ZnT3 Gene Deletion Reduces Colchicine-Induced Dentate Granule Cell Degeneration



Abstract
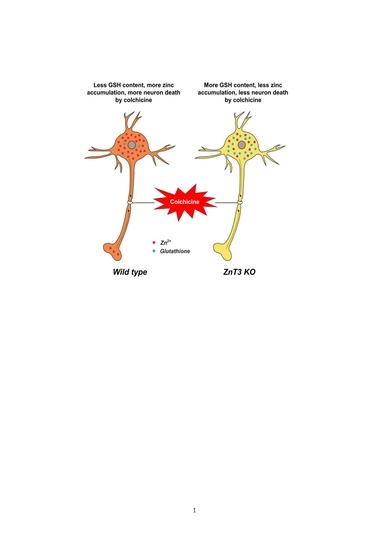
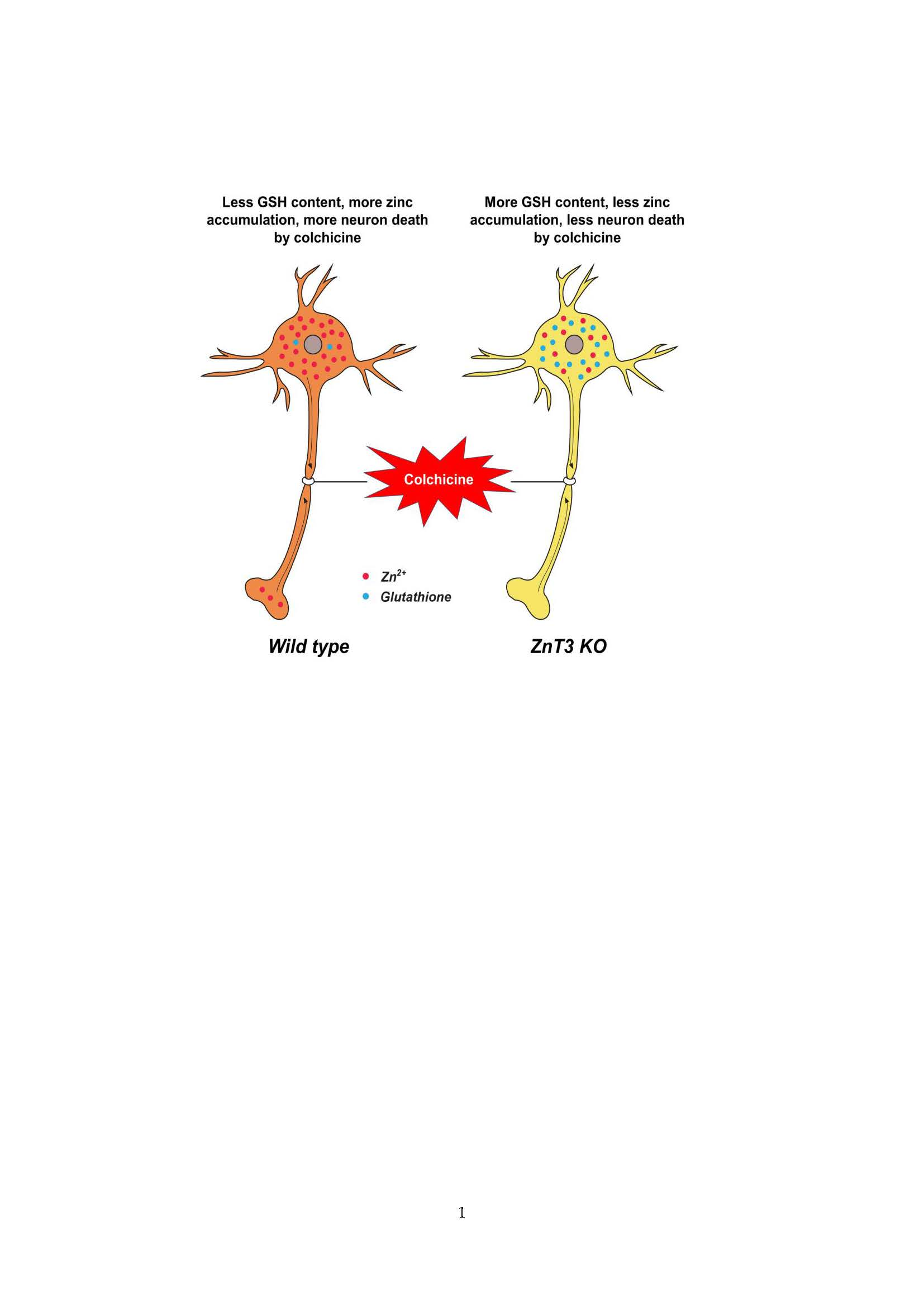
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
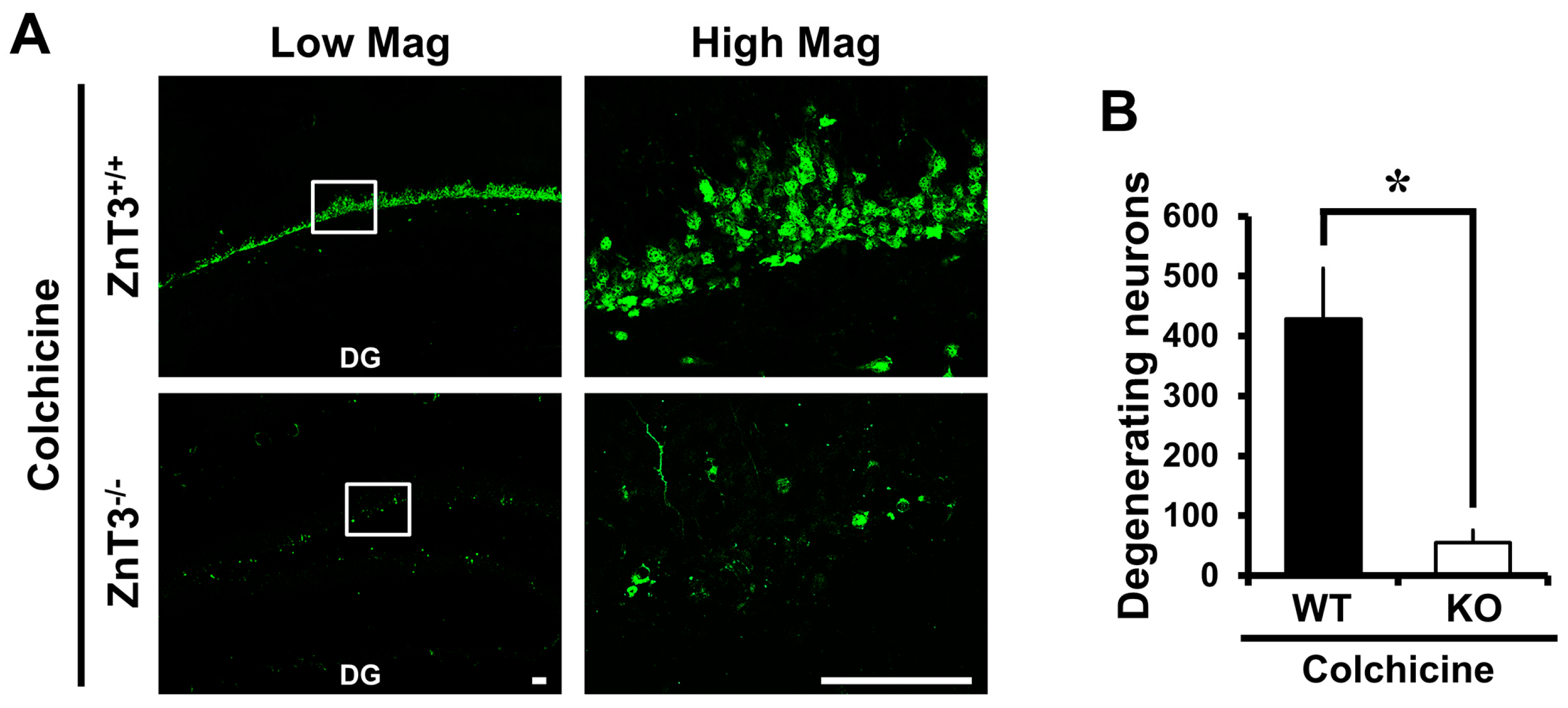
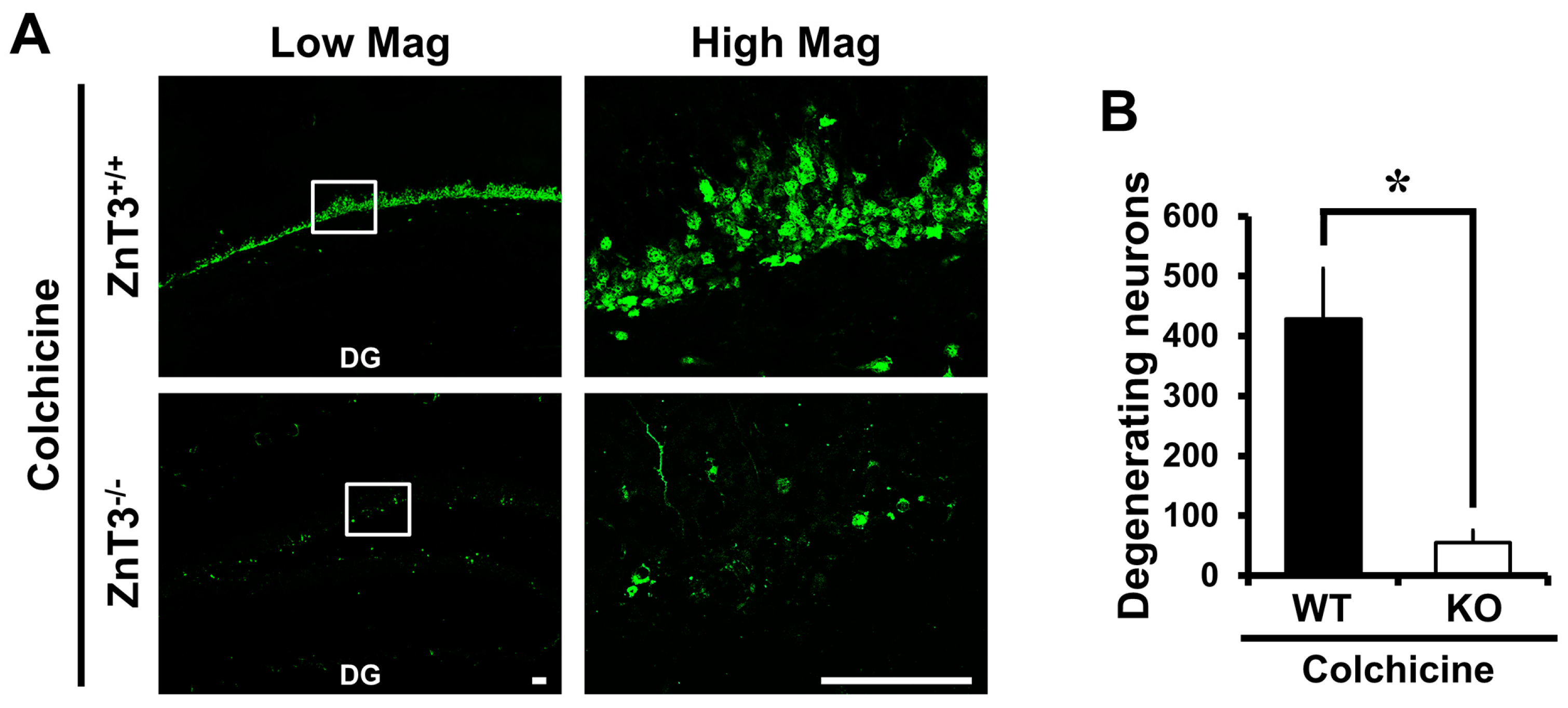
2.1. Colchicine-Induced Dentate Granule Cell Degeneration Is Reduced in ZnT3−/− Mice
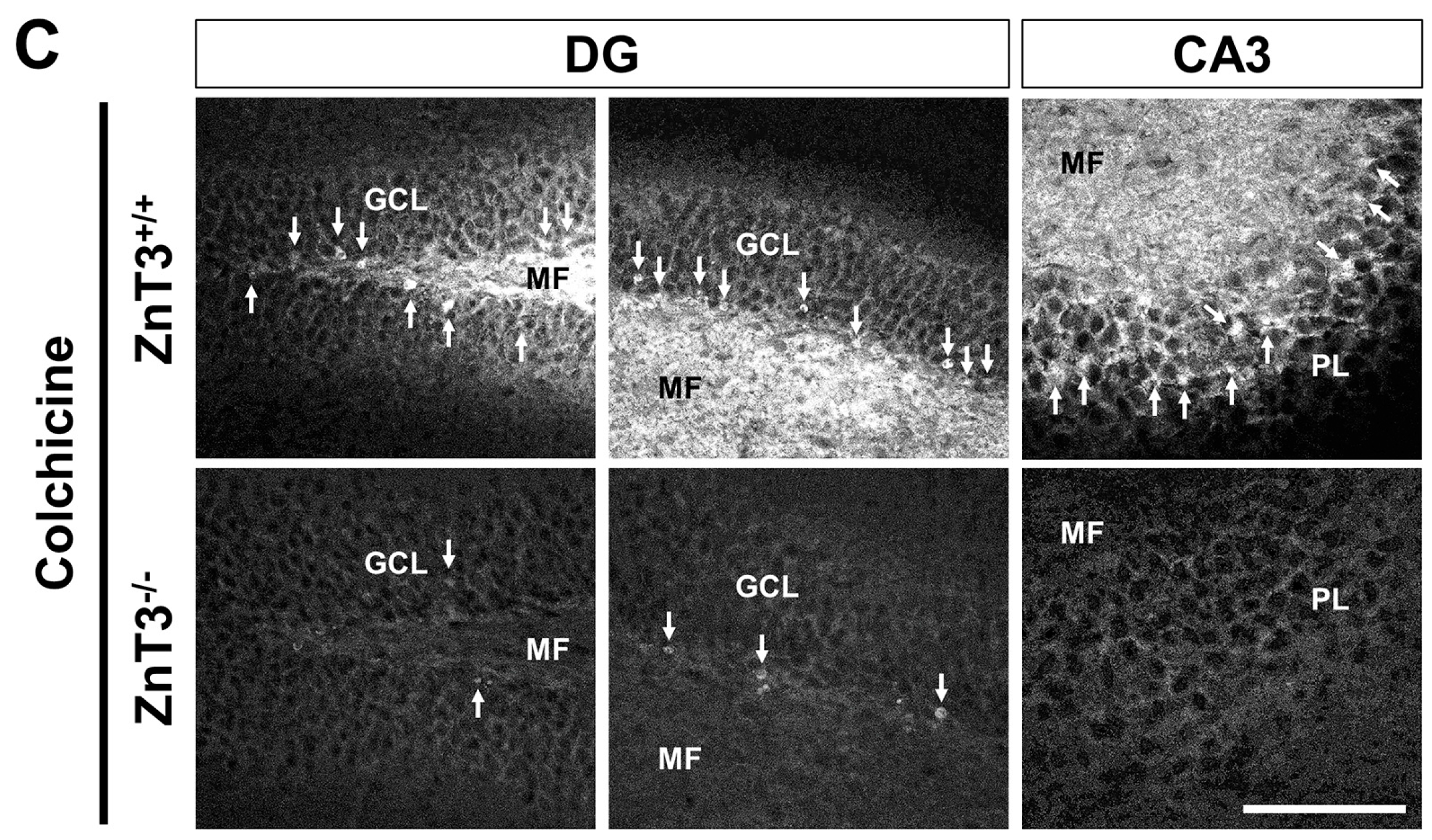
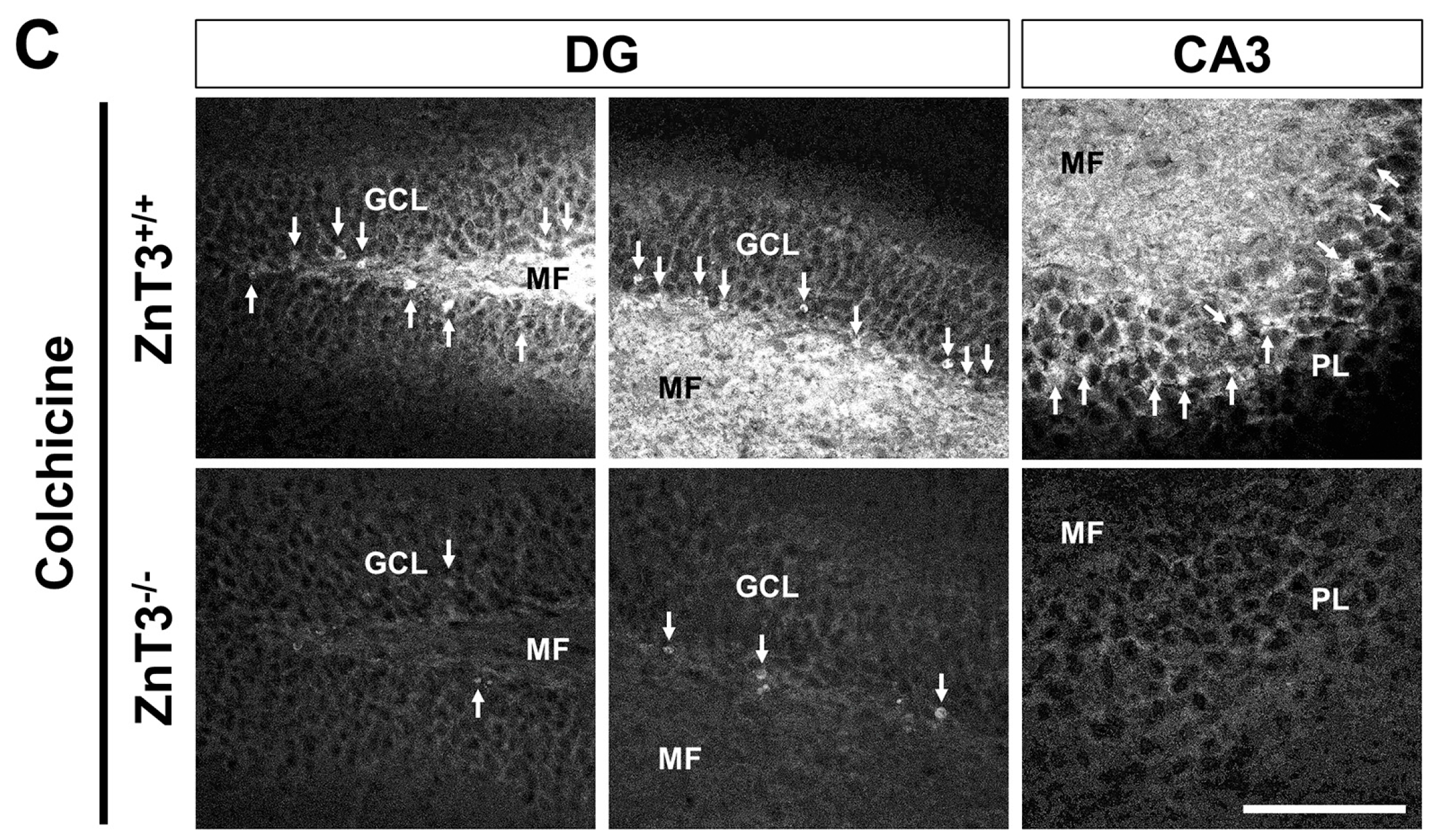
2.2. ZnT3 Gene Deletion Prevents Intracellular Zinc Accumulation in the Dentate Granule Cells after Colchicine Injection
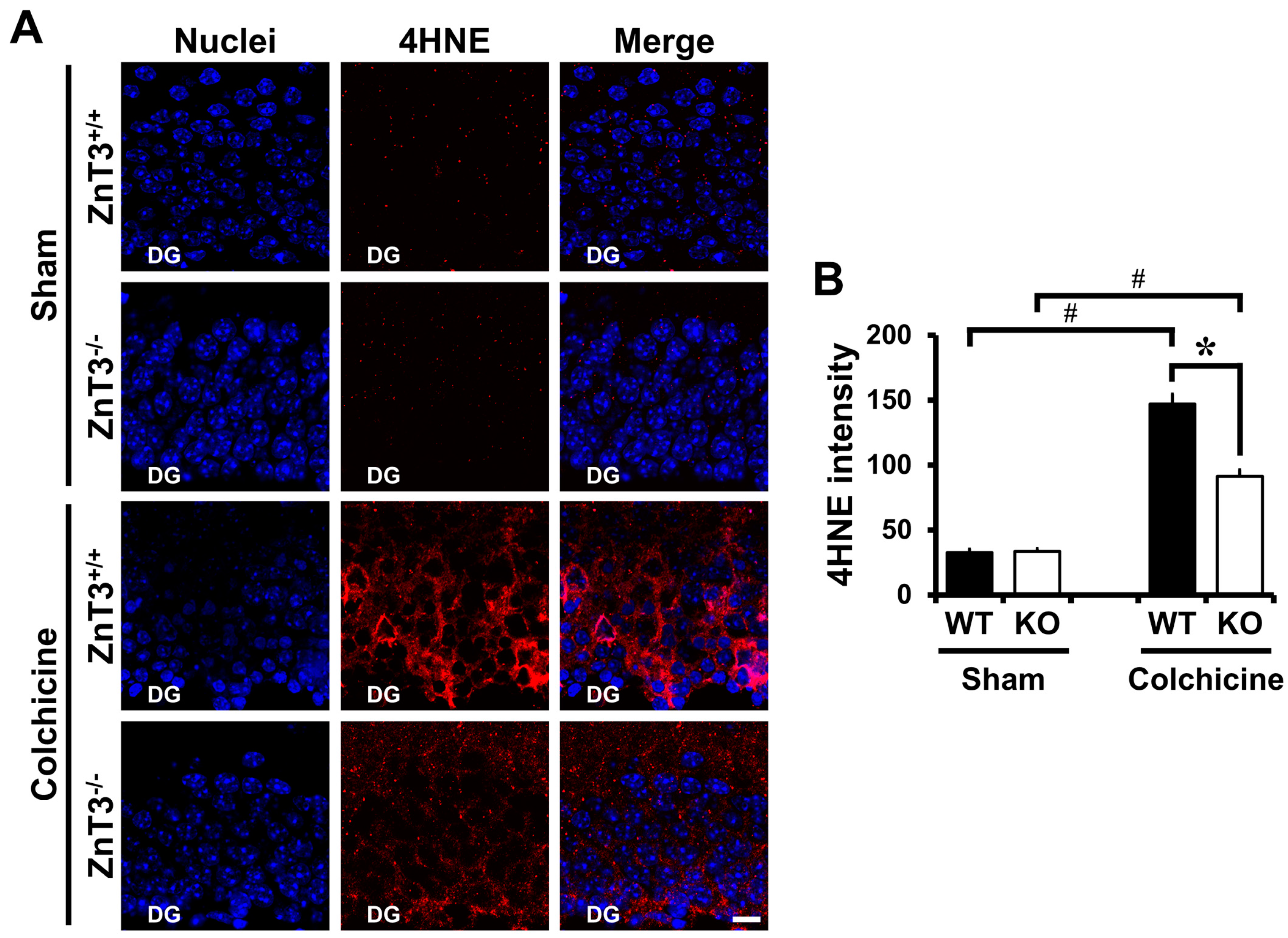
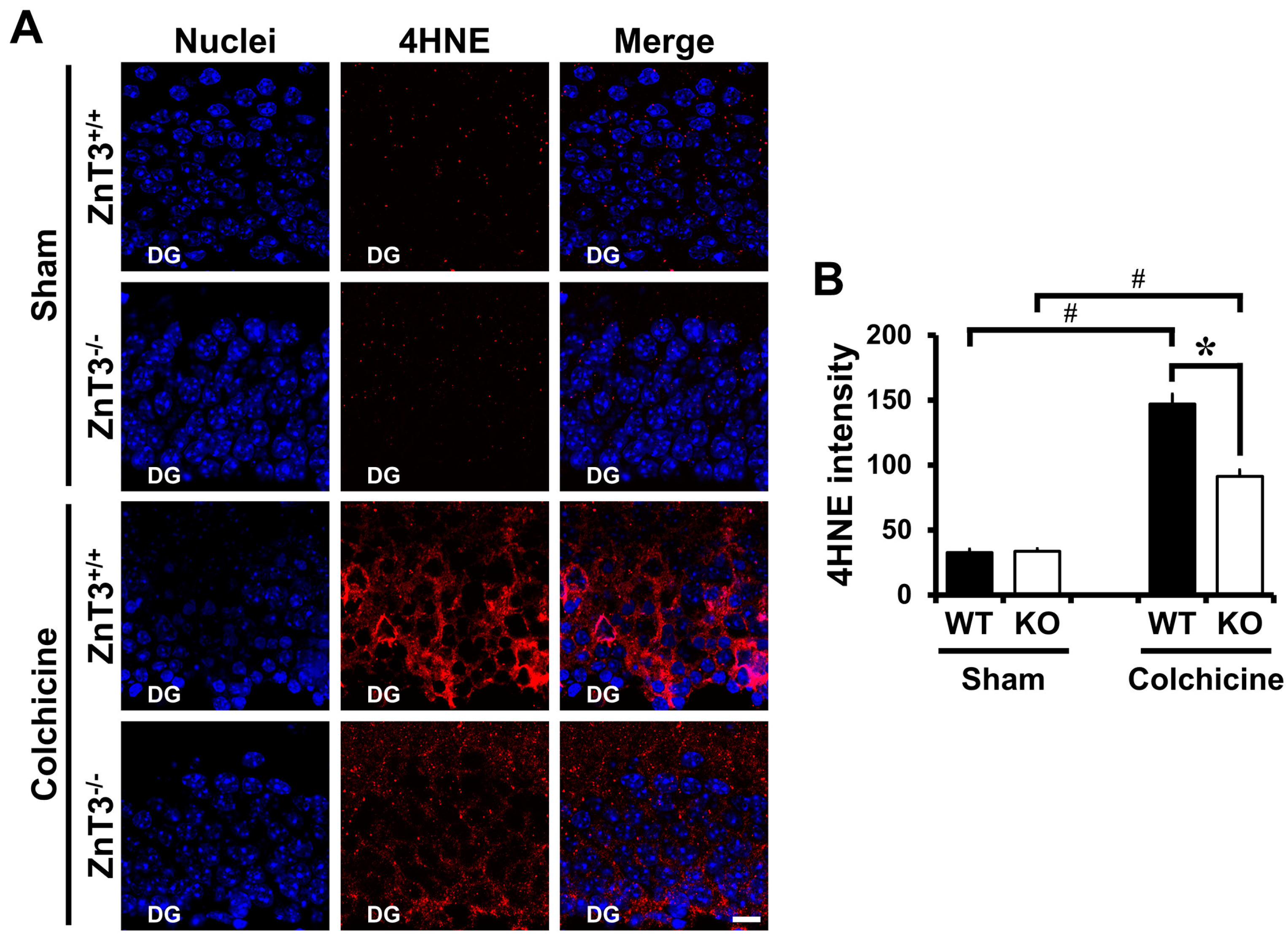
2.3. ZnT3 Gene Deletion Showed Less Oxidative Injury after Colchicine Injection
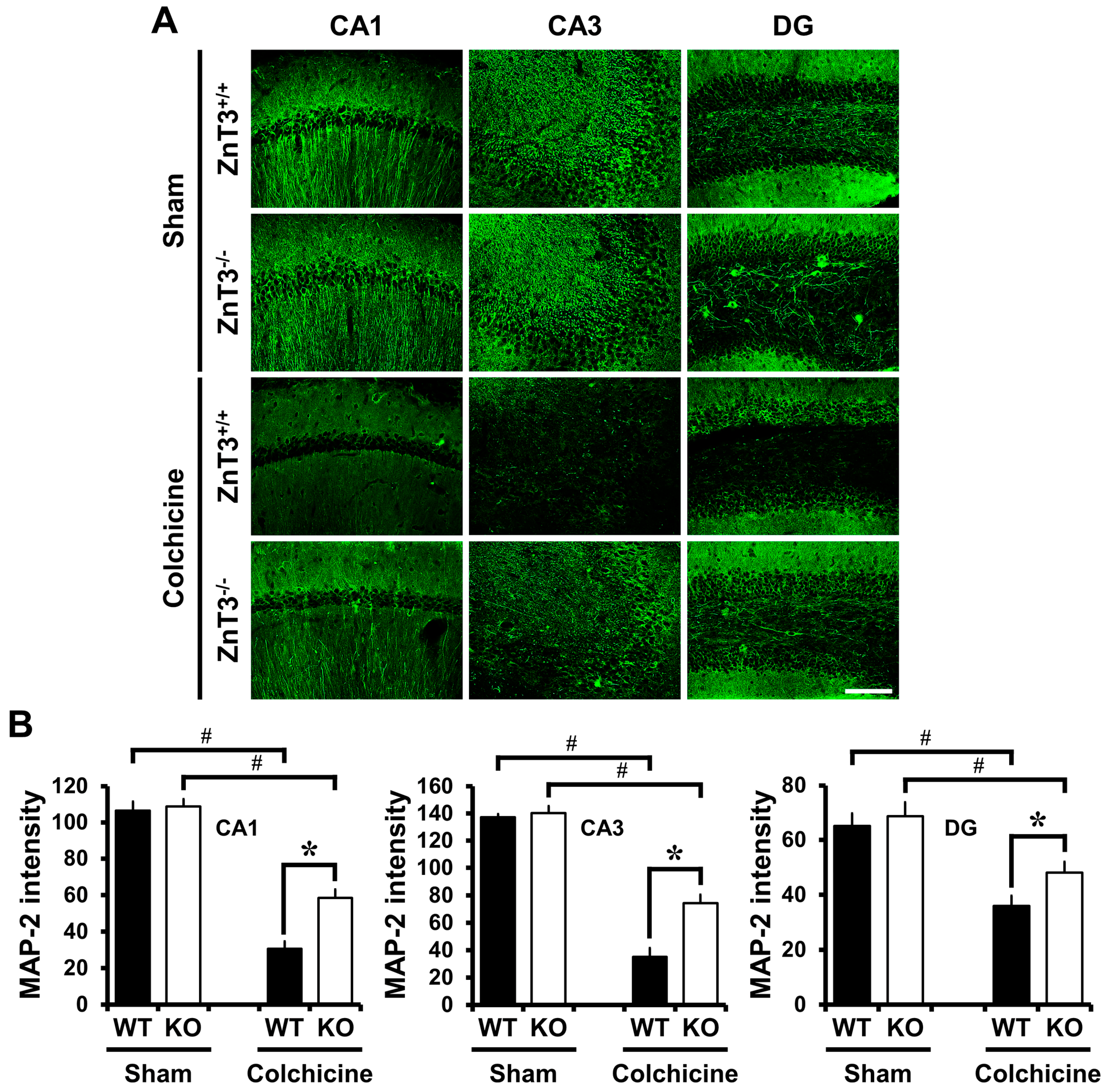
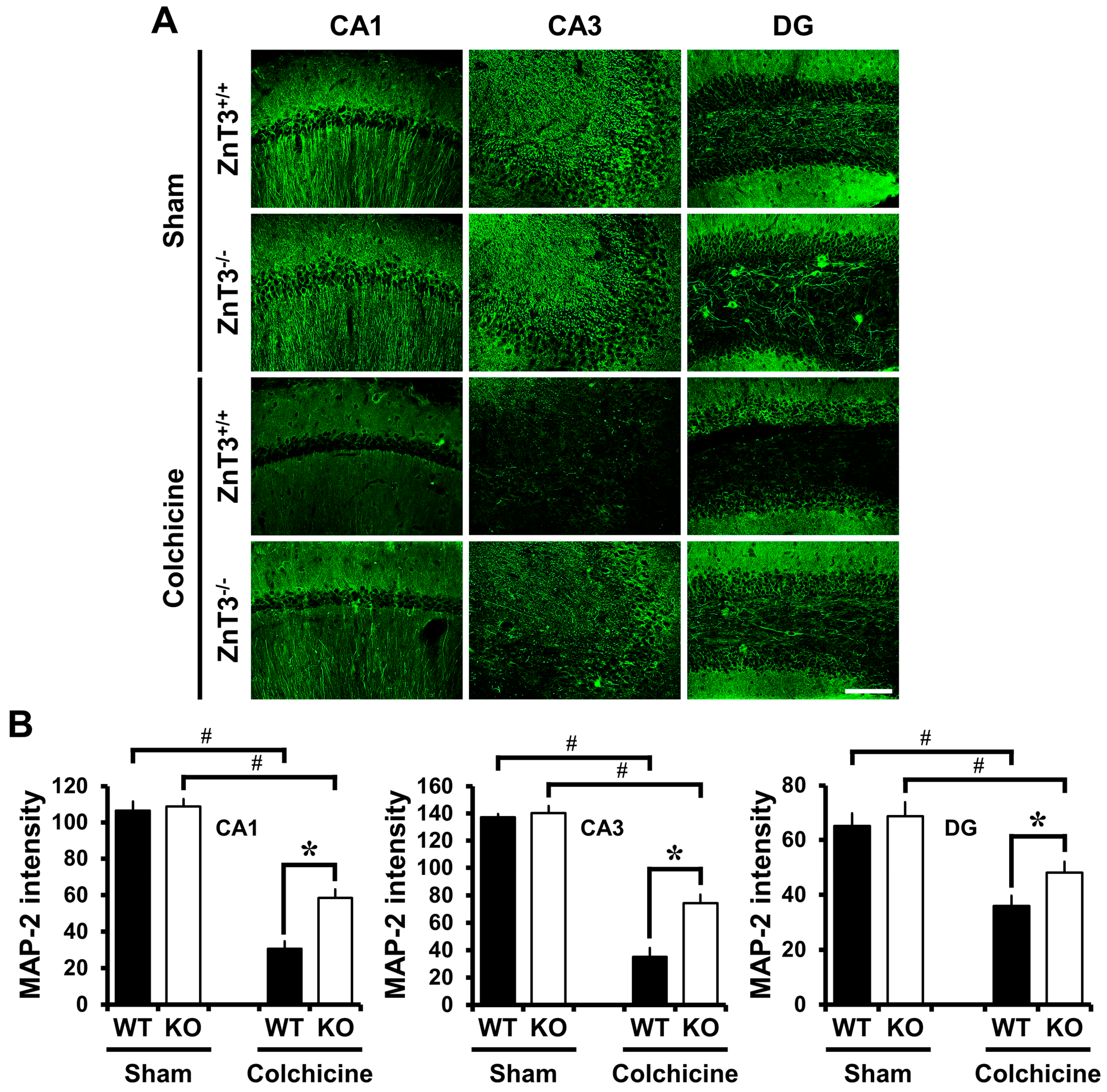
2.4. ZnT3 Gene Deletion Reduced Dendritic Damage after Colchicine Injection
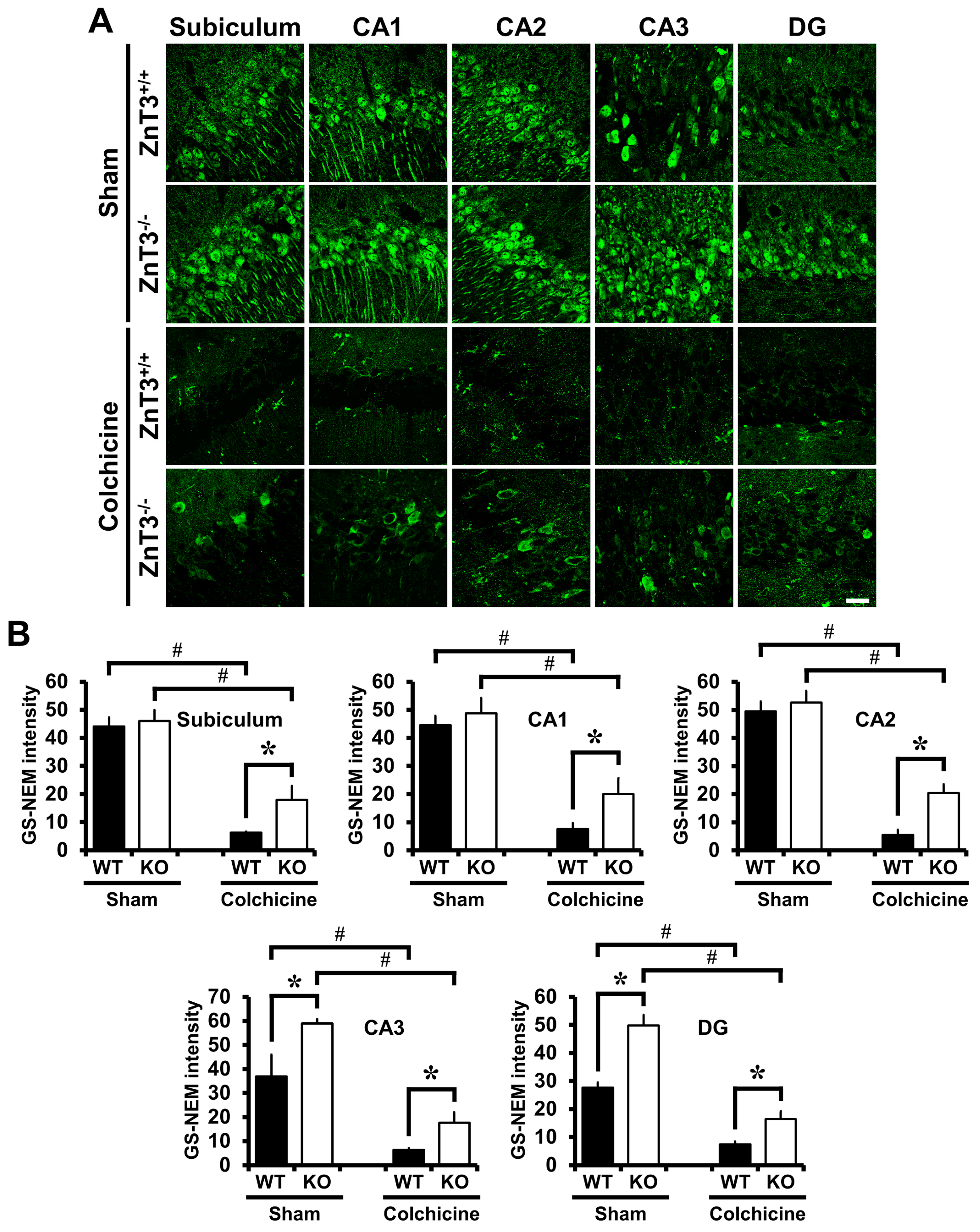
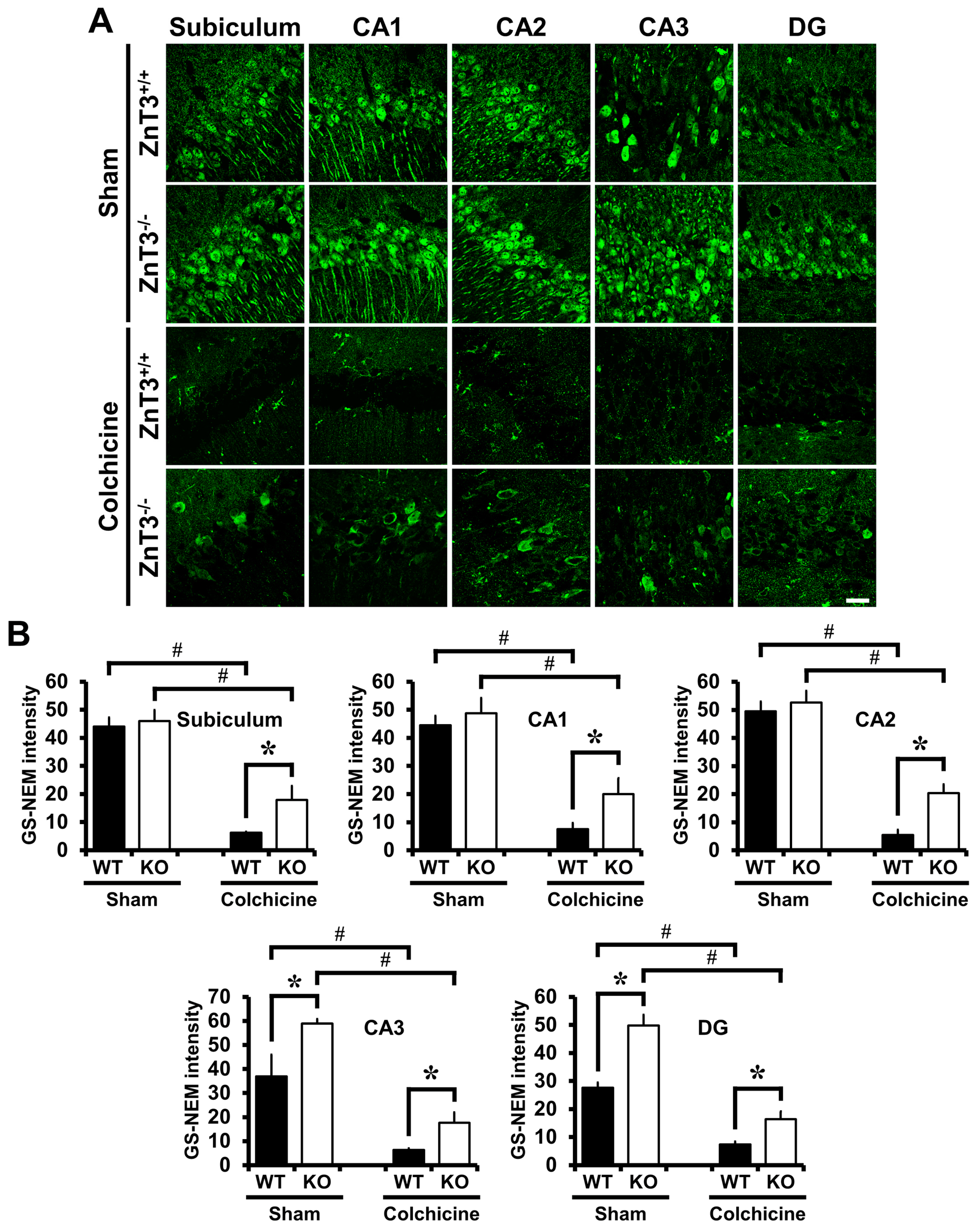
2.5. Colchicine-Induced Neuronal GSH Depletion Is Prevented in ZnT3−/− Mice
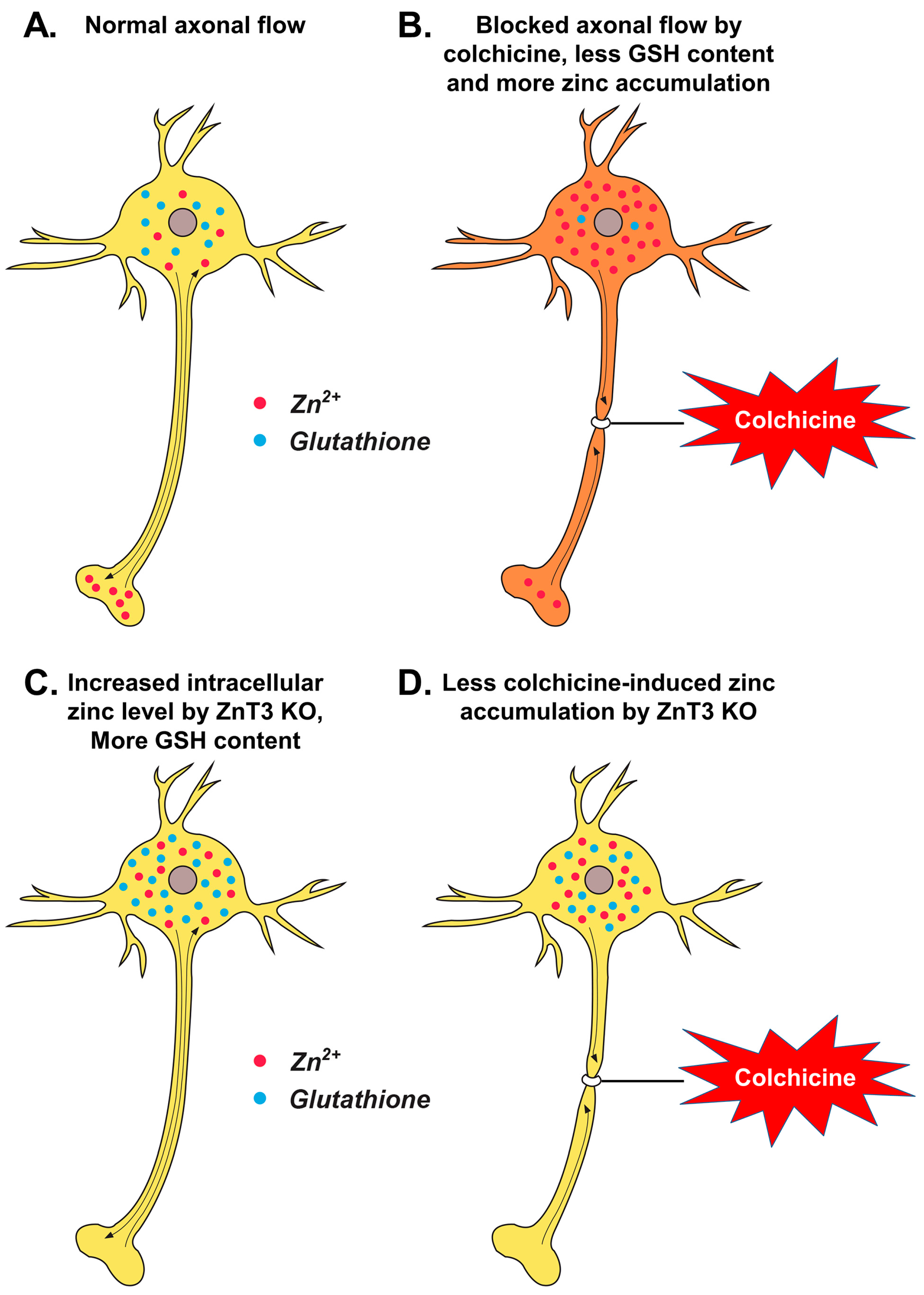
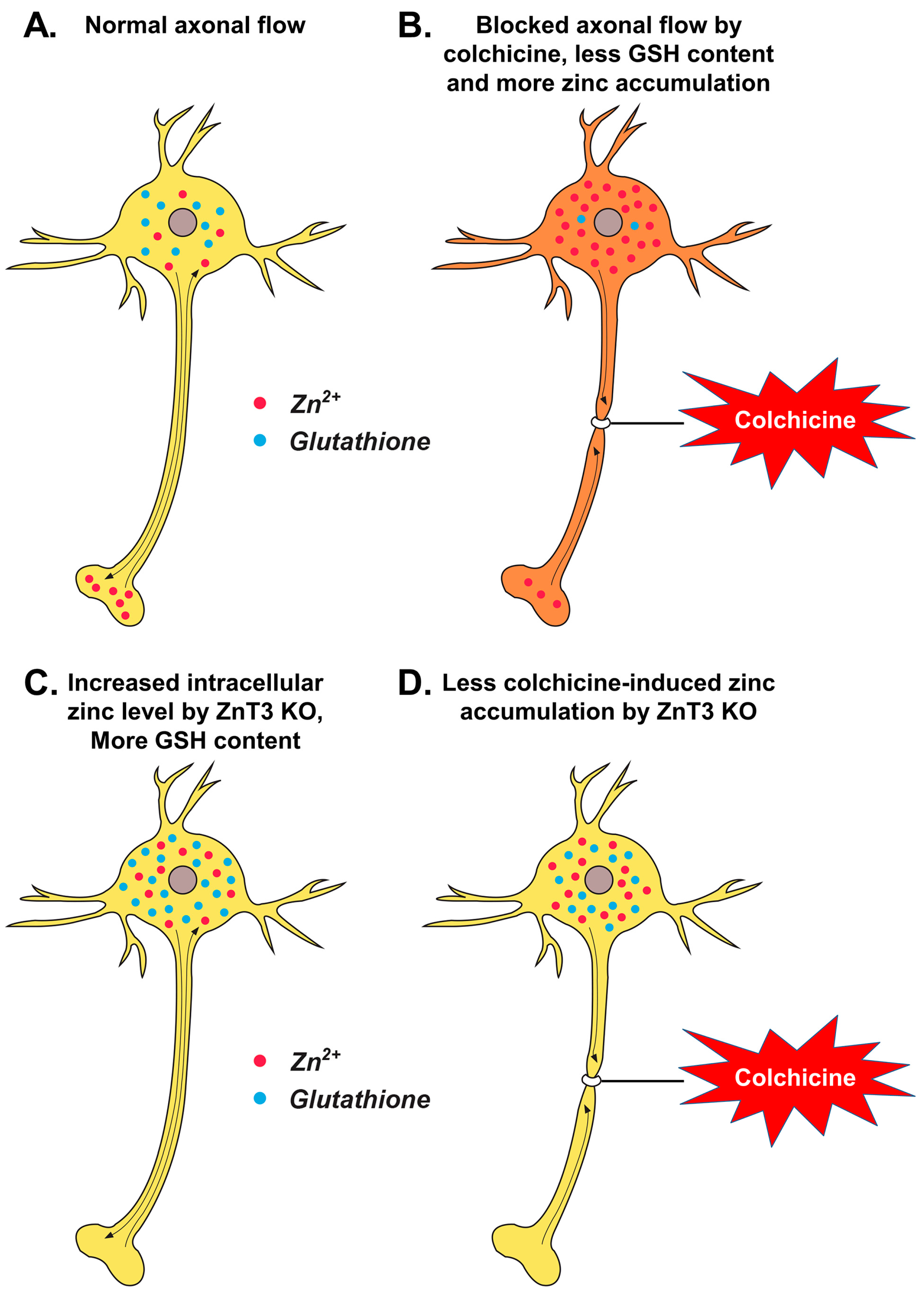
3. Discussion
4. Materials and Methods
4.1. Mouse Colonies
4.2. Colchicine Intrahippocampal Injection and Experimental Design
