Two Distinct Conformations in Bet v 2 Determine Its Proteolytic Resistance to Cathepsin S

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Isolation and Cloning of Bet v 2
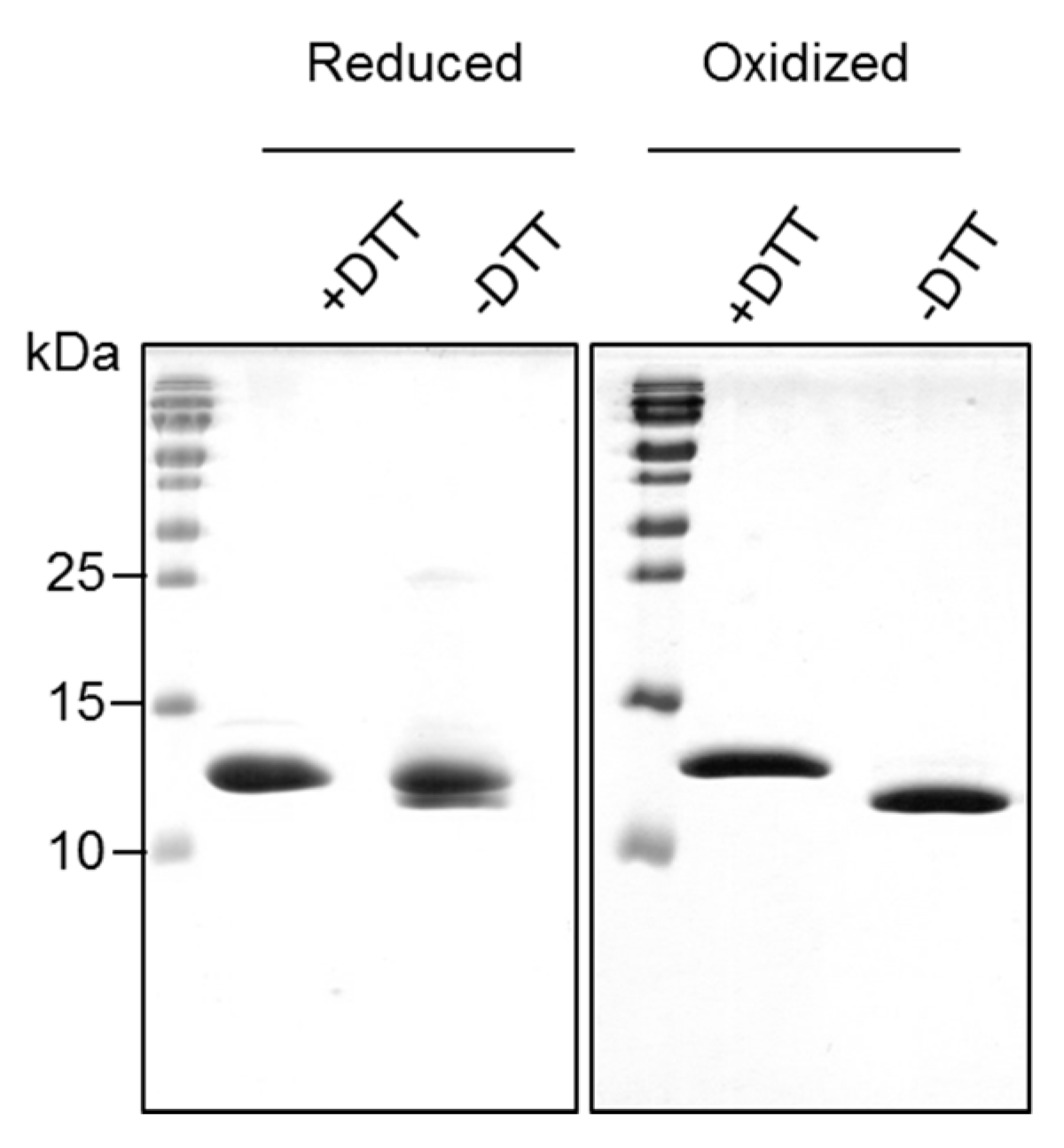
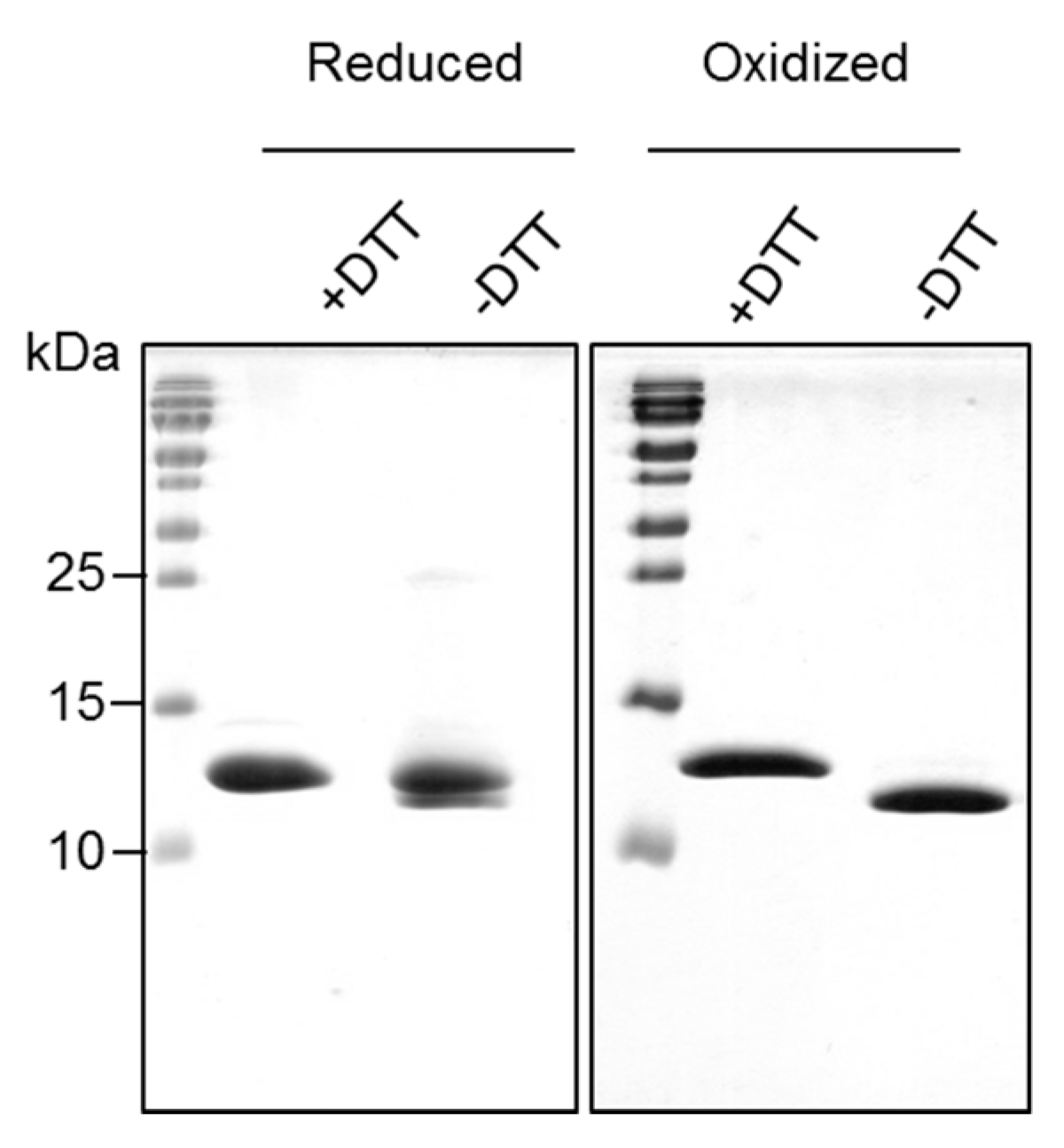
2.2. Recombinantly-Produced Bet v 2 Exists in Two Conformations
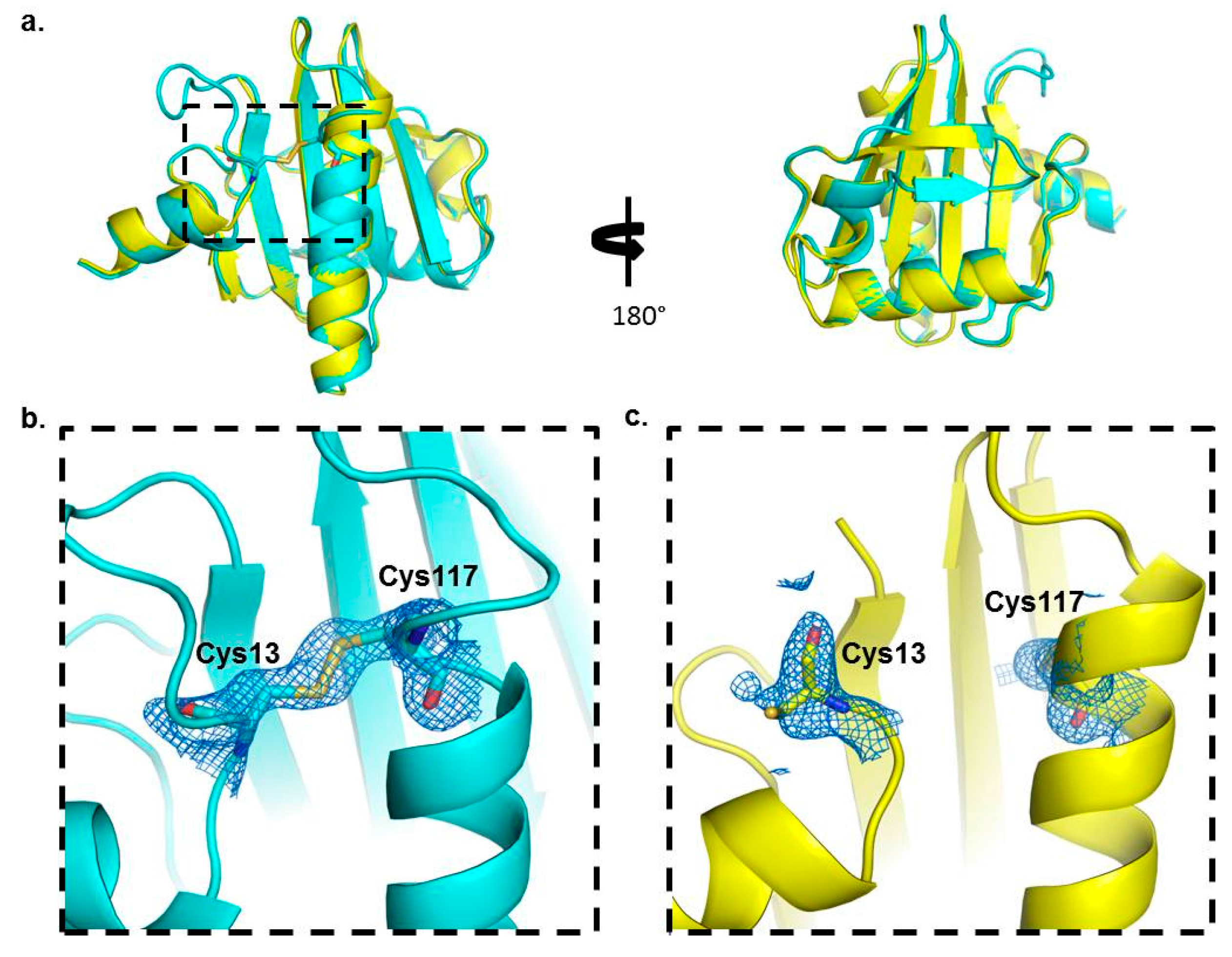
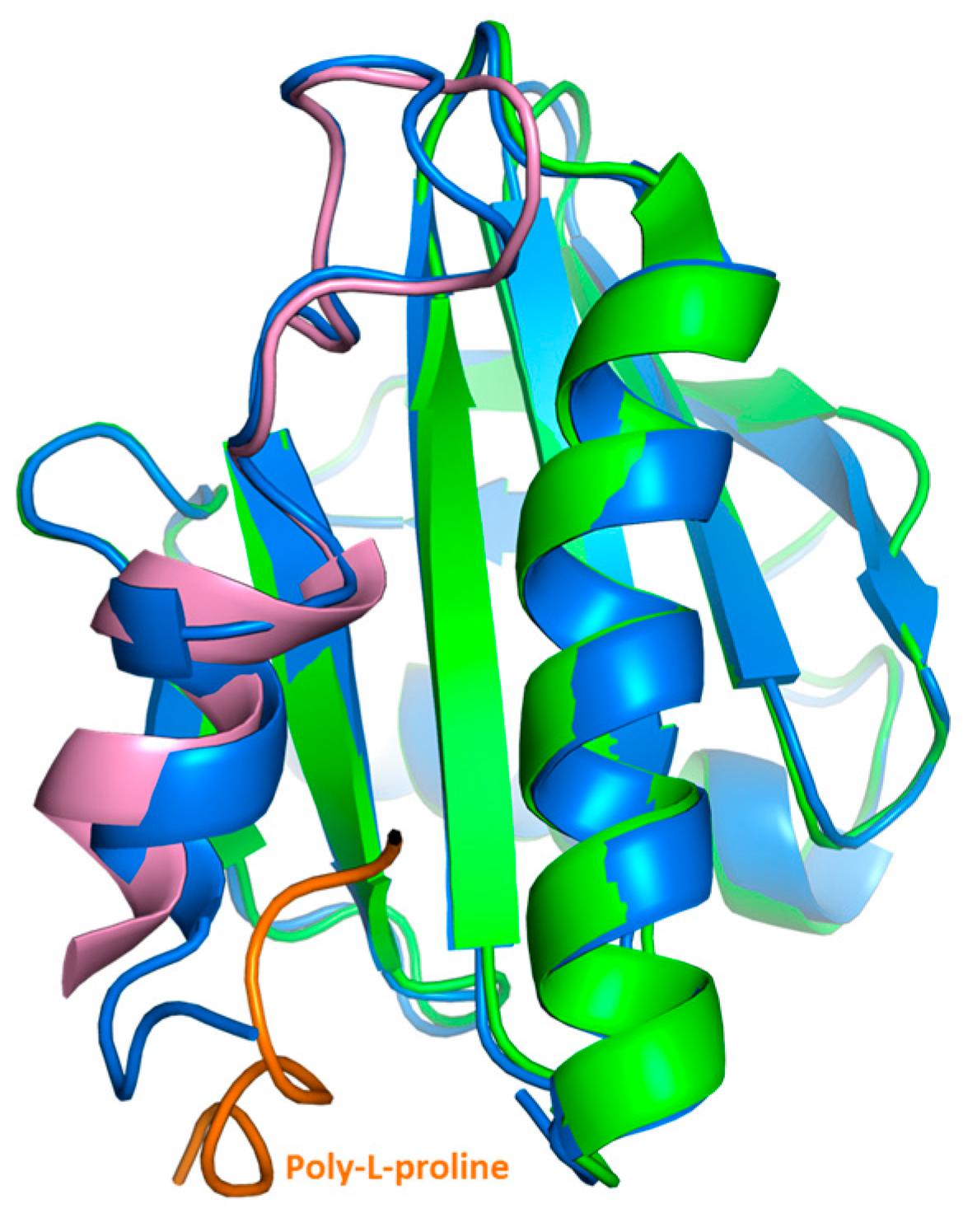
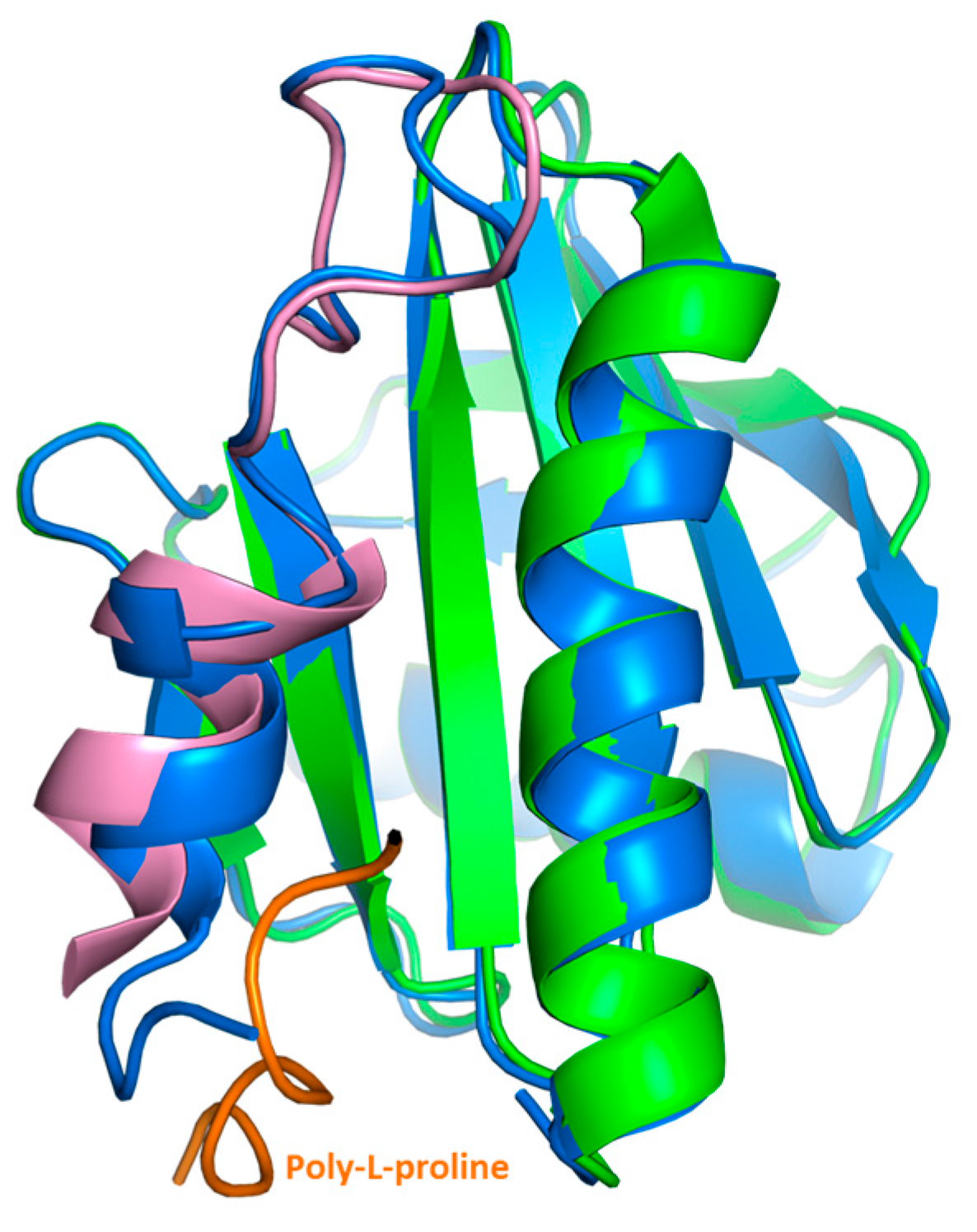
2.3. Crystal Structure Analysis of Bet v 2 in the Presence (Oxidized) and Absence (Reduced) of A Disulfide Bridge
2.4. Structural Studies in Solution
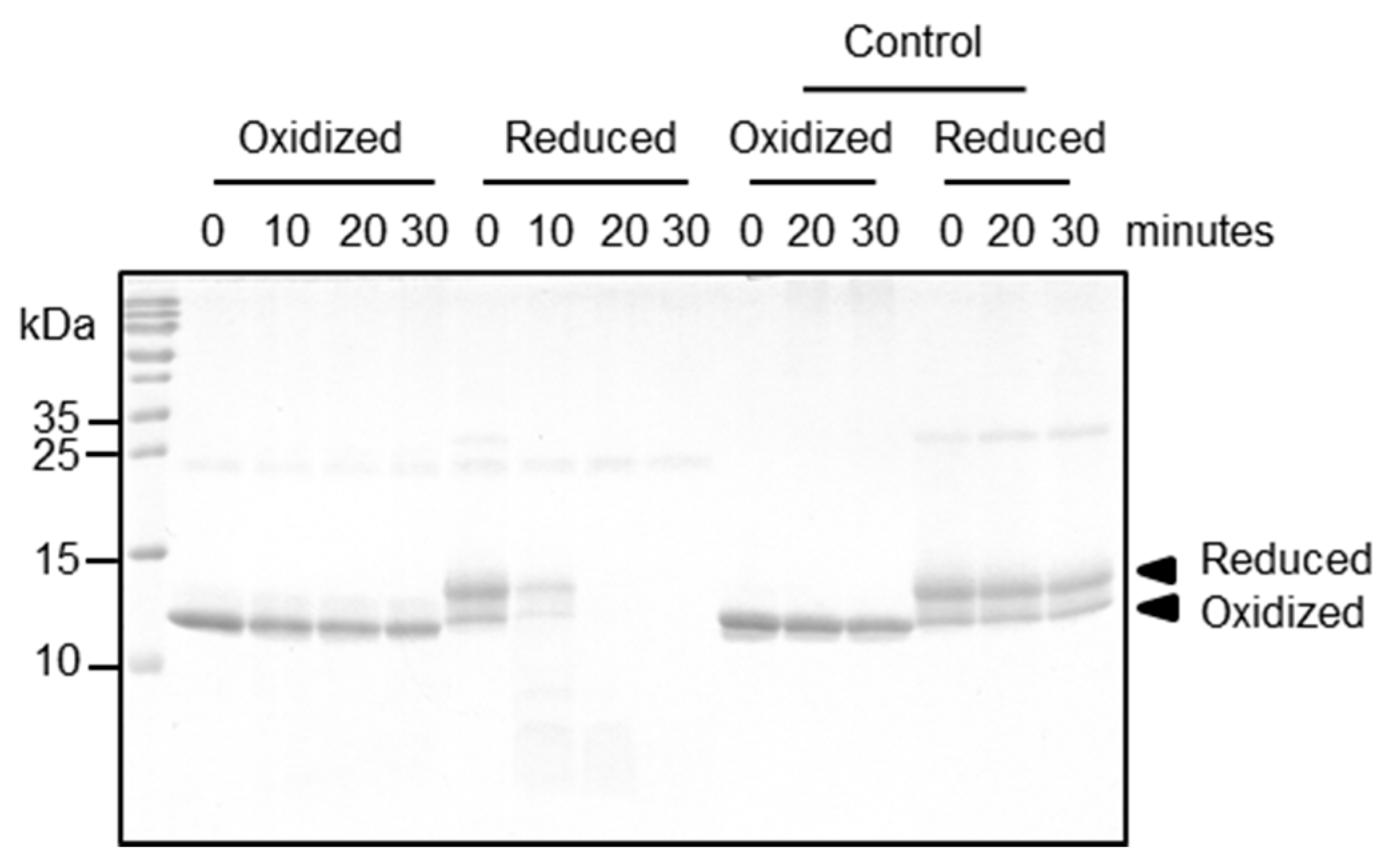
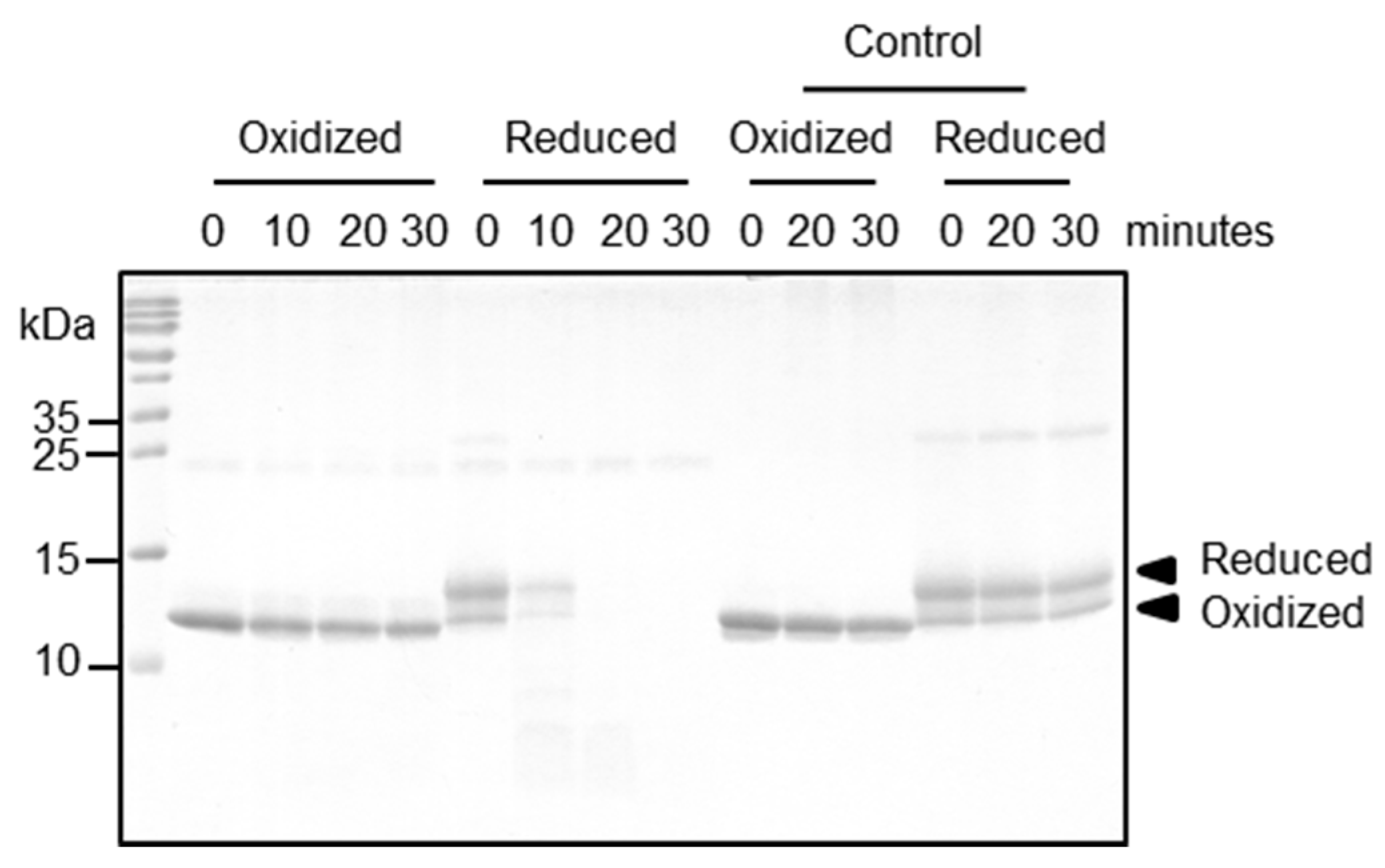
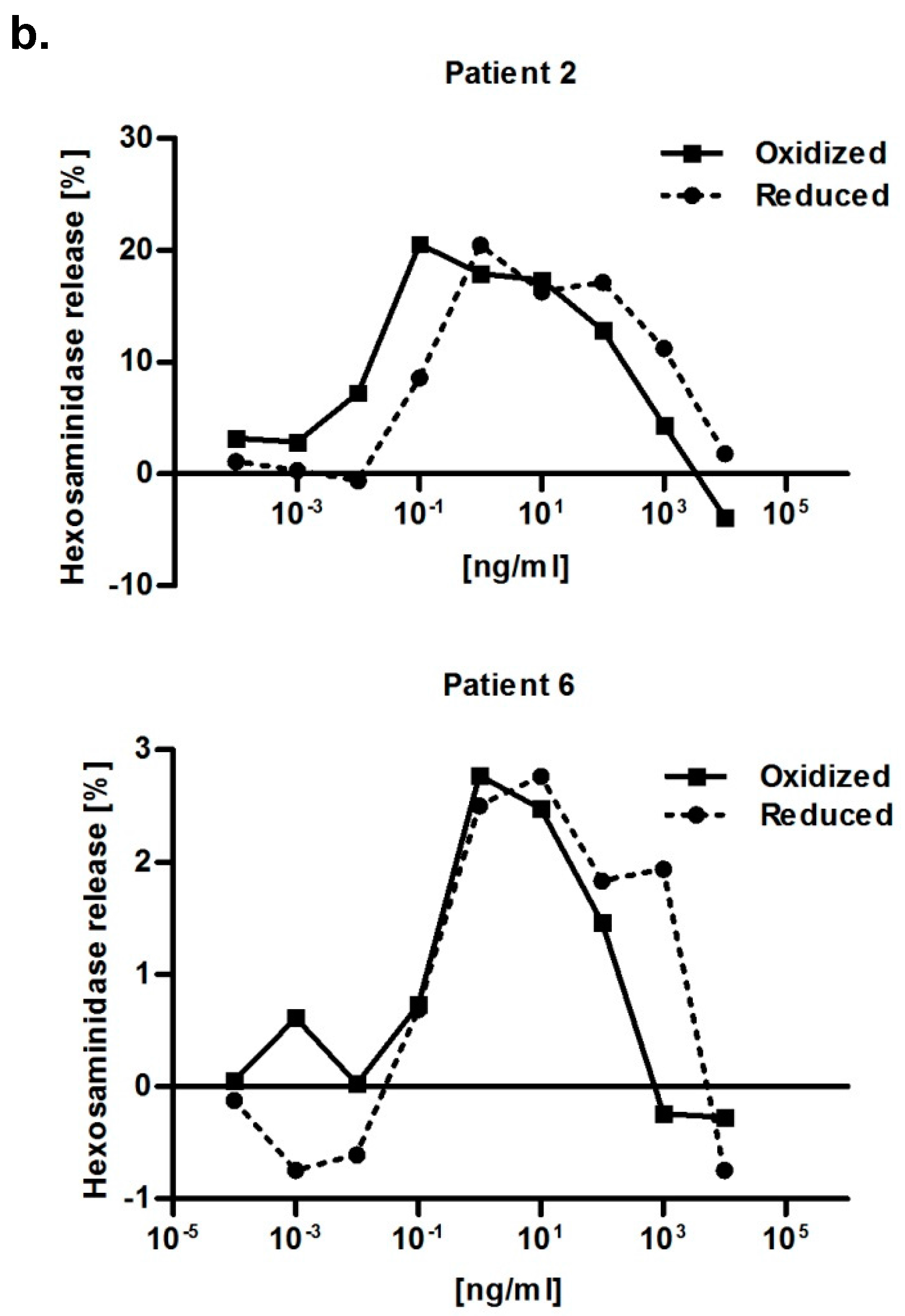
2.5. Functional Relevance of the Disulfide Bridge
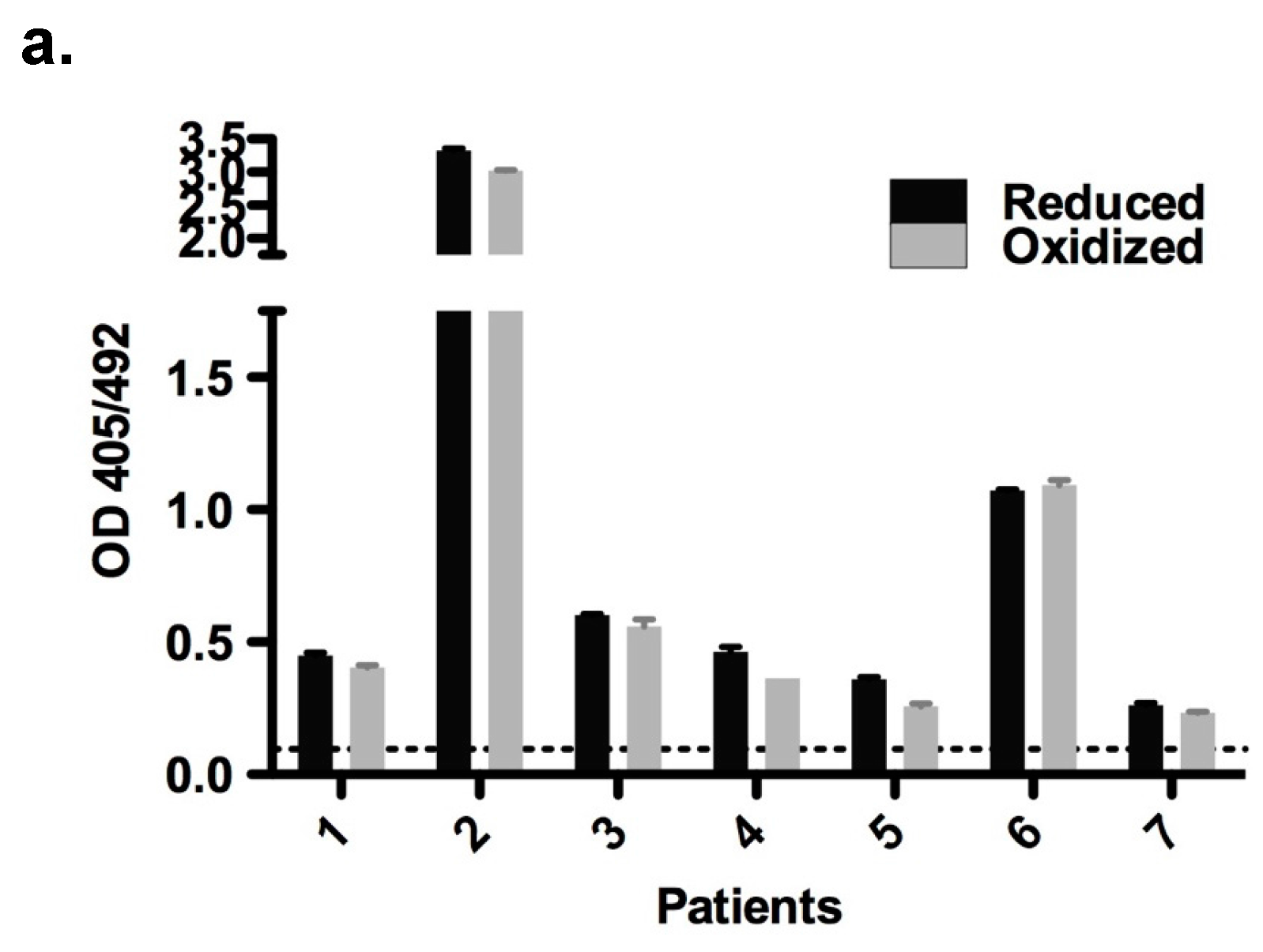
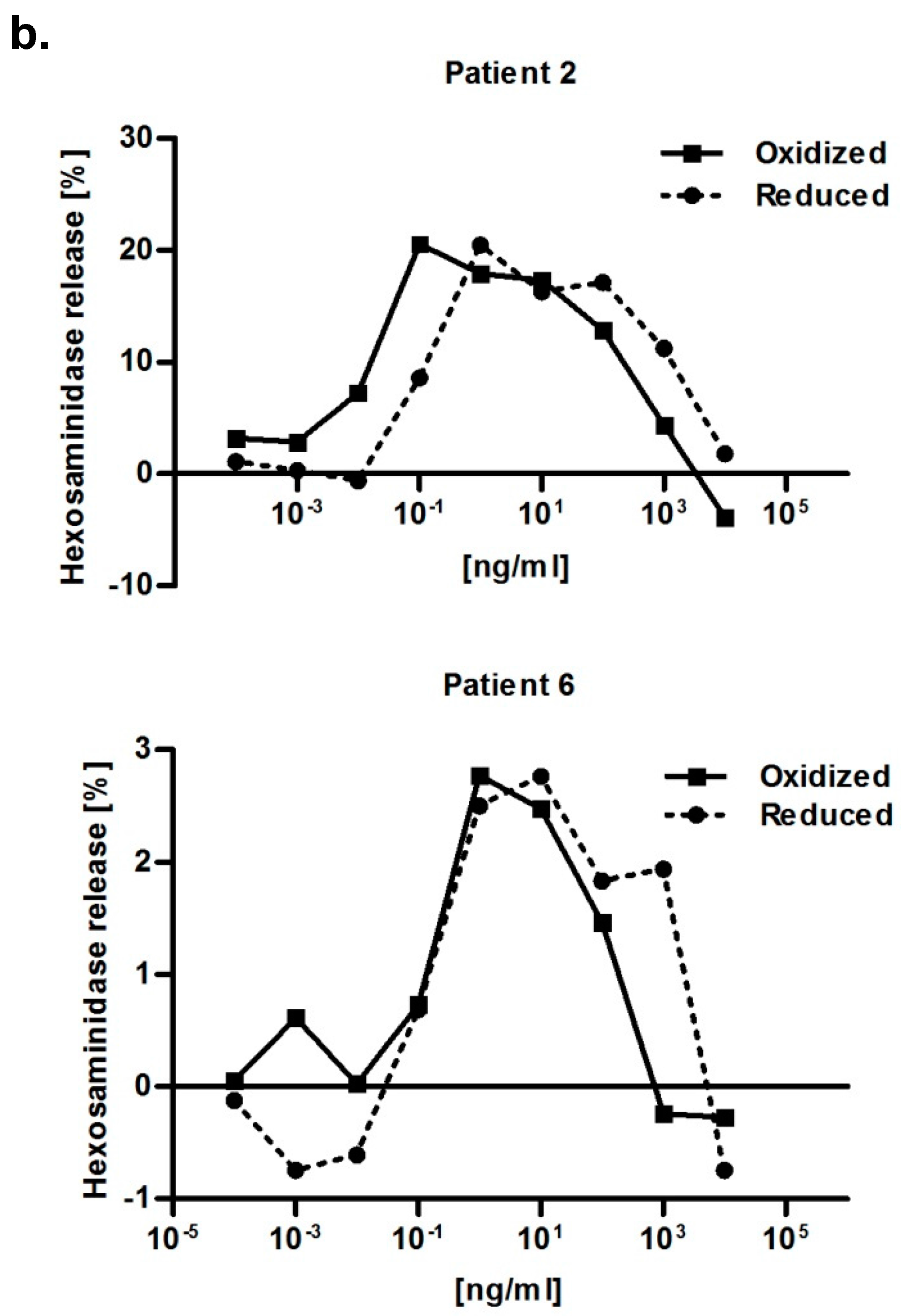
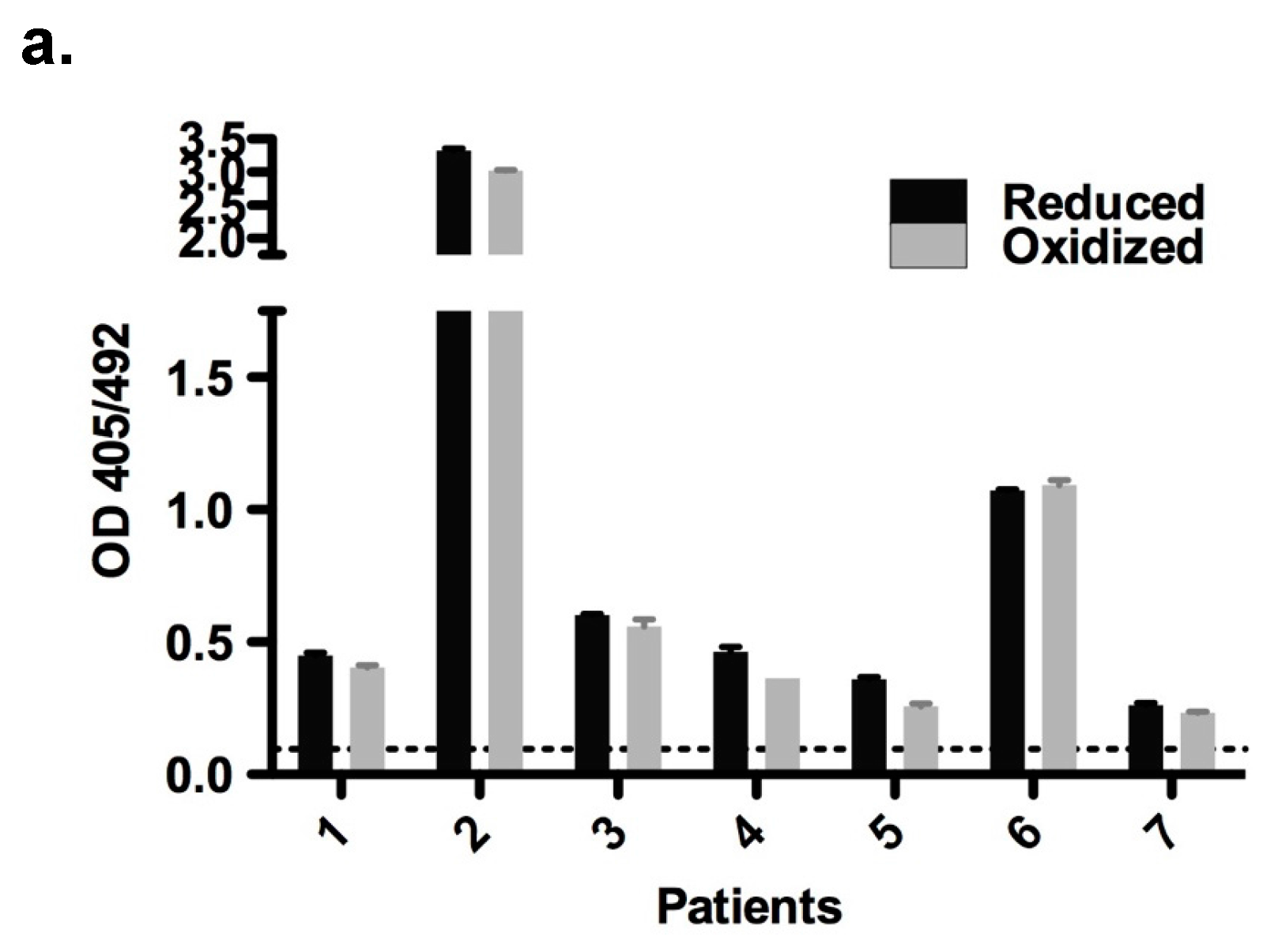
2.6. Immunological Properties of Bet v 2
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression and Purification of Bet v 2
4.2. Mass Spectrometry Analyses
4.3. Crystallization and Structure Determination
4.4. Proteolytic Processing Assay
4.5. Patients and Sera
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. RBL Mediator Release Assay
4.8. NMR One-Dimensional Spectrum
4.9. Circular Dichroism (CD) Spectroscopy
4.10. Fourier Transform Infrared (FTIR) Spectroscopy
4.11. Thermal Shift Assay
