila, N.; Chamorro, A.; Castillo, J.; Davalos, A. Glutamate, interleukin-6, and early clinical worsening in patients with acute stroke. Stroke 2001, 32, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.; Shuaib, A.; Wahlgren, N.G. Neurotransmitters and pathophysiology of stroke: Evidence for the release of glutamate and other transmitters/mediators in animals and humans. J. Stroke Cerebrovasc. Dis. 2000, 9, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.E.; Li, N.; Guo, D.Z.; Pan, S.Y.; Li, H.; Yang, C. High plasma glutamate levels are associated with poor functional outcome in acute ischemic stroke. Cell. Mol. Neurobiol. 2015, 35, 159–165. [Google Scholar] [CrossRef]
- Krivonos, O.V.; Amosova, N.A.; Smolentseva, I.G. Use of the glutamate NMDA receptor antagonist PK-Merz in acute stroke. Neurosci. Behav. Physiol. 2010, 40, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Grotta, J.; Clark, W.; Coull, B.; Pettigrew, L.C.; Mackay, B.; Goldstein, L.B.; Meissner, I.; Murphy, D.; LaRue, L. Safety and tolerability of the glutamate antagonist CGS 19755 (Selfotel) in patients with acute ischemic stroke. Results of a phase IIa randomized trial. Stroke 1995, 26, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Elting, J.W.; Sulter, G.A.; Kaste, M.; Lees, K.R.; Diener, H.C.; Hommel, M.; Versavel, M.; Teelken, A.W.; de Keyser, J. AMPA antagonist ZK200775 in patients with acute ischemic stroke: Possible glial cell toxicity detected by monitoring of S-100B serum levels. Stroke 2002, 33, 2813–2818. [Google Scholar] [CrossRef] [PubMed]
- Higashino, H.; Suzuki, A. A calcium antagonist, NB-818, improves blood flow distribution in the brain and other major organs of stroke-prone spontaneously hypertensive rats (SHRSP). J. Brain Sci. 1997, 23, 79–95. [Google Scholar]
- Shinyama, H.; Kawamura, T.; Iwamoto, M.; Egi, Y.; Tanaka, H.; Kawabata, Y.; Nakamura, N.; Kagitani, Y. Effects of the calcium antagonist AE0047 on the development of neurological deficit and infarction after middle cerebral artery occlusion in stroke-prone spontaneously hypertensive rats. J. Pharm. Pharmacol. 1997, 49, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Sahlen, A.; Hamid, N.; Amanullah, M.R.; Fam, J.M.; Yeo, K.K.; Lau, Y.H.; Lam, C.S.; Ding, Z.P. Impact of aortic root size on left ventricular afterload and stroke volume. Eur. J. Appl. Physiol. 2016, 116, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Zhang, C.F.; Jiang, X.Q.; Zhou, H.Q.; Liu, M. Calcium antagonists for acute ischemic stroke. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Gao, Q.; Cao, C.M.; Bruce, I.C.; Xia, Q. Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning. Life Sci. 2006, 78, 738–745. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Neurodegenerative Disorders | Drug | Target | Clinical Trial ID | Status |
|---|---|---|---|---|
| AD | Losartan/amlodipine | Angiotensin/calcium | NCT02913664 | Phase II |
| SAM-531 | Calcium | NCT00745576 | Phase I | |
| Nilvadipine/Placebo | Calcium | NCT02017340 | Phase III | |
| PD | Isradipine | Calcium channel | NCT00909545 | Phase II |
| Isradipine/Placebo | Calcium channel | NCT02168842 | Phase III | |
| ALS | Fasudil | Calcium | NCT01935518 | Phase II |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Y.; Dong, Y.; Cheng, J. The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders. Int. J. Mol. Sci. 2017, 18, 248. https://doi.org/10.3390/ijms18020248
Liao Y, Dong Y, Cheng J. The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders. International Journal of Molecular Sciences. 2017; 18(2):248. https://doi.org/10.3390/ijms18020248
Chicago/Turabian StyleLiao, Yajin, Yuan Dong, and Jinbo Cheng. 2017. "The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders" International Journal of Molecular Sciences 18, no. 2: 248. https://doi.org/10.3390/ijms18020248
APA StyleLiao, Y., Dong, Y., & Cheng, J. (2017). The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders. International Journal of Molecular Sciences, 18(2), 248. https://doi.org/10.3390/ijms18020248