4.3. Tissue Preparation
4.4. Detection of Cell Death
4.5. Detection of Neuronal Glutathione (GSH)
4.6. Immunofluorescence Staining
4.7. Free Zinc Staining
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goldschmidt, R.B.; Steward, O. Preferential neurotoxicity of colchicine for granule cells of the dentate gyrus of the adult rat. Proc. Natl. Acad. Sci. USA 1980, 77, 3047–3051. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.J.; Geist, M.A.; Veng, L.M.; Willesen, M.G.; Johansen, F.F.; Leist, M.; Vaudano, E. A role for mixed lineage kinases in granule cell apoptosis induced by cytoskeletal disruption. J. Neurochem. 2006, 96, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Veerendra Kumar, M.H.; Gupta, Y.K. Intracerebroventricular administration of colchicine produces cognitive impairment associated with oxidative stress in rats. Pharmacol. Biochem. Behav. 2002, 73, 565–571. [Google Scholar] [CrossRef]
- Hirokawa, N. The neuronal cytoskeleton: Roles in neuronal morphogenesis and organelle transport. Prog. Clin. Biol. Res. 1994, 390, 117–143. [Google Scholar] [PubMed]
- Uppuluri, S.; Knipling, L.; Sackett, D.L.; Wolff, J. Localization of the colchicine-binding site of tubulin. Proc. Natl. Acad. Sci. USA 1993, 90, 11598–11602. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Seghal, N.; Naidu, P.S.; Padi, S.S.; Goyal, R. Colchicines-induced neurotoxicity as an animal model of sporadic dementia of Alzheimer’s type. Pharmacol. Rep. 2007, 59, 274–283. [Google Scholar] [PubMed]
- Kumar, A.; Dogra, S.; Prakash, A. Neuroprotective effects of centella asiatica against intracerebroventricular colchicine-induced cognitive impairment and oxidative stress. Int. J. Alzheimers Dis. 2009. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.P. Neurological disease. Adv. Pharmacol. 1997, 38, 557–580. [Google Scholar] [PubMed]
- Friedman, J. Why is the nervous system vulnerable to oxidative stress? In Oxidative Stress and Free Radical Damage in Neurology. Oxidative Stress in Applied Basic Research and Clinical Practice; Gadoth, N., Göbel, H., Eds.; Humana Press: Clifton, NJ, USA, 2011; pp. 19–27. [Google Scholar]
- Buttke, T.M.; Sandstrom, P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Beaver, J.P.; Waring, P. A decrease in intracellular glutathione concentration precedes the onset of apoptosis in murine thymocytes. Eur. J. Cell Biol. 1995, 68, 47–54. [Google Scholar] [PubMed]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; O, W.