4.12. Sequence Alignment and Other Computational Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DTT | Dithiothreitol |
| MHC | Major histocompatibility complex |
| PDB | Protein data bank |
| RBL | Rat basophil leukemia |
| IgE | Immunoglobulin E |
| MS | Mass spectrometer |
| NMR | Nuclear magnetic resonance spectroscopy |
| CD | Circular dichroism spectroscopy |
| FTIR | Fourier transform infrared spectroscopy |
| RMSD | Root-mean-square deviation |
| ELISA | Enzyme-linked immunosorbent assay |
| IPTG | Isopropyl β-d-1-thiogalactopyranoside |
| TBS | Tris-buffered saline |
| TFA | Trifluoroacetic acid |
| ACN | Acetonitrile |
| ESRF | European synchrotron radiation facility |
| ATCC | American type culture collection |
| FCS | Fetal calf serum |
| BSA | Bovine serum albumin |
| EDTA | Ethylenediaminetetraacetic acid |
| BES | N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid |
References
- Burbach, G.J.; Heinzerling, L.M.; Edenharter, G.; Bachert, C.; Bindslev-Jensen, C.; Bonini, S.; Bousquet, J.; Bousquet-Rouanet, L.; Bousquet, P.J.; Bresciani, M.; et al. GA2LEN skin test study II: Clinical relevance of inhalant allergen sensitizations in Europe. Allergy Eur. J. Allergy Clin. Immunol. 2009, 64, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Breiteneder, H.; Pettenburger, K.; Bito, A.; Valenta, R.; Kraft, D.; Rumpold, H.; Scheiner, O.; Breitenbach, M. The gene coding for the major birch pollen allergen Betv1, is highly homologous to a pea disease resistance response gene. Embo J. 1989, 8, 1935–1938. [Google Scholar] [PubMed]
