The Genetic Basis of Psoriasis

Abstract
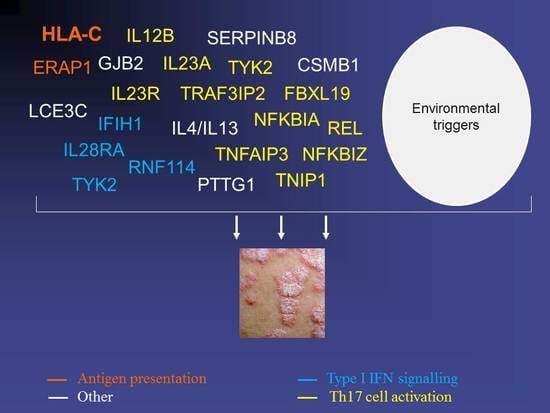
1. The Multifactorial Inheritance of Psoriasis
2. The Conflicting Results of Linkage Studies
3. PSORS1, the Major Genetic Determinant of Psoriasis
4. Additional MHC Associations
5. The PSORS2 Locus
6. The PSORS4 Locus
7. The Advent of Genome-Wide Association Studies
8. Pathogenic Insights Afforded by Large-Scale Association Studies
9. Beyond GWAS
10. Rare Clinical Variants of Psoriasis Have a Distinctive Genetic Basis
11. Conclusions and Outlook
Conflicts of Interest
References
- Duffy, D.L.; Spelman, L.S.; Martin, N.G. Psoriasis in Australian twins. J. Am. Acad. Dermatol. 1993, 29, 428–434. [Google Scholar] [CrossRef]
- Generali, E.; Ceribelli, A.; Stazi, M.A.; Selmi, C. Lessons learned from twins in autoimmune and chronic inflammatory diseases. J. Autoimmun. 2017, 83, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Burden, A.D.; Kirby, B. Psoriasis and related disorders. In Rook’s Textbook of Dermatology; Griffiths, C.E.M., Barker, J.N., Bleiker, T., Chalmers, R.J., Creamer, D., Eds.; Wiley-Blackwell: Chichester, UK, 2016. [Google Scholar]
- International Psoriasis Genetics Consortium. The International Psoriasis Genetics Study: Assessing linkage to 14 candidate susceptibility loci in a cohort of 942 affected sib pairs. Am. J. Hum. Genet. 2003, 73, 430–437. [Google Scholar]
- Enlund, F.; Samuelsson, L.; Enerback, C.; Inerot, A.; Wahlstrom, J.; Yhr, M.; Torinsson, A.; Martinsson, T.; Swanbeck, G. Analysis of three suggested psoriasis susceptibility loci in a large Swedish set of families: Confirmation of linkage to chromosome 6p (HLA region), and to 17q, but not to 4q. Hum. Hered. 1999, 49, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Tomfohrde, J.; Silverman, A.; Barnes, R.; Fernandez-Vina, M.A.; Young, M.; Lory, D.; Morris, L.; Wuepper, K.D.; Stastny, P.; Menter, A.; et al. Gene for familial psoriasis susceptibility mapped to the distal end of human chromosome 17q. Science 1994, 264, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Capon, F.; Novelli, G.; Semprini, S.; Clementi, M.; Nudo, M.; Vultaggio, P.; Mazzanti, C.; Gobello, T.; Botta, A.; Fabrizi, G.; et al. Searching for psoriasis susceptibility genes in Italy: Genome scan and evidence for a new locus on chromosome 1. J. Investig. Dermatol. 1999, 112, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mathur, P.; Dupuis, J.; Tizard, R.; Ticho, B.; Crowell, T.; Gardner, H.; Bowcock, A.M.; Carulli, J. Peptidoglycan recognition proteins Pglyrp3 and Pglyrp4 are encoded from the epidermal differentiation complex and are candidate genes for the PSORS4 locus on chromosome 1q21. Hum. Genet. 2006, 119, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Balendran, N.; Clough, R.L.; Arguello, J.R.; Barber, R.; Veal, C.; Jones, A.B.; Rosbotham, J.L.; Little, A.M.; Madrigal, A.; Barker, J.N.; et al. Characterization of the major susceptibility region for psoriasis at chromosome 6p21.3. J. Investig. Dermatol. 1999, 113, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Stuart, P.E.; Nistor, I.; Hiremagalore, R.; Chia, N.V.; Jenisch, S.; Weichenthal, M.; Abecasis, G.R.; Lim, H.W.; Christophers, E.; et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am. J. Hum. Genet. 2006, 78, 827–851. [Google Scholar] [CrossRef] [PubMed]
- Capon, F.; Munro, M.; Barker, J.; Trembath, R. Searching for the major histocompatibility complex psoriasis susceptibility gene. J. Investig. Dermatol. 2002, 118, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Mallon, E.; Newson, R.; Bunker, C.B. HLA-Cw6 and the genetic predisposition to psoriasis: A meta-analysis of published serologic studies. J. Investig. Dermatol. 1999, 113, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Jonca, N.; Leclerc, E.A.; Caubet, C.; Simon, M.; Guerrin, M.; Serre, G. Corneodesmosomes and corneodesmosin: From the stratum corneum cohesion to the pathophysiology of genodermatoses. Eur. J. Dermatol. 2011, 21 (Suppl. S2), 35–42. [Google Scholar] [PubMed]
