Zinc Signal in Brain Diseases

Abstract
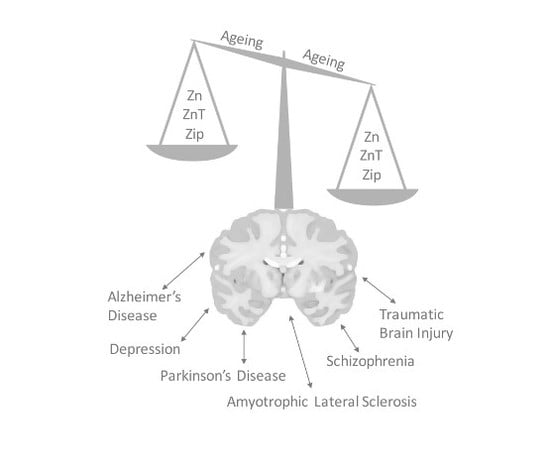
:
{kind=link}
1. Introduction
2. Zinc in the Brain
3. Metallothioneins
4. Zinc- and Iron-like Regulatory Proteins
5. Zinc Transporter Proteins
6. Zinc Signal in Brain Diseases
6.1. Alzheimer’s Disease
6.2. Amyotrophic Lateral Sclerosis
6.3. Traumatic Brain Injury
6.4. Depression
6.5. Schizophrenia
6.6. Parkinson’s Disease
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lao, X.Y.; Sun, T.T.; Ren, L.L.; Kong, X.; Wang, J.L.; Wang, Y.C.; Du, W.; Yu, Y.N.; Weng, Y.R.; et al. Knockdown of ZFX inhibits gastric cancer cell growth in vitro and in vivo via downregulating the ERK-MAPK pathway. Cancer Lett. 2013, 337, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.F.; Briggs, K.K.; Krebs, N.F.; Hambidge, K.M. Zinc supplementation selectively decreases fetal hepatocyte DNA synthesis and insulin-like growth factor II gene expression in primary culture. Pediatr. Res. 1994, 35, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.; Millward, D.J. The effects of severe zinc deficiency on protein turnover in muscle and thymus. Br. J. Nutr. 1987, 57, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.I.; Phillips, T.J. Patients with chronic leg ulcers show diminished levels of vitamins A and E, carotenes, and zinc. Dermatol. Surg. 1999, 25, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.W.; Prasad, A.S.; Kaplan, J.; Fitzgerald, J.T.; Brewer, G.J. Changes in cytokine production and T cell subpopulations in experimentally induced zinc-deficient humans. Am. J. Physiol. 1997, 272, E1002–E1007. [Google Scholar] [PubMed]
- Takeda, A. Significance of Zn(2+) signaling in cognition: Insight from synaptic Zn(2+) dyshomeostasis. J. Trace Elem. Med. Biol. 2014, 28, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Wessells, K.R.; Brown, K.H. Estimating the global prevalence of zinc deficiency: Results based on zinc availability in national food supplies and the prevalence of stunting. PLoS ONE 2012, 7, e50568. [Google Scholar] [CrossRef] [PubMed]
- Bertoni-Freddari, C.; Fattoretti, P.; Casoli, T.; Di Stefano, G.; Giorgetti, B.; Balietti, M. Brain aging: The zinc connection. Exp. Gerontol. 2008, 43, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Giacconi, R.; Cipriano, C.; Muzzioli, M.; Fattoretti, P.; Bertoni-Freddari, C.; Isani, G.; Zambenedetti, P.; Zatta, P. Zinc-bound metallothioneins as potential biological markers of ageing. Brain Res. Bull. 2001, 55, 147–153. [Google Scholar] [CrossRef]
- Nuttall, J.R.; Oteiza, P.I. Zinc and the aging brain. Genes Nutr. 2014, 9, 379. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B. Zinc homeostasis and neurodegenerative disorders. Front. Aging Neurosci. 2013, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration: Role of zinc ion availability. Prog. Neurobiol. 2005, 75, 367–390. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Frederickson, C.J.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [PubMed]
- Wang, Z.Y.; Li, J.Y.; Danscher, G.; Dahlstrom, A. Localization of zinc-enriched neurons in the mouse peripheral sympathetic system. Brain Res. 2002, 928, 165–174. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Cousins, R.J. Mammalian zinc transporters. Annu. Rev. Nutr. 2004, 24, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Manso, Y.; Adlard, P.A.; Carrasco, J.; Vasak, M.; Hidalgo, J. Metallothionein and brain inflammation. J. Biol. Inorg. Chem. 2011, 16, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthy, N.; Sunder, A.S.; Kumar, K.T.M.; Kumar, M.S.; Ganesh, G.N.K.; Chatterjee, M. A Review of Metallothionein Isoforms and their Role in Pathophysiology. World J. Surg. Oncol. 2011, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Cousins, R.J.; Liuzzi, J.P.; Lichten, L.A. Mammalian zinc transport, trafficking, and signals. J. Biol. Chem. 2006, 281, 24085–24089. [Google Scholar] [CrossRef] [PubMed]