- Ciprandi, G.; Comite, P.; Mussap, M.; de Amici, M.; Quaglini, S.; Barocci, F.; Marseglia, G.L.; Scala, E. Profiles of birch sensitization (Bet v 1, Bet v 2, and Bet v 4) and oral allergy syndrome across Italy. J. Investig. Allergol. Clin. Immunol. 2016, 26, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, S.; Ren, H. Profilin as a regulator of the membrane-actin cytoskeleton interface in plant cells. Front. Plant Sci. 2013, 4, 512. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.; Roulias, A.; Ferreira, F.; Egger, M. Panallergens and their impact on the allergic patient. Allergy Asthma Clin. Immunol. 2010, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.; Hauser, M.; Egger, M.; Wallner, M.; Wopfner, N.; Schmidt, G. Molecular properties of plant food allergens: A current classification into protein families. Open Immunol. J. 2008, 1, 1–12. [Google Scholar] [CrossRef]
- Ramachandran, S.; Christensen, H.E.M.; Ishimaru, Y.; Dong, C.-H.; Chao-Ming, W.; Cleary, A.L.; Chua, N.-H. Profilin plays a role in cell elongation, cell shape maintenance, and flowering in Arabidopsis. Plant Physiol. 2000, 124, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Lopez, J.C.; Rodríguez-García, M.I.; Alché, J.D. Analysis of the effects of polymorphism on pollen profilin structural functionality and the generation of conformational, T- and B-cell epitopes. PLoS ONE 2013, 8, e76066. [Google Scholar] [CrossRef] [PubMed]