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Suh, S.W.; Silva, D.; Thompson, R.B. Importance of zinc in the central nervous system: The zinc-containing neuron. J. Nutr. 2000, 130, 1471S–1483S. [Google Scholar] [PubMed]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Kawai, H.; Metherate, R.; Glabe, C.G.; Busciglio, J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 2009, 29, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Cull, R.E. Role of axonal transport in maintaining central synaptic connections. Exp. Brain Res. 1975, 24, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Kodama, Y.; Ohnuma, M.; Okada, S. Zinc transport from the striatum and substantia nigra. Brain Res. Bull. 1998, 47, 103–106. [Google Scholar] [CrossRef]
- Choi, B.Y.; Lee, B.E.; Kim, J.H.; Kim, H.J.; Sohn, M.; Song, H.K.; Chung, T.N.; Suh, S.W. Colchicine induced intraneuronal free zinc accumulation and dentate granule cell degeneration. Metallomics 2014, 6, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.H.; Kim, T.Y.; Yoon, Y.H.; Koh, J.Y. Autism phenotypes in ZnT3 null mice: Involvement of zinc dyshomeostasis, MMP-9 activation and BDNF upregulation. Sci. Rep. 2016, 6, 28548. [Google Scholar] [CrossRef] [PubMed]
- Mills, B.J.; Lindeman, R.D.; Lang, C.A. Effect of zinc deficiency on blood glutathione levels. J. Nutr. 1981, 111, 1098–1102. [Google Scholar] [PubMed]