- Levenson, C.W.; Tassabehji, N.M. Role and Regulation of Copper and Zinc Transport Proteins in the Central Nervous System. In Handbook of Neurochemistry and Molecular Neurobiology; Abel, L., Reith, M.E.A., Eds.; Springer: New York, NY, USA, 2007; pp. 257–284. [Google Scholar]
- Hancock, S.M.; Bush, A.I.; Adlard, P.A. The clinical implications of impaired zinc signaling in the brain. In Zinc Signals in Cellular Functions and Disorders; Springer: Tokyo, Janpan, 2014. [Google Scholar]
- Coudray, N.; Valvo, S.; Hu, M.; Lasala, R.; Kim, C.; Vink, M.; Zhou, M.; Provasi, D.; Filizola, M.; Tao, J.; et al. Inward-facing conformation of the zinc transporter YiiP revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Findley, S.D. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 1995, 14, 639–649. [Google Scholar] [PubMed]
- Tsuda, M.; Imaizumi, K.; Katayama, T.; Kitagawa, K.; Wanaka, A.; Tohyama, M.; Takagi, T. Expression of zinc transporter gene, ZnT-1, is induced after transient forebrain ischemia in the gerbil. J. Neurosci. 1997, 17, 6678–6684. [Google Scholar] [PubMed]
- Lichten, L.A.; Cousins, R.J. Mammalian zinc transporters: Nutritional and physiologic regulation. Annu. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H. Proposed glucocorticoid-mediated zinc signaling in the hippocampus. Metallomics 2012, 4, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.; Chorin, E.; Sekler, I.; Silverman, W.F.; Atkin, S.; Russell, J.T.; Hershfinkel, M. Synaptically Released Zinc Triggers Metabotropic Signaling via a Zinc-Sensing Receptor in the Hippocampus. J. Neurosci. 2009, 29, 2890–2901. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Pan, E.; Xiong, Z.Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy Fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at Glutamatergic Synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, Y.J.; Kim, T.Y.; Koh, J.Y.; Kim, Y.H. Essential Role for Zinc-Triggered p75(NTR) Activation in Preconditioning Neuroprotection. J. Neurosci. 2008, 28, 10919–10927. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Rink, L. Zinc Signaling. In Zinc in Human Health; IOS Press: Amsterdam, The Netherlands, 2011; pp. 94–117. [Google Scholar]
- Fujise, Y.; Kubota, M.; Suzuki, M.; Tamano, H.; Takeda, A. Blockade of intracellular Zn2+ signaling in the basolateral amygdala affects object recognition memory via attenuation of dentate gyrus LTP. Neurochem. Int. 2017, 108, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Koike, Y.; Osaw, M.; Tamano, H. Characteristic of Extracellular Zn2+ Influx in the Middle-Aged Dentate Gyrus and Its Involvement in Attenuation of LTP. Mol. Neurobiol. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Suzuki, M.; Tempaku, M.; Ohashi, K.; Tamano, H. Influx of extracellular Zn(2+) into the hippocampal CA1 neurons is required for cognitive performance via long-term potentiation. Neuroscience 2015, 304, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Hisatsune, M.; Murakami, T.; Nakada, H.; Fujii, H. Maintained LTP and Memory Are Lost by Zn2+ Influx into Dentate Granule Cells, but Not Ca2+ Influx. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Ogawa, T.; Takada, S.; Nakamura, M.; Fujii, H.; Ando, M. Intracellular Zn2+ Signaling in the Dentate Gyrus Is Required for Object Recognition Memory. Hippocampus 2014, 24, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Minamino, T.; Fujii, H.; Takada, S.; Nakamura, M.; Ando, M.; Takeda, A. Blockade of intracellular Zn2+ signaling in the dentate gyrus erases recognition memory via impairment of maintained LTP. Hippocampus 2015, 25, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Beyer, N.; Coulson, D.T.; Heggarty, S.; Ravid, R.; Irvine, G.B.; Hellemans, J.; Johnston, J.A. ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain. Mol. Neurodegener. 2009, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, T.J.; Blehm, C.J.; Bamberg, D.P.; Fonseca, T.L.R.; Tisser, L.A.; de Oliveira, A.A.; de Andrade, F.M.; Fiegenbaum, M. The effects of interactions between selenium and zinc serum concentration and SEP15 and SLC30A3 gene polymorphisms on memory scores in a population of mature and elderly adults. Genes Nutr. 2014, 9, 377. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Tanzi, R.E. The galvanization of β-amyloid in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 7317–7319. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Kawai, H.; Metherate, R.; Glabe, C.G.; Busciglio, J. A role for synaptic zinc in activity-dependent Aβ oligomer formation and accumulation at excitatory synapses. J. Neurosci. 2009, 29, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Maynard, C.J.; Bush, A.I.; Masters, C.L.; Cappai, R.; Li, Q.X. Metals and amyloid-β in Alzheimer’s disease. Int. J. Exp. Pathol. 2005, 86, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Multhaup, G.; Moir, R.D.; Williamson, T.G.; Small, D.H.; Rumble, B.; Pollwein, P.; Beyreuther, K.; Masters, C.L. A novel zinc(II) binding site modulates the function of the β A4 amyloid protein precursor of Alzheimer’s disease. J. Biol. Chem. 1993, 268, 16109–16112. [Google Scholar] [PubMed]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Linkous, D.H.; Adlard, P.A.; Wanschura, P.B.; Conko, K.M.; Flinn, J.M. The effects of enhanced zinc on spatial memory and plaque formation in transgenic mice. J. Alzheimers Dis. 2009, 18, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Railey, A.M.; Groeber, C.M.; Flinn, J.M. The effect of metals on spatial memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 24, 375–381. [Google Scholar] [PubMed]
- Stoltenberg, M.; Bush, A.I.; Bach, G.; Smidt, K.; Larsen, A.; Rungby, J.; Lund, S.; Doering, P.; Danscher, G. Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 2007, 150, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wang, T.; Zheng, W.; Zhao, B.L.; Danscher, G.; Chen, Y.H.; Wang, Z.Y. Zinc overload enhances APP cleavage and Aβ deposition in the Alzheimer mouse brain. PLoS ONE 2010, 5, e15349. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Savva, M.S.; Hung, L.W.; Donnelly, P.S.; Mot, A.I.; Parker, S.J.; Greenough, M.A.; Volitakis, I.; Adlard, P.A.; Cherny, R.A.; et al. The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 2011, 119, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Jeng, J.M.; Malavolta, M.; Mocchegiani, E.; Sensi, S.L. Zinc dyshomeostasis: A key modulator of neuronal injury. J. Alzheimers Dis. 2005, 8, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Clements, A.; Allsop, D.; Walsh, D.M.; Williams, C.H. Aggregation and metal-binding properties of mutant forms of the amyloid Aβ peptide of Alzheimer’s disease. J. Neurochem. 1996, 66, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.; Pereira, C.; Santos, M.S.; Oliveira, C. Effect of zinc ions on the cytotoxicity induced by the amyloid β-peptide. Antioxid. Redox Signal. 2000, 2, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Danscher, G.; Jensen, K.B.; Frederickson, C.J.; Kemp, K.; Andreasen, A.; Juhl, S.; Stoltenberg, M.; Ravid, R. Increased amount of zinc in the hippocampus and amygdala of Alzheimer’s diseased brains: A proton-induced X-ray emission spectroscopic analysis of cryostat sections from autopsy material. J. Neurosci. Methods 1997, 76, 53–59. [Google Scholar] [CrossRef]