- Mares-Mejía, I.; Martínez-Caballero, S.; Garay-Canales, C.; Cano-Sánchez, P.; Torres-Larios, A.; Lara-González, S.; Ortega, E.; Rodríguez-Romero, A. Structural insights into the IgE mediated responses induced by the allergens Hev b 8 and Zea m 12 in their dimeric forms. Sci. Rep. 2016, 6, 32552. [Google Scholar] [CrossRef] [PubMed]
- Offermann, L.R.; Schlachter, C.R.; Perdue, M.L.; Majorek, K.A.; He, J.Z.; Booth, W.T.; Garrett, J.; Kowal, K.; Chruszcz, M. Structural, functional, and immunological characterization of profilin panallergens Amb a 8, Art v 4, and Bet v 2. J. Biol. Chem. 2016, 291, 15447–15459. [Google Scholar] [CrossRef] [PubMed]
- Erler, A.; Hawranek, T.; Krückemeier, L.; Asam, C.; Egger, M.; Ferreira, F.; Briza, P. Proteomic profiling of birch (Betula verrucosa) pollen extracts from different origins. Proteomics 2011, 11, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Machado, Y.; Freier, R.; Scheiblhofer, S.; Thalhamer, T.; Mayr, M.; Briza, P.; Grutsch, S.; Ahammer, L.; Fuchs, J.E.; Wallnoefer, H.G.; et al. Fold stability during endolysosomal acidification is a key factor for allergenicity and immunogenicity of the major birch pollen allergen. J. Allergy Clin. Immunol. 2016, 137, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Dall, E.; Brandstetter, H. Activation of legumain involves proteolytic and conformational events, resulting in a context-and substrate-dependent activity profile. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.; Leonetti, F.; Greenbaum, D.C.; Lecaille, F.; Bogyo, M.; Brömme, D.; Ellman, J.A.; Craik, C.S. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 2006, 281, 12824–12832. [Google Scholar] [CrossRef] [PubMed]
- Mellon, M.B.; Frank, B.T.; Fang, K.C. Mast cell alpha-chymase reduces IgE recognition of birch pollen profilin by cleaving antibody-binding epitopes. J. Immunol. 2002, 168, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Doyle, H.A.; Mamula, M.J. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 2001, 22, 443–449. [Google Scholar] [CrossRef]
- Pierce, R.A.; Field, E.D.; den Haan, J.M.; Caldwell, J.A.; White, F.M.; Marto, J.A.; Wang, W.; Frost, L.M.; Blokland, E.; Reinhardus, C.; et al. Cutting edge: The HLA-A*0101-restricted HY minor histocompatibility antigen originates from DFFRY and contains a cysteinylated cysteine residue as identified by a novel mass spectrometric technique. J. Immunol. 1999, 163, 6360–6364. [Google Scholar] [PubMed]
- Meadows, L.; Wang, W.; den Haan, J.M.M.; Blokland, E.; Reinhardus, C.; Drijfhout, J.W.; Shabanowitz, J.; Pierce, R.; Agulnik, A.I.; Bishop, C.E.; et al. The HLA-A*0201-restricted H-Y antigen contains a posttranslationally modified cysteine that significantly affects T cell recognition. Immunity 1997, 6, 273–281. [Google Scholar] [CrossRef]