- Fernandez, M.A.; O’Dell, B.L. Effect of zinc deficiency on plasma glutathione in the rat. Proc. Soc. Exp. Biol. Med. 1983, 173, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Parat, M.O.; Richard, M.J.; Beani, J.C.; Favier, A. Involvement of zinc in intracellular oxidant/antioxidant balance. Biol. Trace Elem. Res. 1997, 60, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Iszard, M.B.; Liu, J.; Klaassen, C.D. Effect of several metallothionein inducers on oxidative stress defense mechanisms in rats. Toxicology 1995, 104, 25–33. [Google Scholar] [CrossRef]
- Emerich, D.F.; Walsh, T.J. Cholinergic cell loss and cognitive impairments following intraventricular or intradentate injection of colchicine. Brain Res. 1990, 517, 157–167. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nakamura, S.; Kase, Y.; Noguchi, T.; Ishihara, T. Colchicine lesions in the rat hippocampus mimic the alterations of several markers in Alzheimer’s disease. Brain Res. 1987, 408, 57–64. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Zarkovic, N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol. Aspects Med. 2003, 24, 281–291. [Google Scholar] [CrossRef]
- Izant, J.G.; McIntosh, J.R. Microtubule-associated proteins: A monoclonal antibody to MAP2 binds to differentiated neurons. Proc. Natl. Acad. Sci. USA 1980, 77, 4741–4745. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.; Matus, A. Differences in the cellular distributions of two microtubule-associated proteins, MAP1 and MAP2, in rat brain. J. Neurosci. 1984, 4, 151–160. [Google Scholar] [PubMed]
- Park, J.S.; Bateman, M.C.; Goldberg, M.P. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol. Dis. 1996, 3, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar] [CrossRef]
- Yu, Z.; Cheng, G.; Hu, B. Mechanism of colchicine impairment on learning and memory, and protective effect of CGP36742 in mice. Brain Res. 1997, 750, 53–58. [Google Scholar] [CrossRef]
- Hammer, B.; Parker, W.D., Jr.; Bennett, J.P., Jr. NMDA receptors increase OH radicals in vivo by using nitric oxide synthase and protein kinase C. Neuroreport 1993, 5, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Dufourny, L.; Leroy, D.; Warembourg, M. Differential effects of colchicine on the induction of nitric oxide synthase in neurons containing progesterone receptors of the guinea pig hypothalamus. Brain Res. Bull. 2000, 52, 435–443. [Google Scholar] [CrossRef]
