Metaplasia in the Stomach—Precursor of Gastric Cancer?


Abstract
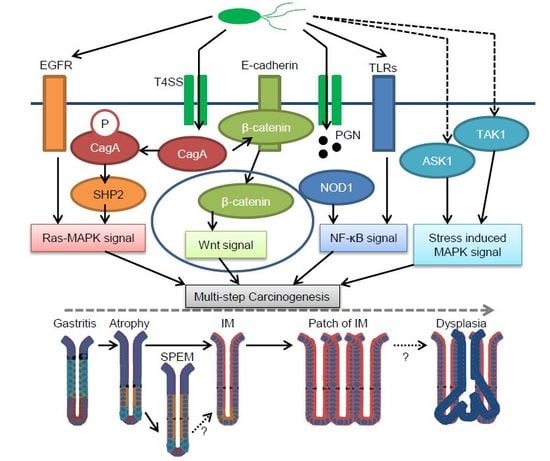
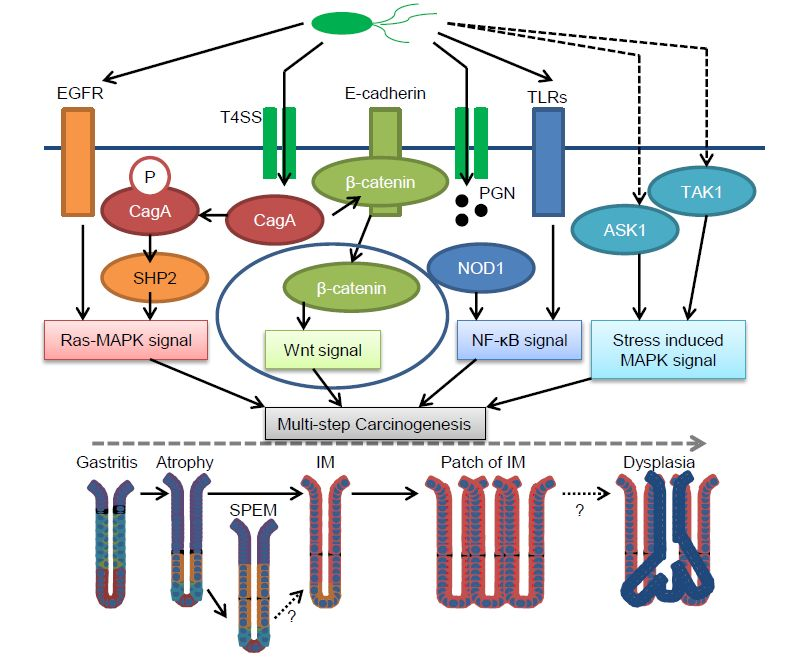
1. Human Gastric Cancer Pathogenesis
2. Mouse Models of Metaplasia
3. Origins of Metaplasia
4. Cellular Responses during H. pylori Infection
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Shiao, Y.H. Phenotypic and genotypic events in gastric carcinogenesis. Cancer Res. 1994, 54, 1941s–1943s. [Google Scholar] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First american cancer society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Satoh, K.; Ido, K.; Taniguchi, Y.; Takimoto, T.; Takemoto, T. Gastritis in the Japanese stomach. Scand. J. Gastroenterol. 1996, 31, 17–20. [Google Scholar] [CrossRef]
- Shichijo, S.; Hirata, Y.; Sakitani, K.; Yamamoto, S.; Serizawa, T.; Niikura, R.; Watabe, H.; Yoshida, S.; Yamada, A.; Yamaji, Y.; et al. Distribution of intestinal metaplasia as a predictor of gastric cancer development. J. Gastroenterol. Hepatol. 2015, 30, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Rugge, M.; de Boni, M.; Pennelli, G.; de Bona, M.; Giacomelli, L.; Fassan, M.; Basso, D.; Plebani, M.; Graham, D.Y. Gastritis olga-staging and gastric cancer risk: A twelve-year clinico-pathological follow-up study. Aliment. Pharmacol. Ther. 2010, 31, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Capelle, L.G.; de Vries, A.C.; Haringsma, J.; Ter Borg, F.; de Vries, R.A.; Bruno, M.J.; van Dekken, H.; Meijer, J.; van Grieken, N.C.; Kuipers, E.J. The staging of gastritis with the olga system by using intestinal metaplasia as an accurate alternative for atrophic gastritis. Gastrointest. Endosc. 2010, 71, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Tominaga, S.; Ito, Y.; Kobayashi, S.; Yoshii, Y.; Matsuura, A.; Kameya, A.; Kano, T.; Ikari, A. A prospective study of atrophic gastritis and stomach cancer risk. Jpn. J. Cancer Res. 1992, 83, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Cassaro, M.; Rugge, M.; Gutierrez, O.; Leandro, G.; Graham, D.Y.; Genta, R.M. Topographic patterns of intestinal metaplasia and gastric cancer. Am. J. Gastroenterol. 2000, 95, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.C.; van Grieken, N.C.; Looman, C.W.; Casparie, M.K.; de Vries, E.; Meijer, G.A.; Kuipers, E.J. Gastric cancer risk in patients with premalignant gastric lesions: A nationwide cohort study in the Netherlands. Gastroenterology 2008, 134, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Sakitani, K.; Hirata, Y.; Watabe, H.; Yamada, A.; Sugimoto, T.; Yamaji, Y.; Yoshida, H.; Maeda, S.; Omata, M.; Koike, K. Gastric cancer risk according to the distribution of intestinal metaplasia and neutrophil infiltration. J. Gastroenterol. Hepatol. 