- Egger, M.; Jürets, A.; Wallner, M.; Briza, P.; Ruzek, S.; Hainzl, S.; Pichler, U.; Kitzmüller, C.; Bohle, B.; Huber, C.G.; et al. Assessing protein immunogenicity with a dendritic cell line-derived endolysosomal degradome. PLoS ONE 2011, 6, e17278. [Google Scholar] [CrossRef] [PubMed]
- Freier, R.; Dall, E.; Brandstetter, H. Protease recognition sites in Bet v 1a are cryptic, explaining its slow processing relevant to its allergenicity. Sci. Rep. 2015, 5, 12707. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.A.; Ball, T.; Mahoney, N.M.; Valenta, R.; Almo, S.C. The molecular basis for allergen cross-reactivity: Crystal structure and IgE-epitope mapping of birch pollen profilin. Structure 1997, 5, 33–45. [Google Scholar] [CrossRef]
- Vrtala, S.; Mayer, P.; Ferreira, F.; Susani, M.; Sehon, A.H.; Kraft, D.; Valenta, R. Induction of IgE antibodies in mice and rhesus monkeys with recombinant birch pollen allergens: Different allergenicity of Bet v 1 and Bet v 2. J. Allergy Clin. Immunol. 1996, 98, 913–921. [Google Scholar] [CrossRef]
- Rinner, O.; Seebacher, J.; Walzthoeni, T.; Mueller, L.; Beck, M.; Schmidt, A.; Mueller, M.; Aebersold, R. Identification of cross-linked peptides from large sequence databases. Nat. Methods 2008, 5, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Dall, E.; Brandstetter, H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 10940–10945. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L.; Luttkopf, D.; Hatahet, L.; Haustein, D.; Vieths, S. Development of a functional in vitro assay as a novel tool for the standardization of allergen extracts in the human system. Allergy 2005, 60, 1021–1028. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bet v 2 | S-S (%) | S-H (%) |
|---|---|---|
| Oxidized | 100 | 0 |
| Reduced | 11 | 89 |
| PDB Code | 5NZB | 5NZC |
|---|---|---|
| Structure | Oxidized Bet v 2 | Reduced Bet v 2 |
| Data collection | ||
| Wavelength (Å) | 0.9677 | 0.9677 |
| Unit cell parameters | ||
| a, b, c (Å) | 31.76, 57.34, 59.04 | 74.40, 90.52, 83.17 |
| α, β, γ (degrees) | α = β = γ = 90 | α = β = γ = 90 |
| Space group | P212121 | C2221 |
| Solvent content (%) | 34.61 | 49.79 |
| Protein chains in AU | 1 | 2 |
| Resolution range (Å) | 41.13–1.69 | 41.59–2.00 |
| Highest resolution shell (Å) | 1.80–1.69 | 2.12–2.00 |
| Unique reflections | 12,513 (1969) | 19,093 (3009) |
| Redundancy | 16.12 (16.47) | 5.67 (5.63) |
| CC1/2 (%) | 99.8 (92.4) | 99.9 (85.5) |
| Completeness (%) | 99.6 (99.1) | 98.4 (97.2) |
| *Rmerge | 0.079 (0.640) | 0.074 (0.547) |
| *Rmeas | 0.082 (0.660) | 0.081 (0.605) |
| Average I/σ(I) | 20.21 (4.37) | 14.35 (3.13) |
| Refinement | ||
| Rwork (%) | 19.48 | 20.58 |
| Rfree (%) | 22.44 | 23.22 |
| Mean B value (Å2) | 29 | 34 |
| B from Wilson plot (Å2) | 23.1 | 30.7 |
| RMSD bond length (Å) | 0.009 | 0.006 |
| RMSD bond angles (degrees) | 0.989 | 0.905 |
| No. of amino acid residues | 132 | 124 |
| No. of water molecules | 99 | 91 |
| Ramachandran plot | ||
| Most favored regions (%) | 96 | 98 |
| Additional allowed regions (%) | 4 | 2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soh, W.T.; Briza, P.; Dall, E.; Asam, C.; Schubert, M.; Huber, S.; Aglas, L.; Bohle, B.; Ferreira, F.; Brandstetter, H. Two Distinct Conformations in Bet v 2 Determine Its Proteolytic Resistance to Cathepsin S. Int. J. Mol. Sci. 2017, 18, 2156. https://doi.org/10.3390/ijms18102156
Soh WT, Briza P, Dall E, Asam C, Schubert M, Huber S, Aglas L, Bohle B, Ferreira F, Brandstetter H. Two Distinct Conformations in Bet v 2 Determine Its Proteolytic Resistance to Cathepsin S. International Journal of Molecular Sciences. 2017; 18(10):2156. https://doi.org/10.3390/ijms18102156
Chicago/Turabian StyleSoh, Wai Tuck, Peter Briza, Elfriede Dall, Claudia Asam, Mario Schubert, Sara Huber, Lorenz Aglas, Barbara Bohle, Fatima Ferreira, and Hans Brandstetter. 2017. "Two Distinct Conformations in Bet v 2 Determine Its Proteolytic Resistance to Cathepsin S" International Journal of Molecular Sciences 18, no. 10: 2156. https://doi.org/10.3390/ijms18102156
APA StyleSoh, W. T., Briza, P., Dall, E., Asam, C., Schubert, M., Huber, S., Aglas, L., Bohle, B., Ferreira, F., & Brandstetter, H. (2017). Two Distinct Conformations in Bet v 2 Determine Its Proteolytic Resistance to Cathepsin S. International Journal of Molecular Sciences, 18(10), 2156. https://doi.org/10.3390/ijms18102156