Lysine-Less Variants of Spinal Muscular Atrophy SMN and SMNΔ7 Proteins Are Degraded by the Proteasome Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
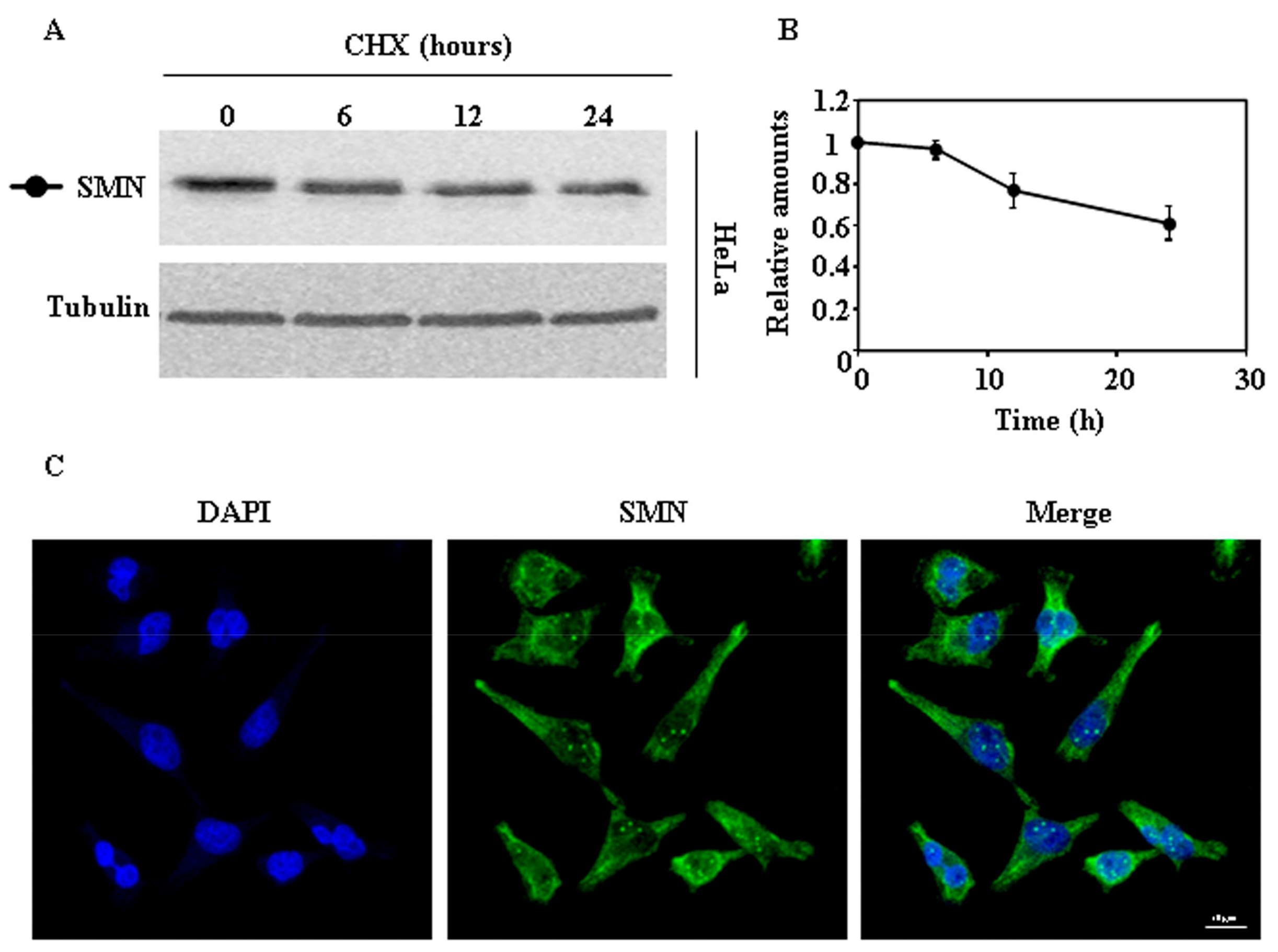
2.1. Endogenous SMN Degradation
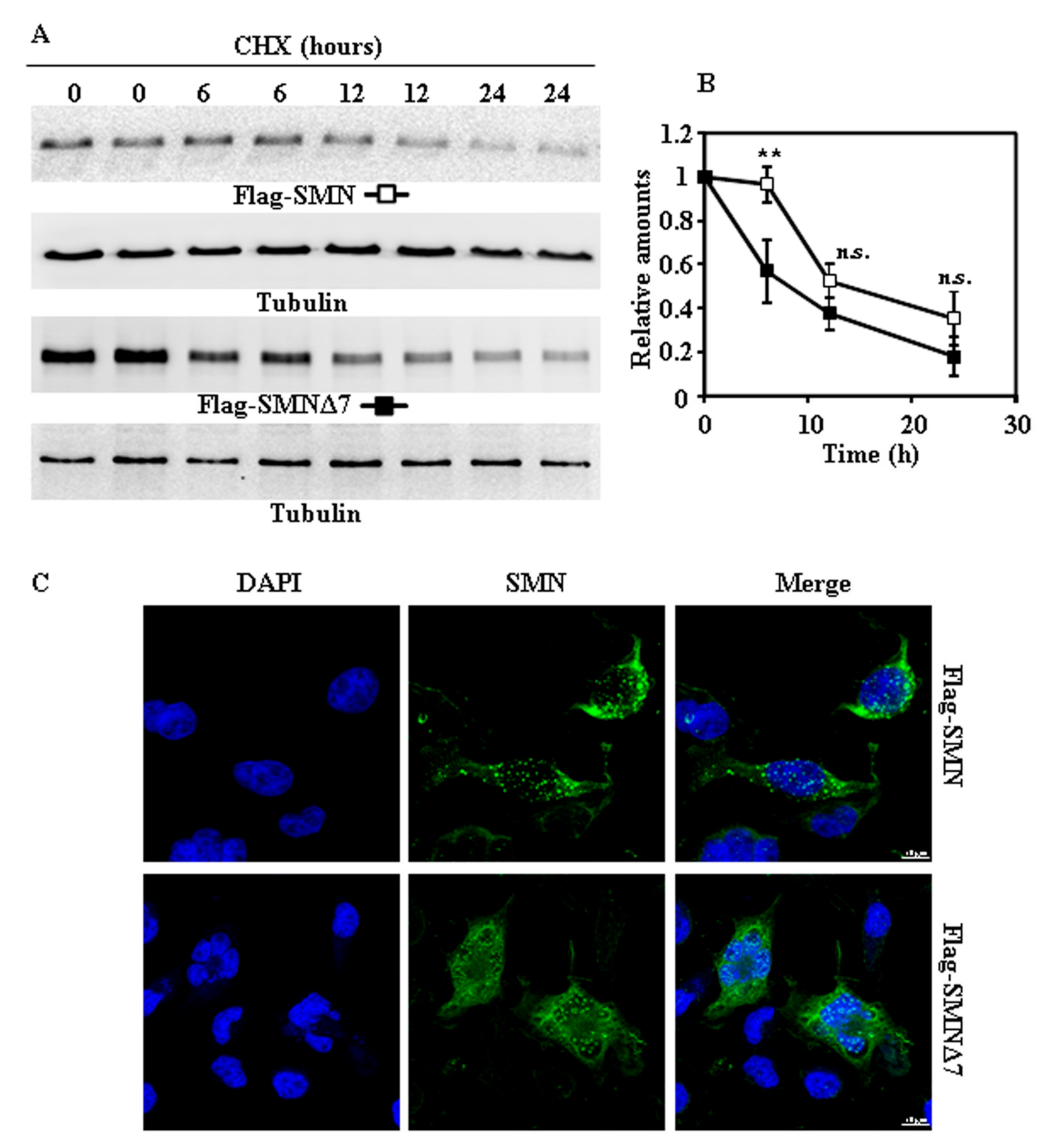
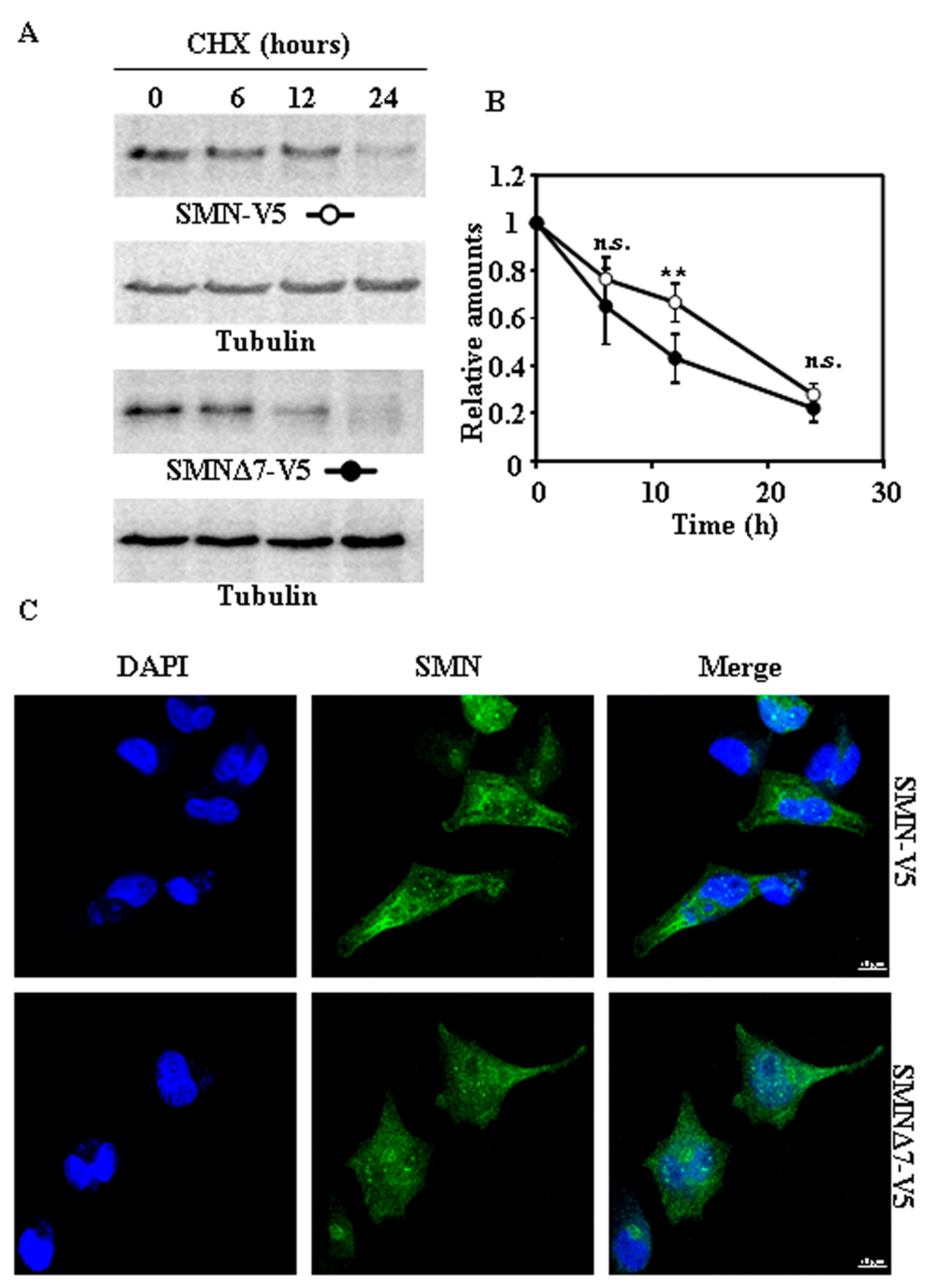
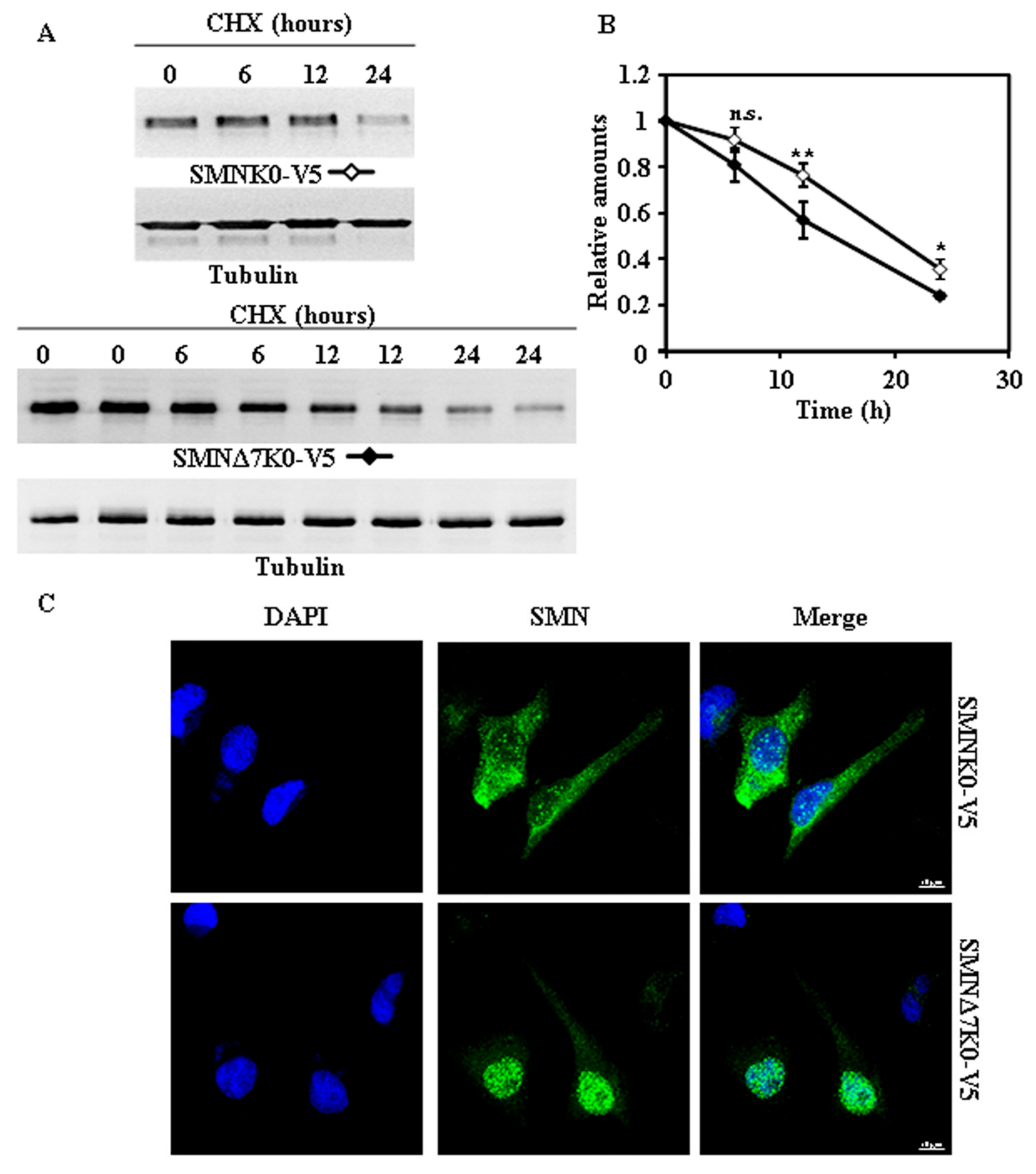
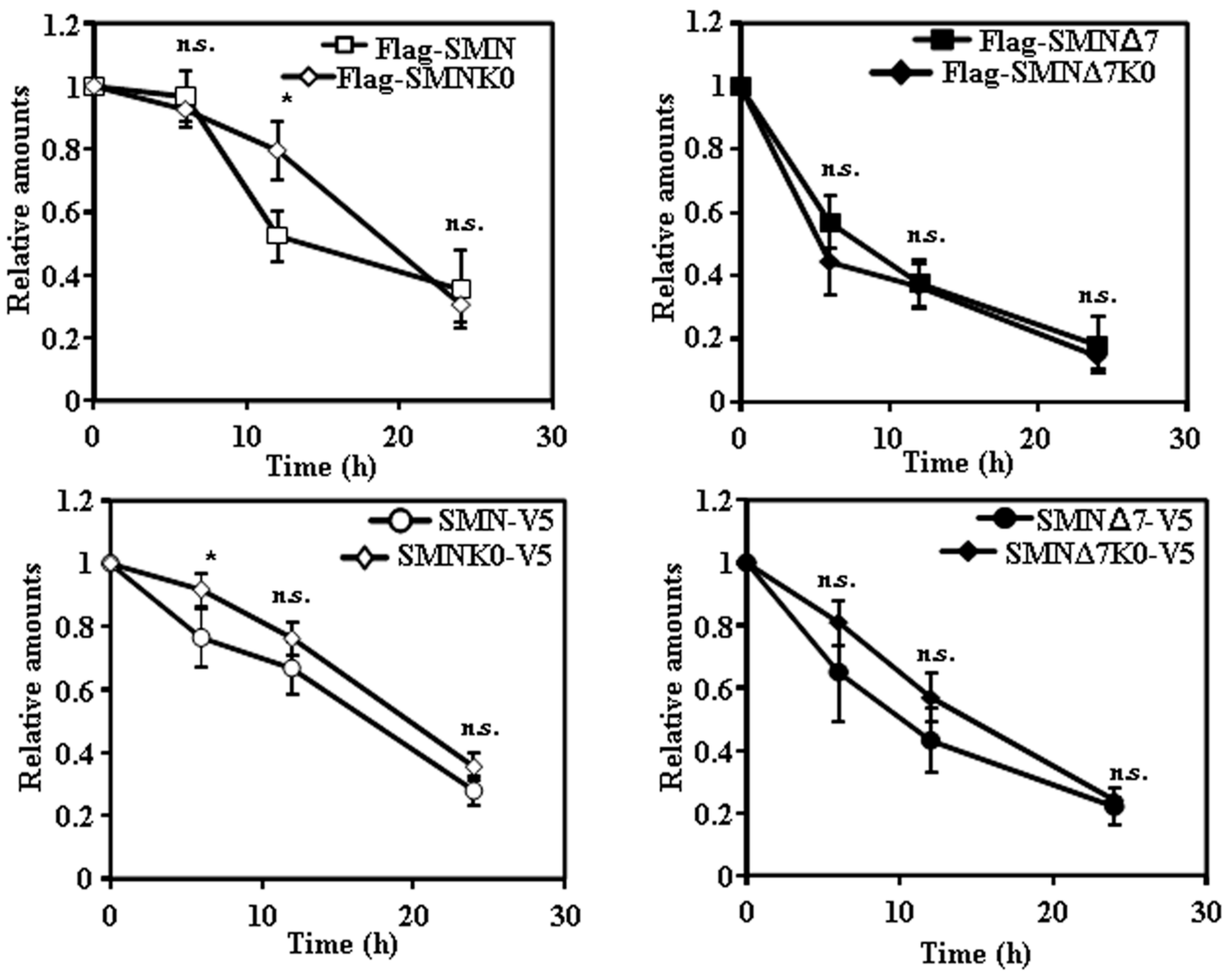
2.2. Tagging SMN and SMNΔ7 at Their N- or C-Terminus and Degradation
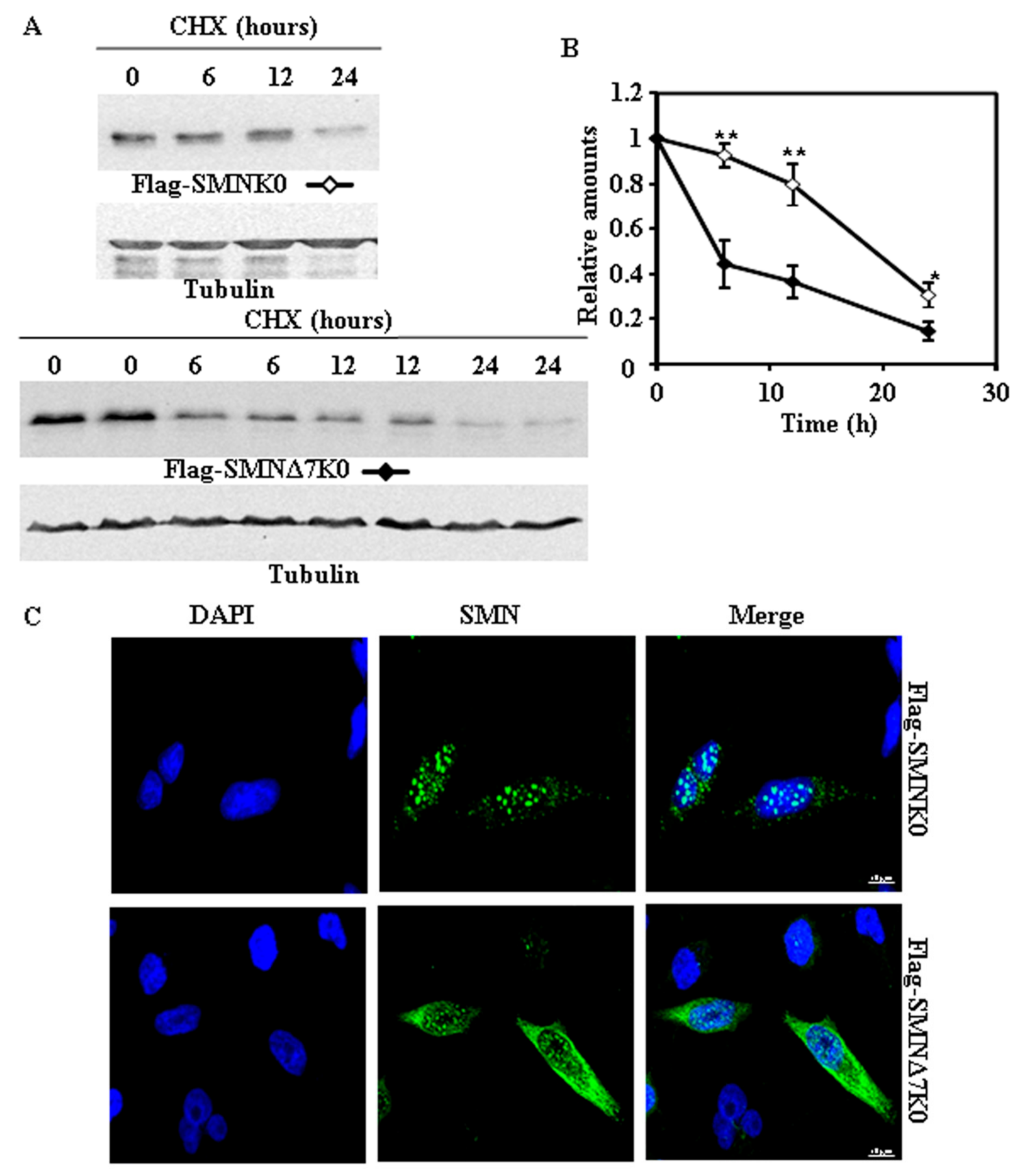
2.3. Requirement of Ubiquitylation of SMN Proteins for Degradation
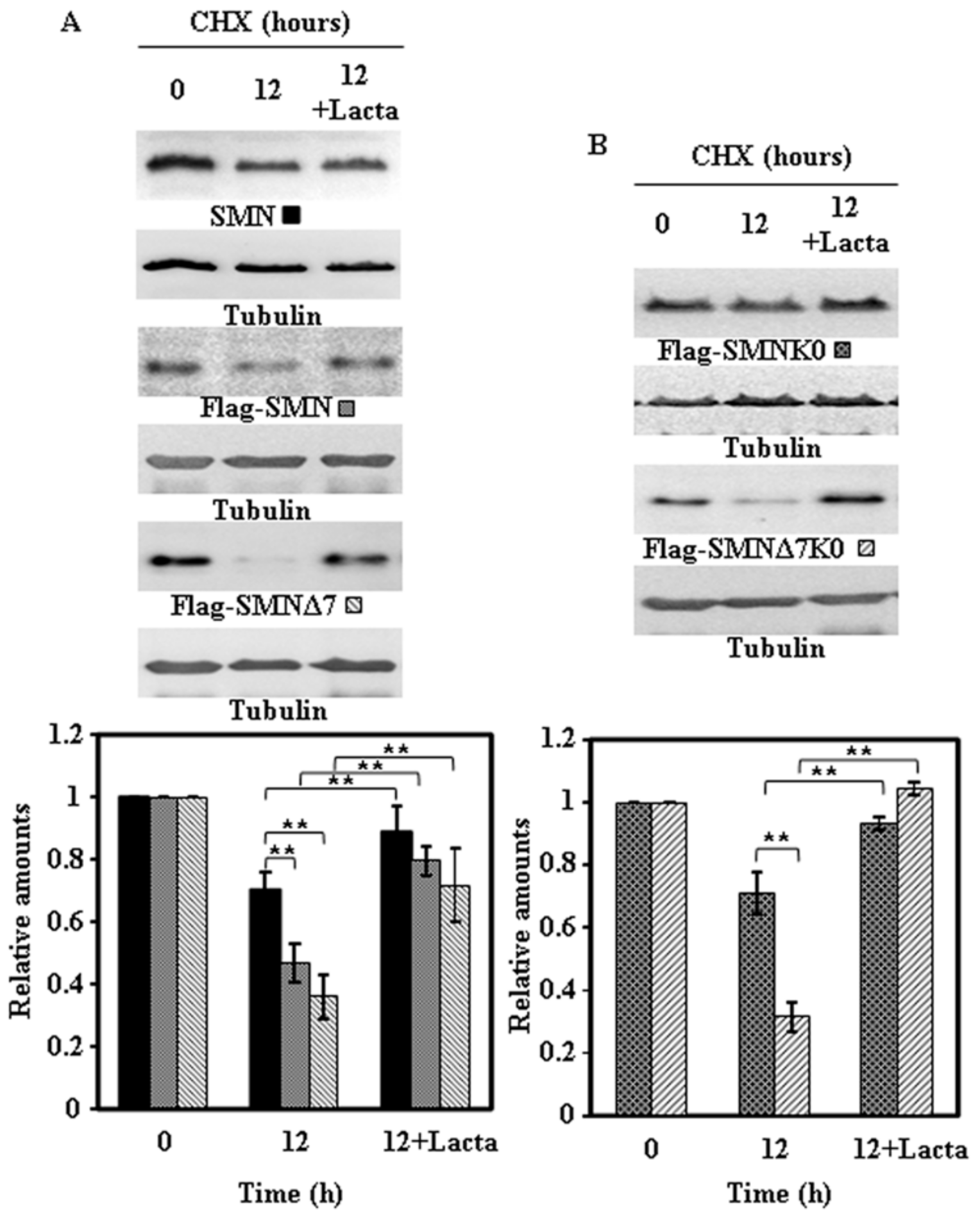
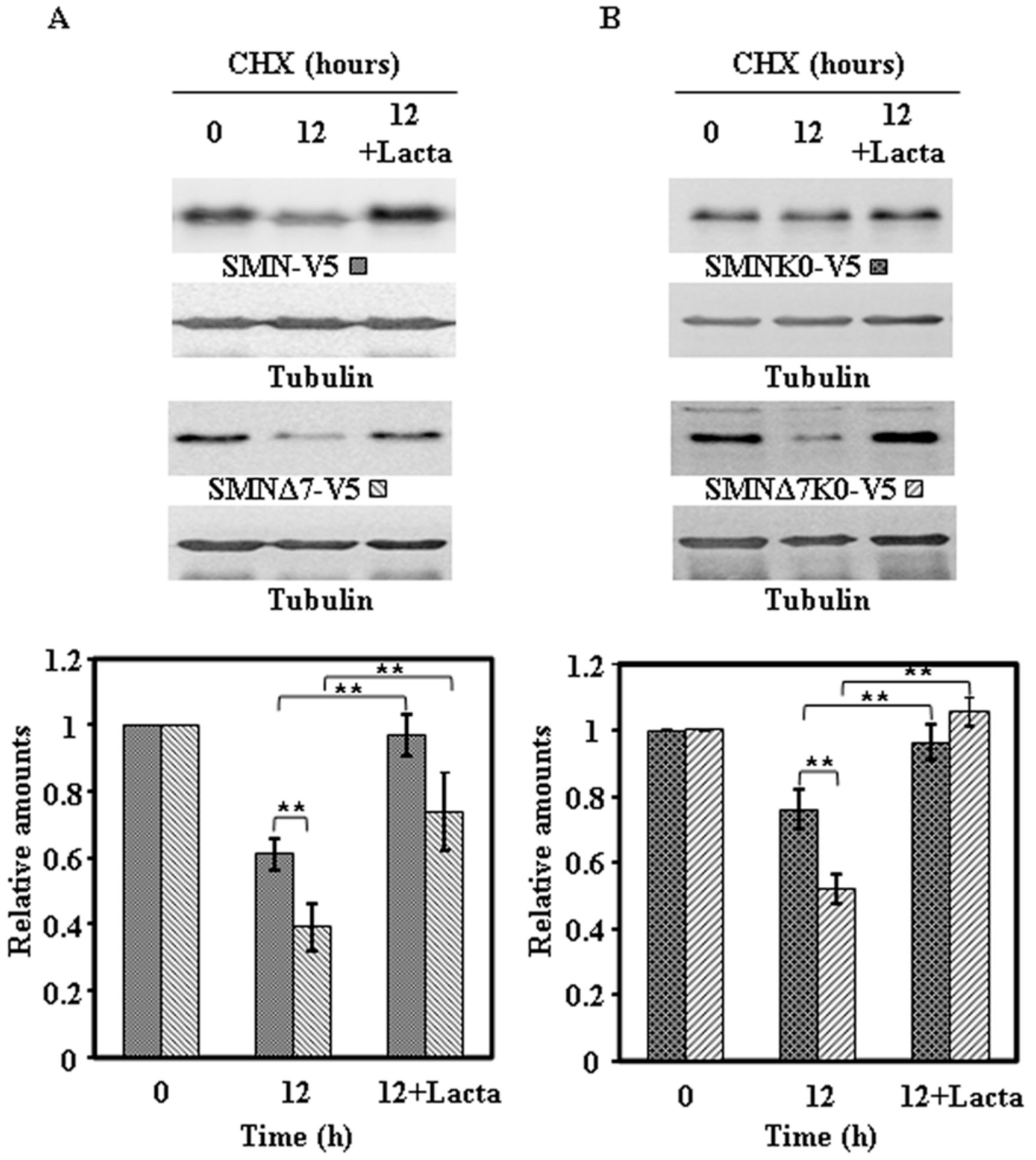
2.4. Effect of Proteasome Inhibition on the Degradation of Endogenous SMN and Ectopically Expressed SMN Constructs
3. Discussion
4. Materials and Methods
4.1. DNA Constructs
4.2. Studies of Endogenous and Ectopically Expressed SMN Protein Degradation
4.3. Immunofluorescence and Confocal Microscopy
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CHX | Cycloheximide |
| DAPI | 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride, 4′,6-Diamidino-2-phenylindole dihydrochloride |
| PMSF | Phenylmethanesulfonyl fluoride |
| PVDF | Polyvinylidene difluoride |
| SMA | Spinal Muscular Atrophy |
References
- Iannaccone, S.T. Modern management of spinal muscular atrophy. J. Child Neurol. 2007, 22, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Hung, W.C.; Chuang, Y.J.; Jong, Y.J. Degradation of survival motor neuron (SMN) protein is mediated via the ubiquitin/proteasome pathway. Neurochem. Int. 2004, 45, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.G.; Munoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of SMN protein stability. Mol. Cell. Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Lai, M.C.; Er, T.K.; Yang, S.N.; Hung, C.H.; Tsai, H.H.; Lin, Y.C.; Chang, J.G.; Lo, Y.C.; Jong, Y.J. Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) regulates the level of SMN expression through ubiquitination in primary spinal muscular atrophy fibroblasts. Clin. Chim. Acta 2010, 411, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Trinkle-Mulcahy, L.; Boulon, S.; Lam, Y.W.; Urcia, R.; Boisvert, F.M.; Vandermoere, F.; Morrice, N.A.; Swift, S.; Rothbauer, U.; Leonhardt, H.; et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 2008, 183, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Han, K.J.; Foster, D.G.; Zhang, N.Y.; Kanisha, K.; Dzieciatkowska, M.; Sclafani, R.A.; Hansen, K.C.; Peng, J.; Liu, C.W. Ubiquitin Specific Protease 9X deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J. Biol. Chem. 2012, 287, 43741–43752. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.Y.; Dimitriadi, M.; Terzic, B.; Cable, C.; Hart, A.C.; Chitnis, A.; Fischbeck, K.H.; Burnett, B.G. The E3 ubiquitin ligase mind bomb 1 ubiquitinates and promotes the degradation of survival of motor neuron protein. Mol. Biol. Cell 2013, 24, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Han, K.J.; Foster, D.; Harhaj, E.W.; Dzieciatkowska, M.; Hansen, K.; Liu, C.W. Monoubiquitination of survival motor neuron regulates its cellular localization and Cajal body integrity. Hum. Mol. Genet. 2016, 25, 1392–1405. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Castelao, B.; Ruiz-Rivas, C.; Castano, J.G. A critical appraisal of quantitative studies of protein degradation in the framework of cellular proteostasis. Biochem. Res. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Mani, D.R.; Eisenhaure, T.; Mertins, P.; Jaffe, J.D.; Clauser, K.R.; Hacohen, N.; Carr, S.A. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell Proteom. 2012, 11, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Cambridge, S.B.; Gnad, F.; Nguyen, C.; Bermejo, J.L.; Kruger, M.; Mann, M. Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. J. Proteome Res. 2011, 10, 5275–5284. [Google Scholar] [CrossRef] [PubMed]
- Dhondt, I.; Petyuk, V.A.; Cai, H.; Vandemeulebroucke, L.; Vierstraete, A.; Smith, R.D.; Depuydt, G.; Braeckman, B.P. FOXO/DAF-16 activation slows down turnover of the majority of proteins in C. elegans. Cell Rep. 2016, 16, 3028–3040. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, D.N.; Giddings, M.R.; Inglis, M.S. Kinetics of degradation of “short-” and “long-lived” proteins in cultured mammalian cells. Cell Biol. Int. Rep. 1980, 4, 1081–1090. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Bourdetsky, D.; Schmelzer, C.E.; Admon, A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. Proc. Natl. Acad. Sci. USA 2014, 111, E1591–E1599. [Google Scholar] [CrossRef] [PubMed]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.; Heck, A.J.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Rodier, G.; Bonneil, E.; Thibault, P.; Meloche, S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol. Cell. Biol. 2004, 24, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.L.; den Besten, W.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Ben-Saadon, R.; Fajerman, I.; Ziv, T.; Hellman, U.; Schwartz, A.L.; Ciechanover, A. The tumor suppressor protein p16INK4a and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system. Direct evidence for ubiquitination at the N-terminal residue. J. Biol. Chem. 2004, 279, 41414–41421. [Google Scholar] [CrossRef] [PubMed]
- Trausch-Azar, J.S.; Lingbeck, J.; Ciechanover, A.; Schwartz, A.L. Ubiquitin-Proteasome-mediated degradation of Id1 is modulated by MyoD. J. Biol. Chem. 2004, 279, 32614–32619. [Google Scholar] [CrossRef] [PubMed]
- Sadeh, R.; Breitschopf, K.; Bercovich, B.; Zoabi, M.; Kravtsova-Ivantsiv, Y.; Kornitzer, D.; Schwartz, A.; Ciechanover, A. The N-terminal domain of MyoD is necessary and sufficient for its nuclear localization-dependent degradation by the ubiquitin system. Proc. Natl. Acad. Sci. USA 2008, 105, 15690–15695. [Google Scholar] [CrossRef] [PubMed]
- Trausch-Azar, J.; Leone, T.C.; Kelly, D.P.; Schwartz, A.L. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1α via the N-terminal pathway. J. Biol. Chem. 2010, 285, 40192–40200. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, K.M.; Basrur, V.; Ashraf, N.S.; Konen, J.R.; Elenitoba-Johnson, K.S.; Todi, S.V.; Paulson, H.L. The ubiquitin-conjugating enzyme (E2) Ube2w ubiquitinates the N-terminus of substrates. J. Biol. Chem. 2013, 288, 18784–18788. [Google Scholar] [CrossRef] [PubMed]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Lasa, M.; Polevoda, B.; Gazquez, C.; Elosegui-Artola, A.; Kim, D.S.; de Juan-Pardo, E.; Demeyer, K.; Hole, K.; Larrea, E.; et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. USA 2012, 109, 12449–12454. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.S.; Shemorry, A.; Varshavsky, A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science 2010, 327, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 2008, 1786, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta 2014, 1843, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lanzas, R.; Castano, J.G. Proteins directly interacting with mammalian 20S proteasomal subunits and ubiquitin-independent proteasomal degradation. Biomolecules 2014, 4, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Coady, T.H.; Lorson, C.L. SMN in spinal muscular atrophy and snRNP biogenesis. Wiley Interdiscip. Rev. RNA 2011, 2, 546–564. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta 2017, 1860, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Pellizzoni, L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007, 8, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, S.; Pellizzoni, L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci. 2015, 35, 8691–8700. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Lanzas, R.; Castaño, J.G. Lysine-Less Variants of Spinal Muscular Atrophy SMN and SMNΔ7 Proteins Are Degraded by the Proteasome Pathway. Int. J. Mol. Sci. 2017, 18, 2667. https://doi.org/10.3390/ijms18122667
Sánchez-Lanzas R, Castaño JG. Lysine-Less Variants of Spinal Muscular Atrophy SMN and SMNΔ7 Proteins Are Degraded by the Proteasome Pathway. International Journal of Molecular Sciences. 2017; 18(12):2667. https://doi.org/10.3390/ijms18122667
Chicago/Turabian StyleSánchez-Lanzas, Raúl, and José G. Castaño. 2017. "Lysine-Less Variants of Spinal Muscular Atrophy SMN and SMNΔ7 Proteins Are Degraded by the Proteasome Pathway" International Journal of Molecular Sciences 18, no. 12: 2667. https://doi.org/10.3390/ijms18122667
APA StyleSánchez-Lanzas, R., & Castaño, J. G. (2017). Lysine-Less Variants of Spinal Muscular Atrophy SMN and SMNΔ7 Proteins Are Degraded by the Proteasome Pathway. International Journal of Molecular Sciences, 18(12), 2667. https://doi.org/10.3390/ijms18122667