- Strange, A.; Capon, F.; Spencer, C.C.; Knight, J.; Weale, M.E.; Allen, M.H.; Barton, A.; Band, G.; Bellenguez, C.; Bergboer, J.G.; et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, A.; Siewert, K.; Stohr, J.; Besgen, P.; Kim, S.M.; Ruhl, G.; Nickel, J.; Vollmer, S.; Thomas, P.; Krebs, S.; et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 2015, 212, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, T.; Hirayama, N. Binding affinity and interaction of LL-37 with HLA-C*06:02 in psoriasis. J. Investig. Dermatol. 2016, 136, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Hamm, D.; Gulati, N.; Lowes, M.A.; Krueger, J.G. Deep sequencing of the T-cell receptor repertoire demonstrates polyclonal T-cell infiltrates in psoriasis. F1000Res. 2015, 4, 460. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Bertoni, A.; Spain, S.L.; Simpson, M.A.; Pullabhatla, V.; Tonda, R.; Hundhausen, C.; Di Meglio, P.; de Jong, P.; Hayday, A.C.; et al. An in-depth characterization of the major psoriasis susceptibility locus identifies candidate susceptibility alleles within an HLA-C enhancer element. PLoS ONE 2013, 8, e71690. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.; Spain, S.L.; Capon, F.; Hayday, A.; Nestle, F.O.; Clop, A.; Barker, J.N.; Weale, M.E.; Trembath, R.C. Conditional analysis identifies three novel major histocompatibility complex loci associated with psoriasis. Hum. Mol. Genet. 2012, 21, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Han, B.; Tsoi, L.C.; Stuart, P.E.; Ellinghaus, E.; Tejasvi, T.; Chandran, V.; Pellett, F.; Pollock, R.; Bowcock, A.M.; et al. Fine mapping major histocompatibility complex associations in psoriasis and its clinical subtypes. Am. J. Hum. Genet. 2014, 95, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Cao, H.; Zuo, X.; Zhang, T.; Zhang, X.; Liu, X.; Xu, R.; Chen, G.; Zhang, Y.; Zheng, X.; et al. Deep sequencing of the MHC region in the Chinese population contributes to studies of complex disease. Nat. Genet. 2016, 48, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Yang, C.F.; Fann, C.S.; Chen, C.L.; Tsai, T.F.; Chien, Y.H.; Chiang, S.C.; Chen, C.H.; Hung, S.I.; Wu, J.Y.; et al. Mapping of psoriasis to 17q terminus. J. Med. Genet. 2005, 42, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Fuchs-Telem, D.; Sarig, O.; van Steensel, M.A.; Isakov, O.; Israeli, S.; Nousbeck, J.; Richard, K.; Winnepenninckx, V.; Vernooij, M.; Shomron, N.; et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet. 2012, 91, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Berki, D.M.; Liu, L.; Choon, S.E.; David Burden, A.; Griffiths, C.E.; Navarini, A.A.; Tan, E.S.; Irvine, A.D.; Ranki, A.; Ogo, T.; et al. Activating CARD14 mutations are associated with generalized pustular psoriasis but rarely account for familial recurrence in psoriasis vulgaris. J. Investig. Dermatol. 2015, 135, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Muto, M.; Akiyama, M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J. Investig. Dermatol. 2014, 134, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, I.; Zotti, T.; Ferravante, A.; Vessichelli, M.; Vito, P.; Stilo, R. Alternative splicing of CARMA2/CARD14 transcripts generates protein variants with differential effect on NF-κB activation and endoplasmic reticulum stress-induced cell death. J. Cell. Physiol. 2011, 226, 3121–3131. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.Y.; de Guzman Strong, C. The molecular revolution in cutaneous biology: EDC and locus control. J. Investig. Dermatol. 2017, 137, e101–e104. [Google Scholar] [CrossRef] [PubMed]
- De Cid, R.; Riveira-Munoz, E.; Zeeuwen, P.L.; Robarge, J.; Liao, W.; Dannhauser, E.N.; Giardina, E.; Stuart, P.E.; Nair, R.; Helms, C.; et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 2009, 41, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Riveira-Munoz, E.; He, S.M.; Escaramis, G.; Stuart, P.E.; Huffmeier, U.; Lee, C.; Kirby, B.; Oka, A.; Giardina, E.; Liao, W.; et al. Meta-analysis confirms the LCE3C_LCE3B deletion as a risk factor for psoriasis in several ethnic groups and finds interaction with HLA-Cw6. J. Investig. Dermatol. 2011, 131, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Cargill, M.; Schrodi, S.J.; Chang, M.; Garcia, V.E.; Brandon, R.; Callis, K.P.; Matsunami, N.; Ardlie, K.G.; Civello, D.; Catanese, J.J.; et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 2007, 80, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, E.; Ellinghaus, D.; Stuart, P.E.; Nair, R.P.; Debrus, S.; Raelson, J.V.; Belouchi, M.; Fournier, H.; Reinhard, C.; Ding, J.; et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010, 42, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Stuart, P.E.; Nair, R.P.; Ellinghaus, E.; Ding, J.; Tejasvi, T.; Gudjonsson, J.E.; Li, Y.; Weidinger, S.; Eberlein, B.; Gieger, C.; et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010, 42, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Stuart, P.E.; Tian, C.; Gudjonsson, J.E.; Das, S.; Zawistowski, M.; Ellinghaus, E.; Barker, J.N.; Chandran, V.; Dand, N.; et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat. Commun. 2017, 8, 15382. [Google Scholar] [CrossRef] [PubMed]
- Dand, N.; Mucha, S.; Tsoi, L.C.; Mahil, S.K.; Stuart, P.E.; Arnold, A.; Baurecht, H.; Burden, A.D.; Duffin, K.C.; Chandran, V.; et al. Exome-wide association study reveals novel psoriasis susceptibility locus at TNFSF15 and rare protective alleles in genes contributing to type I IFN signalling. Hum. Mol. Genet. 2017, 26, 4301–4313. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, P.; di Cesare, A.; Laggner, U.; Chu, C.C.; Napolitano, L.; Villanova, F.; Tosi, I.; Capon, F.; Trembath, R.C.; Peris, K.; et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE 2011, 6, e17160. [Google Scholar] [CrossRef] [PubMed]
- Capon, F.; Bijlmakers, M.J.; Wolf, N.; Quaranta, M.; Huffmeier, U.; Allen, M.; Timms, K.; Abkevich, V.; Gutin, A.; Smith, R.; et al. Identification of ZNF313/RNF114 as a novel psoriasis susceptibility gene. Hum. Mol. Genet. 2008, 17, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Spain, S.L.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Knight, J.; Tejasvi, T.; Kang, H.M.; Allen, M.H.; Lambert, S.; et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat. Commun. 2015, 6, 7001. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Krueger, J.G.; Guttman-Yassky, E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J. Allergy Clin. Immunol. 2015, 135, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Jabbar-Lopez, Z.K.; Yiu, Z.Z.N.; Ward, V.; Exton, L.S.; Mohd Mustapa, M.F.; Samarasekera, E.; Burden, A.D.; Murphy, R.; Owen, C.M.; Parslew, R.; et al. Quantitative Evaluation of Biologic Therapy Options for Psoriasis: A Systematic Review and Network Meta-Analysis. J. Investig. Dermatol. 2017, 137, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.A.; Mistry, V.; Bockett, N.A.; Ahmad, T.; Ban, M.; Barker, J.N.; Barrett, J.C.; Blackburn, H.; Brand, O.; Burren, O.; et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 2013, 498, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Hollox, E.J.; Huffmeier, U.; Zeeuwen, P.L.; Palla, R.; Lascorz, J.; Rodijk-Olthuis, D.; van de Kerkhof, P.C.; Traupe, H.; de Jongh, G.; den Heijer, M.; et al. Psoriasis is associated with increased β-defensin genomic copy number. Nat. Genet. 2008, 40, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Asumalahti, K.; Ameen, M.; Suomela, S.; Hagforsen, E.; Michaelsson, G.; Evans, J.; Munro, M.; Veal, C.; Allen, M.; Leman, J.; et al. Genetic analysis of PSORS1 distinguishes guttate psoriasis and palmoplantar pustulosis. J. Investig. Dermatol. 2003, 120, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Marsland, A.M.; Chalmers, R.J.; Hollis, S.; Leonardi-Bee, J.; Griffiths, C.E. Interventions for chronic palmoplantar pustulosis. Cochrane Database Syst. Rev. 2006, CD001433. [Google Scholar] [CrossRef]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D.; et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Berki, D.M.; Choon, S.E.; Burden, A.D.; Allen, M.H.; Arostegui, J.I.; Chaves, A.; Duckworth, M.; Irvine, A.D.; Mockenhaupt, M.; et al. IL36RN mutations define a severe auto-inflammatory phenotype of generalized pustular psoriasis. J. Allergy Clin. Immunol. 2015, 135, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.E.; et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Twelves, S.; Farkas, K.; Setta-Kaffetzi, N.; Burden, A.D.; Gach, J.E.; Irvine, A.D.; Kepiro, L.; Mockenhaupt, M.; Oon, H.H.; et al. AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J. Investig. Dermatol. 2016, 136, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Navarini, A.A.; Burden, A.D.; Capon, F.; Mrowietz, U.; Puig, L.; Koks, S.; Kingo, K.; Smith, C.; Barker, J.N.; network, E. European consensus statement on phenotypes of pustular psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Catapano, M.; di Meglio, P.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Trans. Med. 2017, 9, eaan2514. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt, L.J.; van den Reek, J.; Coenen, M.J.H.; de Jong, E. A systematic review of pharmacogenetic studies on the response to biologics in psoriasis patients. Br. J. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Collins, R. What makes UK Biobank special? Lancet 2012, 379, 1173–1174. [Google Scholar] [CrossRef]
- Check Hayden, E. The rise and fall and rise again of 23andMe. Nature 2017, 550, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Biasci, D.; Roberts, R.; Gearry, R.B.; Mansfield, J.C.; Ahmad, T.; Prescott, N.J.; Satsangi, J.; Wilson, D.C.; Jostins, L.; et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat. Genet. 2017, 49, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaci, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288. [Google Scholar] [CrossRef]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Name (Trade Name) | Target (Genetic Association) | Development Stage |
|---|---|---|
| Ustekinumab (Stelara) | p40 subunit shared by IL-12 and IL-23 (SNPs in IL12B, which encodes p40, are associated with psoriasis susceptibility [14,33]) | Approved for the treatment of moderate to severe psoriasis |
| Secukinumab (Cosentyx) | IL-17A (No risk alleles in IL17A, but psoriasis associated SNPs have been identified in TRAF3IP2, which encodes an IL-17 receptor adaptor [14,34]) | Approved for the treatment of moderate to severe psoriasis |
| Ixekizumab (Taltz) | IL-17A (No risk alleles in IL17A, but psoriasis associated SNPs have been identified in TRAF3IP2, which encodes an IL-17 receptor adaptor [14,34]) | Approved for the treatment of moderate to severe psoriasis |
| Broadalumab (Kyntheum) | IL-17 receptor subunit A (IL-17RA) (No risk alleles in IL17RA, but psoriasis associated SNPs have been identified in TRAF3IP2, which encodes an IL-17 receptor adaptor [14,34]) | Approved for the treatment of moderate to severe psoriasis |
| Guselkumab (Tremfya) | p19 subunit unique to IL-23 (SNPs in IL23A, which encodes p19, are associated with psoriasis susceptibility [14,35]) | Approved for the treatment of moderate to severe psoriasis |
| Tildrakizumab | p19 subunit unique to IL-23 (SNPs in IL23A, which encodes p19, are associated with psoriasis susceptibility [14,35]) | Phase III trial successfully completed [60] |
| Risankizumab | p19 subunit unique to IL-23 (SNPs in IL23A, which encodes p19, are associated with psoriasis susceptibility [14,35]) | Phase II trial successfully completed [61] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capon, F. The Genetic Basis of Psoriasis. Int. J. Mol. Sci. 2017, 18, 2526. https://doi.org/10.3390/ijms18122526
Capon F. The Genetic Basis of Psoriasis. International Journal of Molecular Sciences. 2017; 18(12):2526. https://doi.org/10.3390/ijms18122526
Chicago/Turabian StyleCapon, Francesca. 2017. "The Genetic Basis of Psoriasis" International Journal of Molecular Sciences 18, no. 12: 2526. https://doi.org/10.3390/ijms18122526
APA StyleCapon, F. (2017). The Genetic Basis of Psoriasis. International Journal of Molecular Sciences, 18(12), 2526. https://doi.org/10.3390/ijms18122526