2011, 26, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Kim, N.; Lee, H.S.; Choe, G.; Jo, S.Y.; Chon, I.; Choi, C.; Yoon, H.; Shin, C.M.; Park, Y.S.; et al. Correlation between endoscopic and histological diagnoses of gastric intestinal metaplasia. Gut Liver 2013, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Shichijo, S.; Hirata, Y.; Niikura, R.; Hayakawa, Y.; Yamada, A.; Ushiku, T.; Fukayama, M.; Koike, K. Histological intestinal metaplasia and endoscopic atrophy are predictors of gastric cancer development after Helicobacter pylori eradication. Gastrointest. Endosc. 2016, 84, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef]
- Ogura, K.; Hirata, Y.; Yanai, A.; Shibata, W.; Ohmae, T.; Mitsuno, Y.; Maeda, S.; Watabe, H.; Yamaji, Y.; Okamoto, M.; et al. The effect of Helicobacter pylori eradication on reducing the incidence of gastric cancer. J. Clin. Gastroenterol. 2008, 42, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Mera, R.M.; Bravo, L.E.; Camargo, M.C.; Bravo, J.C.; Delgado, A.G.; Romero-Gallo, J.; Yepez, M.C.; Realpe, J.L.; Schneider, B.G.; Morgan, D.R.; et al. Dynamics of Helicobacter pylori infection as a determinant of progression of gastric precancerous lesions: 16-year follow-up of an eradication trial. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, L.; Shi, R.; Huang, X.; Li, S.W.; Huang, Z.; Zhang, G. Gastric atrophy and intestinal metaplasia before and after Helicobacter pylori eradication: A meta-analysis. Digestion 2011, 83, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Murakami, K.; Okimoto, T.; Sato, R.; Uchida, M.; Abe, T.; Shiota, S.; Nakagawa, Y.; Mizukami, K.; Fujioka, T. Ten-year prospective follow-up of histological changes at five points on the gastric mucosa as recommended by the updated sydney system after Helicobacter pylori eradication. J. Gastroenterol. 2012, 47, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Fontham, E.T.; Bravo, J.C.; Bravo, L.E.; Ruiz, B.; Zarama, G.; Realpe, J.L.; Malcom, G.T.; Li, D.; Johnson, W.D.; et al. Chemoprevention of gastric dysplasia: Randomized trial of antioxidant supplements and anti-Helicobacter pylori therapy. J. Natl. Cancer Inst. 2000, 92, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Rinaldi, V.; Hassan, C.; Diana, F.; Winn, S.; Castagna, G.; Attili, A.F. Ascorbic acid and intestinal metaplasia in the stomach: A prospective, randomized study. Aliment. Pharmacol. Ther. 2000, 14, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.J.; Yi, H.G.; Dai, J.C.; Wei, M.X. Histological changes of gastric mucosa after Helicobacter pylori eradication: A systematic review and meta-analysis. World J. Gastroenterol. 2014, 20, 5903–5911. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, K.L.; Hopker, W.W. Loss of differentiation in intestinal metaplasia in cancerous stomachs. A comparative morphologic study. Pathol. Res. Pract. 1979, 164, 249–258. [Google Scholar] [CrossRef]