- Wallis, R.A.; Panizzon, K.L.; Henry, D.; Wasterlain, C.G. Neuroprotection against nitric oxide injury with inhibitors of ADP-ribosylation. Neuroreport 1993, 5, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Cuajungco, M.P.; LaBuda, C.J.; Suh, S.W. Nitric oxide causes apparent release of zinc from presynaptic boutons. Neuroscience 2002, 115, 471–474. [Google Scholar] [CrossRef]
- Song, Y.; Leonard, S.W.; Traber, M.G.; Ho, E. Zinc deficiency affects DNA damage, oxidative stress, antioxidant defenses, and DNA repair in rats. J. Nutr. 2009, 139, 1626–1631. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2012, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Liblau, R.S.; Tisch, R.; Shokat, K.; Yang, X.; Dumont, N.; Goodnow, C.C.; McDevitt, H.O. Intravenous injection of soluble antigen induces thymic and peripheral T-cells apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 3031–3036. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Yoo, J.H.; Song, H.K.; Sohn, M.; Won, S.J.; Suh, S.W. Pyruvate administration reduces recurrent/moderate hypoglycemia-induced cortical neuron death in diabetic rats. PLoS ONE 2013, 8, e81523. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.N.; Berman, A.E.; Swanson, R.A.; Yenari, M.A. Digitally quantifying cerebral hemorrhage using Photoshop and Image J. J. Neurosci. Methods 2010, 190, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. EAAC1 gene deletion increases neuronal death and blood brain barrier disruption after transient cerebral ischemia in female mice. Int. J. Mol. Sci. 2014, 15, 19444–19457. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Kasarskis, E.J.; Ringo, D.; Frederickson, R.E. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 1987, 20, 91–103. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Hong, D.K.; Suh, S.W. ZnT3 Gene Deletion Reduces Colchicine-Induced Dentate Granule Cell Degeneration. Int. J. Mol. Sci. 2017, 18, 2189. https://doi.org/10.3390/ijms18102189
Choi BY, Hong DK, Suh SW. ZnT3 Gene Deletion Reduces Colchicine-Induced Dentate Granule Cell Degeneration. International Journal of Molecular Sciences. 2017; 18(10):2189. https://doi.org/10.3390/ijms18102189
Chicago/Turabian StyleChoi, Bo Young, Dae Ki Hong, and Sang Won Suh. 2017. "ZnT3 Gene Deletion Reduces Colchicine-Induced Dentate Granule Cell Degeneration" International Journal of Molecular Sciences 18, no. 10: 2189. https://doi.org/10.3390/ijms18102189
APA StyleChoi, B. Y., Hong, D. K., & Suh, S. W. (2017). ZnT3 Gene Deletion Reduces Colchicine-Induced Dentate Granule Cell Degeneration. International Journal of Molecular Sciences, 18(10), 2189. https://doi.org/10.3390/ijms18102189