- Andrasi, E.; Farkas, E.; Gawlik, D.; Rosick, U.; Bratter, P. Brain Iron and Zinc Contents of German Patients with Alzheimer Disease. J. Alzheimers Dis. 2000, 2, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.M.; Reynolds, G.P.; Ward, N.I. Hippocampal tin, aluminum and zinc in Alzheimer’s disease. Biometals 1993, 6, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Rulon, L.L.; Robertson, J.D.; Lovell, M.A.; Deibel, M.A.; Ehmann, W.D.; Markesber, W.R. Serum zinc levels and Alzheimer’s disease. Biol. Trace Elem. Res. 2000, 75, 79–85. [Google Scholar] [CrossRef]
- Kim, I.; Park, E.J.; Seo, J.; Ko, S.J.; Lee, J.; Kim, C.H. Zinc stimulates tau S214 phosphorylation by the activation of Raf/mitogen-activated protein kinase-kinase/extracellular signal-regulated kinase pathway. Neuroreport 2011, 22, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.Y.; Zhu, Y.Z.; Zhu, H.L.; Fan, J.B.; Chen, J.; Liang, Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 2009, 284, 34648–34657. [Google Scholar] [CrossRef] [PubMed]
- An, W.L.; Bjorkdahl, C.; Liu, R.; Cowburn, R.F.; Winblad, B.; Pei, J.J. Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3β in SH-SY5Y neuroblastoma cells. J. Neurochem. 2005, 92, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; An, W.L.; Zhou, X.W.; Nishimura, T.; Norberg, J.; Benedikz, E.; Gotz, J.; Winblad, B. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006, 580, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Wei, Y.P.; Xiong, Y.; Wang, X.C.; Xie, A.J.; Wang, X.L.; Yang, Y.; Wang, Q.; Lu, Y.M.; Liu, R.W.J. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J. Biol. Chem. 2012, 287, 11174–11182. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Jing, X.P.; Zhou, X.W.; Wang, X.L.; Yang, Y.; Sun, X.Y.; Qiu, M.; Cao, F.Y.; Lu, Y.M.; Liu, R.; et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol. Aging 2013, 34, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, Z.; Cao, Y.; Lang, M.; Lu, B.; Zhou, B. Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation. Cell Rep. 2014, 8, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Tanzi, R.E. The role of zinc in the cerebral deposition of Aβ amyloid in Alzheimer’s disease. In Research Advances in Alzheimer’s Disease and Related Disorders, 1st ed.; Khalid, I., James, M., Bengt, W., Henry, W., Eds.; Wiley: Ann Arbor, MI, USA, 1995; pp. 607–618. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Tainer, J.A.; Getzoff, E.D.; Malencik, D.A.; Anderson, S.R.; Bomben, V.C.; Meyers, K.R.; Karplus, P.A.; Beckman, J.S. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol. 2007, 373, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Noguchi, T.; Ikegami, S.; Sakurai, T.; Kakita, A.; Toyoshima, Y.; Kambe, T.; Yamada, M.; Inden, M.; Hara, H.; et al. Zinc transporters ZnT3 and ZnT6 are downregulated in the spinal cords of patients with sporadic amyotrophic lateral sclerosis. J. Neurosci. Res. 2015, 93, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Veenith, T.V.; Carter, E.L.; Geeraerts, T.; Grossac, J.; Newcombe, V.F.; Outtrim, J.; Gee, G.S.; Lupson, V.; Smith, R.; Aigbirhio, F.I.; et al. Pathophysiologic Mechanisms of Cerebral Ischemia and Diffusion Hypoxia in Traumatic Brain Injury. JAMA Neurol. 2016, 73, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Tonder, N.; Johansen, F.F.; Frederickson, C.J.; Zimmer, J.; Diemer, N.H. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci. Lett. 1990, 109, 247–252. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Hernandez, M.D.; McGinty, J.F. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res. 1989, 480, 317–321. [Google Scholar] [CrossRef]
- Coles, J.P. Regional ischemia after head injury. Curr. Opin. Crit. Care 2004, 10, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Peets, A.D.; Berthiaume, L.R.; Bagshaw, S.M.; Federico, P.; Doig, C.J.; Zygun, D.A. Prolonged refractory status epilepticus following acute traumatic brain injury: A case report of excellent neurological recovery. Crit. Care 2005, 9, R725–R728. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Chen, J.W.; Motamedi, M.; Bell, B.; Listiak, K.; Pons, N.F.; Danscher, G.; Frederickson, C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000, 852, 268–273. [Google Scholar] [CrossRef]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Doering, P.; Stoltenberg, M.; Penkowa, M.; Rungby, J.; Larsen, A.; Danscher, G. Chemical blocking of zinc ions in CNS increases neuronal damage following traumatic brain injury (TBI) in mice. PLoS ONE 2010, 5, e10131. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, H.L.; Frederickson, C.J.; DeWitt, D.S.; Saban, R.; Parsley, M.O.; Stephenson, R.; Velasco, M.; Uchida, T.; Shimamura, M.; Prough, D.S. Protective effects of zinc chelation in traumatic brain injury correlate with upregulation of neuroprotective genes in rat brain. Neurosci. Lett. 2004, 355, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Cope, E.C.; Morris, D.R.; Scrimgeour, A.G.; VanLandingham, J.W.; Levenson, C.W. Zinc supplementation provides behavioral resiliency in a rat model of traumatic brain injury. Physiol. Behav. 2011, 104, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Cope, E.C.; Morris, D.R.; Scrimgeour, A.G.; Levenson, C.W. Scrimgeour; Levenson, C.W. Use of Zinc as a Treatment for Traumatic Brain Injury in the Rat: Effects on Cognitive and Behavioral Outcomes. Neurorehabilit. Neural Repair 2012, 26, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Rowan, M.; Garner, C.C. Brain-Delivery of Zinc-Ions as Potential Treatment for Neurological Diseases: Mini Review. Drug Deliv. Lett. 2011, 1, 13–23. [Google Scholar] [PubMed]
- Maes, M.; D’Haese, P.C.; Scharpe, S.; D’Hondt, P.; Cosyns, P.; de Broe, M.E. Hypozincemia in depression. J. Affect. Disord. 1994, 31, 135–140. [Google Scholar] [CrossRef]
- Maes, M.; Vandoolaeghe, E.; Neels, H.; Demedts, P.; Wauters, A.; Meltzer, H.Y.; Altamura, C.; Desnyder, R. Lower serum zinc in major depression is a sensitive marker of treatment resistance and of the immune/inflammatory response in that illness. Biol. Psychiatry 1997, 42, 349–358. [Google Scholar] [CrossRef]
- McLoughlin, I.J.; Hodge, J.S. Zinc in depressive disorder. Acta Psychiatr. Scand. 1990, 82, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Low, C.M.; Zheng, F.; Lyuboslavsky, P.; Traynelis, S.F. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-d-aspartate NR1/NR2A receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 11062–11067. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B.; Poleszak, E.; Sowa-Kucma, M.; Siwek, M.; Dudek, D.; Ryszewska-Pokrasniewicz, B.; Radziwon-Zaleska, M.; Opoka, W.; Czekaj, J.; Pilc, A.; et al. Antidepressant activity of zinc and magnesium in view of the current hypotheses of antidepressant action. Pharmacol. Rep. 2008, 60, 588–589. [Google Scholar] [PubMed]
- Szewczyk, B.; Poleszak, E.; Sowa-Kucma, M.; Wrobel, A.; Slotwinski, S.; Listos, J.; Wlaz, P.; Cichy, A.; Siwek, A.; Dybala, M.; et al. The involvement of NMDA and AMPA receptors in the mechanism of antidepressant-like action of zinc in the forced swim test. Amino Acids 2010, 39, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Cichy, A.; Sowa-Kucma, M.; Legutko, B.; Pomierny-Chamiolo, L.; Siwek, A.; Piotrowska, A.; Szewczyk, B.; Poleszak, E.; Pilc, A.; Nowak, G. Zinc-induced adaptive changes in NMDA/glutamatergic and serotonergic receptors. Pharmacol. Rep. 2009, 61, 1184–1191. [Google Scholar] [CrossRef]
- Szewczyk, B.; Poleszak, E.; Wlaz, P.; Wrobel, A.; Blicharska, E.; Cichy, A.; Dybala, M.; Siwek, A.; Pomierny-Chamiolo, L.; Piotrowska, A.; et al. The involvement of serotonergic system in the antidepressant effect of zinc in the forced swim test. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Doboszewska, U.; Wlaz, P.; Nowak, G.; Radziwon-Zaleska, M.; Cui, R.J.; Mlyniec, K. Zinc in the Monoaminergic Theory of Depression: Its Relationship to Neural Plasticity. Neural Plast. 2017. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, M.A.; Kranz, T.M.; Kleinhaus, K.; Joe, P.; Getz, M.; Johnson, P.; Chao, M.V.; Malaspina, D. The Emerging Role for Zinc in Depression and Psychosis. Front. Pharmacol. 2017, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H. Insight into zinc signaling from dietary zinc deficiency. Brain Res. Rev. 2009, 62, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Kumura, J. Preliminary reports on the metabolismof trace elements in neuro psychiatric diseases. I. Zinc in schizophrenia. Proc. Jpn. Acad. Sci. 1965, 41, 943–947. [Google Scholar]
- McLardy, T. Hippocampal zinc in chronic alcoholism and schizophrenia. IRCS Med. Sci. 1973, 2, 1010. [Google Scholar]
- Carrera, N.; Arrojo, M.; Sanjuan, J.; Ramos-Rios, R.; Paz, E.; Suarez-Rama, J.J.; Paramo, M.; Agra, S.; Brenlla, J.; Martinez, S.; et al. Association study of nonsynonymous single nucleotide polymorphisms in schizophrenia. Biol. Psychiatry 2012, 71, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Perez-Becerril, C.; Morris, A.G.; Mortimer, A.; McKenna, P.J.; de Belleroche, J. Allelic variants in the zinc transporter-3 gene, SLC30A3, a candidate gene identified from gene expression studies, show gender-specific association with schizophrenia. Eur. Psychiatry 2014, 29, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Perez-Becerril, C.; Morris, A.G.; Mortimer, A.; McKenna, P.J.; de Belleroche, J. Common variants in the chromosome 2p23 region containing the SLC30A3 (ZnT3) gene are associated with schizophrenia in female but not male individuals in a large collection of European samples. Psychiatry Res. 2016, 246, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Udawela, M.; Greenough, M.A.; Neo, J.; Suk Seo, M.; Money, T.T.; Upadhyay, A.; Bush, A.I.; Everall, I.P.; Thomas, E.A.; et al. Increased cortical expression of the zinc transporter SLC39A12 suggests a breakdown in zinc cellular homeostasis as part of the pathophysiology of schizophrenia. NPJ Schizophr. 2016, 2, 16002. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Kanzer, S.H.; Zimmerman, E.A.; Molho, E.S.; Celmins, D.F.; Heckman, S.M.; Dick, R. Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimers Dis. Dement. 2010, 25, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Forsleff, L.; Schauss, A.G.; Bier, I.D.; Stuart, S. Evidence of functional zinc deficiency in Parkinson’s disease. J. Altern. Complement. Med. 1999, 5, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Schaffner, W. Zinc supplement greatly improves the condition of parkin mutant Drosophila. Biol. Chem. 2010, 391, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Krainc, D. Zn(2+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and α-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 27912801. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114 Pt 4, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portbury, S.D.; Adlard, P.A. Zinc Signal in Brain Diseases. Int. J. Mol. Sci. 2017, 18, 2506. https://doi.org/10.3390/ijms18122506
Portbury SD, Adlard PA. Zinc Signal in Brain Diseases. International Journal of Molecular Sciences. 2017; 18(12):2506. https://doi.org/10.3390/ijms18122506
Chicago/Turabian StylePortbury, Stuart D., and Paul A. Adlard. 2017. "Zinc Signal in Brain Diseases" International Journal of Molecular Sciences 18, no. 12: 2506. https://doi.org/10.3390/ijms18122506
APA StylePortbury, S. D., & Adlard, P. A. (2017). Zinc Signal in Brain Diseases. International Journal of Molecular Sciences, 18(12), 2506. https://doi.org/10.3390/ijms18122506