- Filipe, M.I.; Munoz, N.; Matko, I.; Kato, I.; Pompe-Kirn, V.; Jutersek, A.; Teuchmann, S.; Benz, M.; Prijon, T. Intestinal metaplasia types and the risk of gastric cancer: A cohort study in Slovenia. Int. J. Cancer 1994, 57, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, A.; Iishi, H.; Ishiguro, S.; Tatsuta, M.; Nakae, Y.; Merchant, J.L. Epithelial cell turnover in relation to ongoing damage of the gastric mucosa in patients with early gastric cancer: Increase of cell proliferation in paramalignant lesions. J. Gastroenterol. 2005, 40, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Pittayanon, R.; Rerknimitr, R.; Klaikaew, N.; Sanpavat, A.; Chaithongrat, S.; Mahachai, V.; Kullavanijaya, P.; Barkun, A. The risk of gastric cancer in patients with gastric intestinal metaplasia in 5-year follow-up. Aliment. Pharmacol. Ther. 2017, 46, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.A.; Sanz-Anquela, J.M.; Companioni, O.; Bonet, C.; Berdasco, M.; Lopez, C.; Mendoza, J.; Martin-Arranz, M.D.; Rey, E.; Poves, E.; et al. Incomplete type of intestinal metaplasia has the highest risk to progress to gastric cancer: Results of the spanish follow-up multicenter study. J. Gastroenterol. Hepatol. 2016, 31, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Wei, W.Y.; Kong, F.B.; Lian, C.; Luo, W.; Xiao, Q.; Xie, Y.B. Prognostic significance of Cdx2 immunohistochemical expression in gastric cancer: A meta-analysis of published literatures. J. Exp. Clin. Cancer Res. 2012, 31, 98. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.; Piazuelo, M.B.; Wilson, K.T. Pathology of gastric intestinal metaplasia: Clinical implications. Am. J. Gastroenterol. 2010, 105, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Rokkas, T.; Filipe, M.I.; Sladen, G.E. Detection of an increased incidence of early gastric cancer in patients with intestinal metaplasia type III who are closely followed up. Gut 1991, 32, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Gonzalez, L.; Graham, T.A.; Rodriguez-Justo, M.; Leedham, S.J.; Novelli, M.R.; Gay, L.J.; Ventayol-Garcia, T.; Green, A.; Mitchell, I.; Stoker, D.L.; et al. The clonal origins of dysplasia from intestinal metaplasia in the human stomach. Gastroenterology 2011, 140, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, A.; Yamauchi, Y.; Hirohashi, S. P53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. Int. J. Cancer 1996, 69, 28–33. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Sethi, N.; Sepulveda, A.R.; Bass, A.J.; Wang, T.C. Oesophageal adenocarcinoma and gastric cancer: Should we mind the gap? Nat. Rev. Cancer 2016, 16, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Yu, J.; To, K.F.; Go, M.Y.; Ma, P.K.; Chan, F.K.; Sung, J.J. Apoptosis and proliferation in Helicobacter pylori-associated gastric intestinal metaplasia. Aliment. Pharmacol. Ther. 2001, 15, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Fox, J.G.; Gonda, T.; