Around and beyond 53BP1 Nuclear Bodies



Abstract

1. Introduction
2. Composition of p53 Binding Protein 1 (53BP1) Nuclear Bodies (NBs)
3. Dynamics of 53BP1 NBs
4. Replication Stress and 53BP1 NBs
4.1. Sources of Replication Stress (RS)
4.2. 53BP1 NBs Originate from RS
4.3. Response to RS Regulates 53BP1 NBs
5. Common Fragile Sites (CFS) and 53BP1 NBs
6. Late DNA Synthesis for Under-Replicated Regions
6.1. Mitotic DNA Synthesis (MiDAS)
6.2. Anaphase Bridges Are the Last Opportunity to Properly Segregate DNA
7. Major Cellular Processes and 53BP1 NBs
7.1. Transcription
7.2. Double-Strand Breaks (DSB) Signaling and DNA Repair
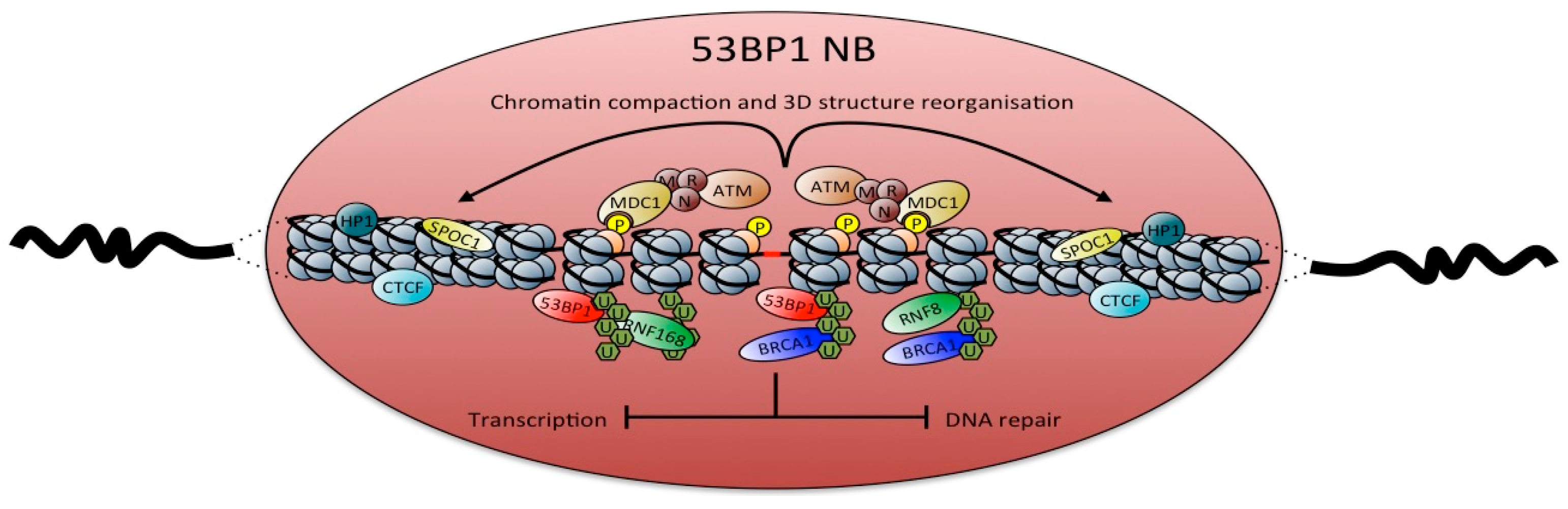
7.3. Chromatin Structure and Chromosome Segregation
7.4. Connection with the Cell Cycle
8. 53BP1 in Different Types of NBs
9. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 53BP1 | p53 Binding protein 1 |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related |
| BRCA1 | Breast cancer 1 |
| CDC | Cell division cycle |
| CFS | Common fragile sites |
| CIN | Chromosomal instability |
| DDR | DNA damage response |
| DSB | DNA double-strand break |
| ERCC1 | Excision repair cross-complementation group 1 protein |
| FA | Fanconi anemia |
| FANC | Fanconi anemia complementation group |
| HR | Homologous recombination |
| MCM | Mini chromosome maintenance |
| MDC1 | Mediator of DNA damage checkpoint protein 1 |
| MiDAS | Mitotic DNA synthesis |
| MRN | MRE11–RAD50–NBS1 complex |
| NBs | Nuclear bodies |
| NHEJ | Non-homologous end-joining |
| PML | Promyelocytic leukemia |
| PTF | Proximal sequence element (PSE)-binding transcription factor |
| RF | Replication fork |
| RNF | Ring finger protein |
| RPA | Replication protein A |
| RS | Replication stress |
| SPOC1 | Survival time associated PHD finger protein in Ovarian Cancer 1 |
| ssDNA | Single-stranded DNA |
| SUMO | Small ubiquitin-like modifier |
| TIFs | Telomere dysfunction-induced foci |
| UFBs | Ultra-fine bridges |
| XPG | Xeroderma pigmentosum complementation group G protein |
| XRCC | X-ray repair cross complementing |
References
- Pederson, T. The nucleolus. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Stanek, D.; Fox, A.H. Nuclear bodies: News insights into structure and function. Curr. Opin. Cell Biol. 2017, 46, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.S.; Zhang, B.; Spector, D.L. Biogenesis and function of nuclear bodies. Trends Genet. 2011, 27, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, S.P.; Dundr, M. Nucleation of nuclear bodies by RNA. Nat. Cell Biol. 2011, 13, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Szostecki, C.; Guldner, H.H.; Will, H. Autoantibodies against “nuclear dots” in primary biliary cirrhosis. Semin. Liver Dis. 1997, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.A.; Maul, G.G.; Miller, W.H., Jr.; Chen, J.D.; Kakizuka, A.; Evans, R.M. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 1994, 76, 333–343. [Google Scholar] [CrossRef]
- Weis, K.; Rambaud, S.; Lavau, C.; Jansen, J.; Carvalho, T.; Carmo-Fonseca, M.; Lamond, A.; Dejean, A. Retinoic acid regulates aberrant nuclear localization of PML-RAR α in acute promyelocytic leukemia cells. Cell 1994, 76, 345–356. [Google Scholar] [CrossRef]
- Chang, H.R.; Munkhjargal, A.; Kim, M.J.; Park, S.Y.; Jung, E.; Ryu, J.H.; Yang, Y.; Lim, J.S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Matunis, M.J.; Dejean, A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 1998, 17, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Muller, S.; Ronchetti, S.; Freemont, P.S.; Dejean, A.; Pandolfi, P.P. Role of SUMO-1-modified PML in nuclear body formation. Blood 2000, 95, 2748–2752. [Google Scholar] [PubMed]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [PubMed]
- Salomoni, P.; Pandolfi, P.P. The role of PML in tumor suppression. Cell 2002, 108, 165–170. [Google Scholar] [CrossRef]
- Dellaire, G.; Ching, R.W.; Ahmed, K.; Jalali, F.; Tse, K.C.; Bristow, R.G.; Bazett-Jones, D.P. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, CHK2, and ATR. J. Cell Biol. 2006, 175, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Hu, P.; Ye, T.Z.; Stan, R.; Ellis, N.A.; Pandolfi, P.P. A role for PML and the nuclear body in genomic stability. Oncogene 1999, 18, 7941–7947. [Google Scholar] [CrossRef] [PubMed]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. P53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Ward, I.M.; Minn, K.; Van Deursen, J.; Chen, J. P53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003, 23, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Smogorzewska, A.; De Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Rodier, F.; Munoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppe, J.P.; Campeau, E.; Beausejour, C.M.; Kim, S.H.; et al. DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Pombo, A.; Cuello, P.; Schul, W.; Yoon, J.B.; Roeder, R.G.; Cook, P.R.; Murphy, S. Regional and temporal specialization in the nucleus: A transcriptionally-active nuclear domain rich in PTF, Oct1 and PIKA antigens associates with specific chromosomes early in the cell cycle. EMBO J. 1998, 17, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Roy, K.; Katyal, S.; Sun, X.; Bleoo, S.; Godbout, R. Dynamic nature of cleavage bodies and their spatial relationship to DDX1 bodies, Cajal bodies, and gems. Mol. Biol. Cell 2006, 17, 1126–1140. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Lukas, C.; Melander, F.; Bartek, J.; Lukas, J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by MDC1/NFBD1. J. Cell Biol. 2005, 170, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grofte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. Mdc1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Diaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.Y.; Obuse, C.; Nishi, R.; et al. BRCA1 directs the repair pathway to Homologous Recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017, 803–805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Godinez, V.; Wu, T.; Sherman, A.J.; Lee, C.S.; Liaw, L.H.; Zhongsheng, Y.; Yokomori, K.; Berns, M.W. Analysis of DNA double-strand break response and chromatin structure in mitosis using laser microirradiation. Nucleic Acids Res. 2010, 38, e202. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human recq helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, C.P.; Carr, A.M.; Oliver, A.W. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 2014, 22, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grofte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Yuce, O.; West, S.C. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 2013, 33, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Germain, D.R.; Poon, H.Y.; Hildebrandt, M.R.; Monckton, E.A.; McDonald, D.; Hendzel, M.J.; Godbout, R. DEAD box 1 facilitates removal of RNA and homologous recombination at DNA double-strand breaks. Mol. Cell. Biol. 2016, 36, 2794–2810. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Monckton, E.A.; Godbout, R. A role for DEAD box 1 at DNA double-strand breaks. Mol. Cell. Biol. 2008, 28, 6413–6425. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Li, X.; Zheng, W.; Li, Z.; Lu, D.; Chen, G.; Gong, D.; Yang, L.; Fu, J.; Shi, P.; et al. CTCF prevents genomic instability by promoting homologous recombination-directed DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2017, 114, 10912–10917. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kuo, C.Y.; Stark, J.M.; Shih, H.M.; Ann, D.K. HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res. 2013, 41, 5784–5798. [Google Scholar] [CrossRef] [PubMed]
- Mund, A.; Schubert, T.; Staege, H.; Kinkley, S.; Reumann, K.; Kriegs, M.; Fritsch, L.; Battisti, V.; Ait-Si-Ali, S.; Hoffbeck, A.S.; et al. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res. 2012, 40, 11363–11379. [Google Scholar] [CrossRef] [PubMed]
- Kafer, G.R.; Li, X.; Horii, T.; Suetake, I.; Tajima, S.; Hatada, I.; Carlton, P.M. 5-Hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep. 2016, 14, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome maintenance in response to DNA damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [PubMed]
- Kantidze, O.L.; Razin, S.V. 5-Hydroxymethylcytosine in DNA repair: A new player or a red herring? Cell Cycle 2017, 16, 1499–1501. [Google Scholar] [CrossRef] [PubMed]
- Foltankova, V.; Legartova, S.; Kozubek, S.; Hofer, M.; Bartova, E. DNA-damage response in chromatin of ribosomal genes and the surrounding genome. Gene 2013, 522, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Legartova, S.; Sehnalova, P.; Malyskova, B.; Kuntziger, T.; Collas, P.; Cmarko, D.; Raska, I.; Sorokin, D.V.; Kozubek, S.; Bartova, E. Localized movement and levels of 53BP1 protein are changed by γ-irradiation in PML deficient cells. J. Cell. Biochem. 2016, 117, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novak, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 14728. [Google Scholar] [CrossRef] [PubMed]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and CRB2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Belotserkovskaya, R.; Jackson, S.P. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010, 190, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Fradet-Turcotte, A.; Noordermeer, S.M.; Canny, M.D.; Brun, C.M.; Strecker, J.; Escribano-Diaz, C.; Durocher, D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014, 344, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Acharya, S.S.; Kwon, M.; Drane, P.; Guan, Y.; Adelmant, G.; Kalev, P.; Shah, J.; Pellman, D.; Marto, J.A.; et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 2014, 54, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Mackay, D.R.; Ullman, K.S. ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Mol. Biol. Cell 2015, 26, 2217–2226. [Google Scholar] [CrossRef] [PubMed]
- Cescutti, R.; Negrini, S.; Kohzaki, M.; Halazonetis, T.D. Topbp1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J. 2010, 29, 3723–3732. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Michelena, J.; Teloni, F.; Imhof, R.; Altmeyer, M. Replication-coupled dilution of H4K20me2 guides 53BP1 to pre-replicative chromatin. Cell Rep. 2017, 19, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CTIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Rohban, S.; Campaner, S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. Biophys. Acta 2015, 1849, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Luebben, S.W.; Kawabata, T.; Johnson, C.S.; O’Sullivan, M.G.; Shima, N. A concomitant loss of dormant origins and FANCC exacerbates genome instability by impairing DNA replication fork progression. Nucleic Acids Res. 2014, 42, 5605–5615. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J., Jr.; Fornara, O.; Merchut-Maya, J.M.; Maya-Mendoza, A.; Rahbar, A.; Stragliotto, G.; Broholm, H.; Svensson, M.; Sehested, A.; Soderberg Naucler, C.; et al. Replication stress, DNA damage signalling, and cytomegalovirus infection in human medulloblastomas. Mol. Oncol. 2017, 11, 945–964. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Raschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Schwab, R.A.; Nieminuszczy, J.; Deans, A.J.; West, S.C.; Niedzwiedz, W. The DNA translocase activity of fancm protects stalled replication forks. Hum. Mol. Genet. 2012, 21, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Reynolds, J.J.; Winczura, A.; Blackford, A.N.; Borel, V.; Miller, E.S.; Zlatanou, A.; Nieminuszczy, J.; Ryan, E.L.; Davies, N.J.; et al. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell 2015, 59, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Luebben, S.W.; Kawabata, T.; Akre, M.K.; Lee, W.L.; Johnson, C.S.; O’Sullivan, M.G.; Shima, N. Helq acts in parallel to Fancc to suppress replication-associated genome instability. Nucleic Acids Res. 2013, 41, 10283–10297. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Tsantoulis, P.; Kotsinas, A.; Michalopoulos, I.; Townsend, P.; Gorgoulis, V.G. Are common fragile sites merely structural domains or highly organized “functional” units susceptible to oncogenic stress? Cell. Mol. Life Sci. CMLS 2014, 71, 4519–4544. [Google Scholar] [CrossRef] [PubMed]
- Le Tallec, B.; Koundrioukoff, S.; Wilhelm, T.; Letessier, A.; Brison, O.; Debatisse, M. Updating the mechanisms of common fragile site instability: How to reconcile the different views? Cell. Mol. Life Sci. 2014, 71, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Minocherhomji, S.; Chan, K.L.; Palmai-Pallag, T.; Chu, W.K.; Wass, T.; Mankouri, H.W.; Liu, Y.; Hickson, I.D. MUS81 promotes common fragile site expression. Nat. Cell Biol. 2013, 15, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Andersen, P.L.; Qin, Z.; Xiao, W. Rev3, the catalytic subunit of polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res. 2013, 41, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.; Wang, X.; Lee, M.Y.; Eckert, K.A. Mechanism of replicative DNA polymerase δ pausing and a potential role for DNA polymerase κ in common fragile site replication. J. Mol. Biol. 2013, 425, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.S.; Wittschieben, J.P.; Wood, R.D. DNA polymerase ζ is required for proliferation of normal mammalian cells. Nucleic Acids Res. 2012, 40, 4473–4482. [Google Scholar] [CrossRef] [PubMed]
- Despras, E.; Sittewelle, M.; Pouvelle, C.; Delrieu, N.; Cordonnier, A.M.; Kannouche, P.L. RAD18-dependent SUMOylation of human specialized DNA polymerase η is required to prevent under-replicated DNA. Nat. Commun. 2016, 7, 13326. [Google Scholar] [CrossRef] [PubMed]
- Betous, R.; Pillaire, M.J.; Pierini, L.; Van der Laan, S.; Recolin, B.; Ohl-Seguy, E.; Guo, C.; Niimi, N.; Gruz, P.; Nohmi, T.; et al. DNA polymerase kappa-dependent DNA synthesis at stalled replication forks is important for CHK1 activation. EMBO J. 2013, 32, 2172–2185. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.F.; Bertolin, A.P.; Bergoglio, V.; Pillaire, M.J.; Gonzalez Besteiro, M.A.; Luzzani, C.; Miriuka, S.G.; Cazaux, C.; Hoffmann, J.S.; Gottifredi, V. Cyclin kinase-independent role of p21CDKN1A in the promotion of nascent DNA elongation in unstressed cells. eLife 2016, 5, e18020. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Colnaghi, L.; Huang, T.T. Dysregulation of DNA polymerase kappa recruitment to replication forks results in genomic instability. EMBO J. 2012, 31, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Ogrunc, M.; Martinez-Zamudio, R.I.; Sadoun, P.B.; Dore, G.; Schwerer, H.; Pasero, P.; Lemaitre, J.M.; Dejean, A.; Bischof, O. USP1 regulates cellular senescence by controlling genomic integrity. Cell Rep. 2016, 15, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Xu, Y.J. The cell killing mechanisms of hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Palmai-Pallag, T.; Ying, S.; Hickson, I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009, 11, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The fanc pathway and blm collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Guervilly, J.H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Garner, E.; Hallet, A.; Nguyen, H.D.; Rickman, K.A.; Gill, G.; Smogorzewska, A.; Zou, L. Noncovalent interactions with SUMO and ubiquitin orchestrate distinct functions of the SLX4 complex in genome maintenance. Mol. Cell 2015, 57, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, S.; Hasanova, Z.; Kanagaraj, R.; Chappidi, N.; Altmannova, V.; Menon, S.; Sedlackova, H.; Langhoff, J.; Surendranath, K.; Huhn, D.; et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol. Cell 2017, 66, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, H.D.; Sarbajna, S.; Matos, J.; West, S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for holliday junction resolution in human cells. Mol. Cell 2013, 52, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Hickson, I.D. On the origins of ultra-fine anaphase bridges. Cell Cycle 2009, 8, 3065–3066. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, R.C.; De Boer, H.R.; Schoonen, P.M.; De Vries, E.G.; Lens, S.M.; Van Vugt, M.A. Rif1 is required for resolution of ultrafine DNA bridges in anaphase to ensure genomic stability. Dev. Cell 2015, 34, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. Conflict resolution in the genome: How transcription and replication make it work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Moudry, P.; Lukas, C.; Macurek, L.; Neumann, B.; Heriche, J.K.; Pepperkok, R.; Ellenberg, J.; Hodny, Z.; Lukas, J.; Bartek, J. Nucleoporin NUP153 guards genome integrity by promoting nuclear import of 53BP1. Cell Death Differ. 2012, 19, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Wiedner, M.; Velimezi, G.; Prochazkova, J.; Owusu, M.; Bauer, S.; Loizou, J.I. Atmin is required for the ATM-mediated signaling and recruitment of 53BP1 to DNA damage sites upon replication stress. DNA Repair 2014, 24, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Kondratova, A.; Watanabe, T.; Marotta, M.; Cannon, M.; Segall, A.M.; Serre, D.; Tanaka, H. Replication fork integrity and intra-S phase checkpoint suppress gene amplification. Nucleic Acids Res. 2015, 43, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Macurek, L.; Benada, J.; Mullers, E.; Halim, V.A.; Krejcikova, K.; Burdova, K.; Pechackova, S.; Hodny, Z.; Lindqvist, A.; Medema, R.H.; et al. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle 2013, 12, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Jasin, M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Mollgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J., Jr.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef] [PubMed]
- Somyajit, K.; Saxena, S.; Babu, S.; Mishra, A.; Nagaraju, G. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 2015, 43, 9835–9855. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; He, L.; Wu, H.; Pan, F.; Wu, X.; Zhao, J.; Hu, Z.; Sekhar, C.; Li, H.; Zheng, L.; et al. The FEN1 L209P mutation interferes with long-patch base excision repair and induces cellular transformation. Oncogene 2017, 36, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Herrera-Moyano, E.; Ortega, P.; Aguilera, A. Differential effect of the overexpression of RAD2/XPG family endonucleases on genome integrity in yeast and human cells. DNA Repair 2017, 57, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Maskey, R.S.; Kim, M.S.; Baker, D.J.; Childs, B.; Malureanu, L.A.; Jeganathan, K.B.; Machida, Y.; Van Deursen, J.M.; Machida, Y.J. SPARTAN deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 2014, 5, 5744. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Kops, G.J.; Medema, R.H. Targeting the mitotic checkpoint to kill tumor cells. Horm. Cancer 2011, 2, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.; Karemore, G.; Gudjonsson, T.; Rask, M.B.; Neumann, B.; Heriche, J.K.; Pepperkok, R.; Ellenberg, J.; Gerlich, D.W.; Lukas, J.; et al. Profiling DNA damage response following mitotic perturbations. Nat. Commun. 2016, 7, 13887. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nahse-Kumpf, V.; Larsen, M.S.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuasen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.K.; Jodkowska, K.; Teloni, F.; Bizard, A.H.; Zellweger, R.; Herrador, R.; Ortega, S.; Hickson, I.D.; Altmeyer, M.; Mendez, J.; et al. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 2016, 7, 10660. [Google Scholar] [CrossRef] [PubMed]
- Mullers, E.; Silva Cascales, H.; Burdova, K.; Macurek, L.; Lindqvist, A. Residual CDK1/2 activity after DNA damage promotes senescence. Aging Cell 2017, 16, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Overton, K.W.; Spencer, S.L.; Noderer, W.L.; Meyer, T.; Wang, C.L. Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proc. Natl. Acad. Sci. USA 2014, 111, E4386–E4393. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Moser, J.; Phadke, H.; Basha, A.A.; Spencer, S.L. Endogenous replication stress in mother cells leads to quiescence of daughter cells. Cell Rep. 2017, 19, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Lezaja, A.; Altmeyer, M. Inherited DNA lesions determine G1 duration in the next cell cycle. Cell Cycle 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Hales, C.N.; Ozanne, S.E. DNA damage, cellular senescence and organismal ageing: Causal or correlative? Nucleic Acids Res. 2007, 35, 7417–7428. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; Beausejour, C.; d’Adda di Fagagna, F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 2017, 16, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, B.; Ben-Zimra, M.; Simon, I. Perturbations in the replication program contribute to genomic instability in cancer. Int. J. Mol. Sci. 2017, 18, 1138. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Naim, V. Rescue from replication stress during mitosis. Cell Cycle 2017, 16, 613–633. [Google Scholar] [CrossRef] [PubMed]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Miura, S.; Kurashige, T.; Suzuki, K.; Kondo, H.; Ihara, M.; Nakajima, H.; Masuzaki, H.; Nakashima, M. Significance of p53-binding protein 1 nuclear foci in uterine cervical lesions: Endogenous DNA double strand breaks and genomic instability during carcinogenesis. Histopathology 2011, 59, 441–451. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Misregulation | Gene | Functions Related to 53BP1 NBs Regulation | References |
|---|---|---|---|
| RNAi | ANLN | Cytokinesis regulation | [125] |
| RNAi; KO | ATMIN | ATM activation | [112] |
| RNAi; inhibitor | ATR | Kinase; RS | [20,23,77] |
| RNAi | BLM | RF stability and restart; HR | [23,108] |
| RNAi | BOD1L | RF protection | [80] |
| RNAi | BRCA1 | HR regulation | [116] |
| RNAi; KO | BRCA2 | HR, protection of stalled RF | [23,106,115] |
| RNAi | CDKN1A | Checkpoint protein | [93] |
| RNAi | CDT1 | Initiation of replication | [38] |
| RNAi | CHK1 | RS signaling | [23] |
| RNAi | CTCF | Insulator protein; regulator of chromatin structure | [49] |
| RNAi | EME1 | Regulatory subunit of the MUS81-EME1 nuclease; RF restart, HR, MiDAS | [86,97] |
| RNAi | ERCC1 | Regulatory subunit of the ERCC1-XPF nuclease; NER, ICL repair, SSA, HR | [86] |
| KO | ETAA1 | ATR activation | [78] |
| RNAi | FANCA | FA pathway | [80] |
| KO | FANCC | FA pathway | [73,81] |
| RNAi | FANCM | FA pathway | [79] |
| RNAi | FBXO5 | Ubiquitin ligase complex; mitotic regulator | [23] |
| Overexpression, L209P substitution | FEN1 | Nuclease; BER, processing of Okasaki fragments | [120,121] |
| RNAi | GEN1 | Resolvase; HR | [103] |
| RNAi | HP1 | Heterochromatin factor | [50] |
| Overexpression | HRAS | Control of cell proliferation | [109] |
| F345I substitution | MCM4 | Initiation of replication | [73] |
| RNAi | MCM5 | Initiation of replication | [38] |
| RNAi | MCM10 | Initiation of replication | [23] |
| RNAi | MEX3C | RNA-binding ubiquitin E3 ligase; control of mRNA translation and degradation | [69] |
| RNAi | MRE11 | Nuclease; DSB signaling and repair, RF restart | [113] |
| RNAi | MUS81 | Nuclease; HR, RF restart, MiDAS | [85,86,97,103,104,106] |
| RNAi | PALB2 | HR | [23] |
| RNAi | PICH | DNA translocase; UFB resolution | [108] |
| RNAi | PIGN | Biosynthetic pathway of the glycosylphosphatidylinositol (GPI) anchor | [69] |
| Inhibitor | PLK1 | Kinase; cell cycle regulator, control of chromosome segregation | [97] |
| RNAi | POLD3 | Replication, BER, MMR, DSBR, NER, MiDAS | [97] |
| RNAi | POLH | TLS, SHM, BER, replication | [87,91] |
| RNAi | POLK | TLS, NER, replication | [92] |
| RNAi | RAD18 | TLS | [91] |
| RNAi | RAD51 | HR, RF protection | [23] |
| RNAi | RAD51C | RAD51 paralog; HR, RF protection | [117] |
| RNAi | RAD52 | ssDNA annealing, backup HR pathway, MiDAS | [100] |
| RNAi | RECQ5 | Helicase; HR regulator, RAD51 nucleofilament disruption | [104] |
| KO | REV3 | Pol catalytic subunit; TLS, DSBR, FA pathway, SHM | [90] |
| RNAi | RIF1 | NHEJ, replication timing | [108] |
| RNAi | RPA | Replication, RS signaling, HR, DNA repair | [23] |
| Inhibitor | RRM2 | Ribonucleotide reductase | [116] |
| RNAi | SLX1 | Resolvase; HR, RF restart, ICL repair | [103] |
| RNAi, KO | SLX4 | Docking platform for nucleases; ICL repair, RF restart, MiDAS | [97,102,103] |
| RNAi (in G2/M) | SMC2 | Regulator of chromosome condensation | [97] |
| KO | SPARTAN | Protease; DNA-protein crosslink repair, TLS | [123] |
| RNAi | TOP2A | Topoisomerase; relief of torsional stress, chromatid separation | [23] |
| RNAi | TOPBP1 | Initiation of replication, ATR activation, HR regulation | [23,41] |
| RNAi | TRIP12 | E3 ubiquitin ligase; proteolysis, regulator of RNF168-mediated chromatin ubiquitylation | [42] |
| Inhibitor | UBA1 | E1 ubiquitin ligase; regulator of chromatin ubiquitylation | [111] |
| RNAi | UBR5 | E3 ligase; proteolysis, regulator of RNF168-mediated chromatin ubiquitylation | [42] |
| RNAi | USP1 | Regulator of TLS, FA pathway, HR | [94,95] |
| RNAi | WALP | Regulator of sister chromatid arm cohesion | [97] |
| Inhibitor | WEE1 | Kinase; G2/M checkpoint activation, regulator of replication initiation | [126] |
| Overexpression | XPG | Nuclease; NER | [121] |
| RNAi | XRCC2 | RAD51 paralog, HR, RF protection | [117] |
| RNAi | XRCC3 | RAD51 paralog, HR, RF protection | [117] |
| RNAi | ZNF516 | Transcription activation and repression | [69] |
| Misregulation | Gene | Functions Related to 53BP1 NBs Regulation | References |
|---|---|---|---|
| RNAi | 53BP1 | DSBR mediator | [23] |
| RNAi; inhibitor | ATM | Kinase; DSB signaling | [20,23] |
| RNAi | ATR | Kinase; RS signaling | [41] |
| RNAi; KO | ATMIN | ATM activation | [112] |
| Overexpression | CDC6 | Initiation of replication | [38] |
| KO | H2AX | Histone variant; DSB signaling | [20] |
| RNAi | NUP153 | Nucleoporin; 53BP1 nuclear import | [111] |
| Overexpression | RNASEH1 | R-loop degradation | [45] |
| RNAi | SMC2 | Regulator of chromosome condensation | [23] |
| Overexpression | WIP1 | PP2C family phosphatase; DDR regulator | [45] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Vidal, A.; Vignard, J.; Mirey, G. Around and beyond 53BP1 Nuclear Bodies. Int. J. Mol. Sci. 2017, 18, 2611. https://doi.org/10.3390/ijms18122611
Fernandez-Vidal A, Vignard J, Mirey G. Around and beyond 53BP1 Nuclear Bodies. International Journal of Molecular Sciences. 2017; 18(12):2611. https://doi.org/10.3390/ijms18122611
Chicago/Turabian StyleFernandez-Vidal, Anne, Julien Vignard, and Gladys Mirey. 2017. "Around and beyond 53BP1 Nuclear Bodies" International Journal of Molecular Sciences 18, no. 12: 2611. https://doi.org/10.3390/ijms18122611
APA StyleFernandez-Vidal, A., Vignard, J., & Mirey, G. (2017). Around and beyond 53BP1 Nuclear Bodies. International Journal of Molecular Sciences, 18(12), 2611. https://doi.org/10.3390/ijms18122611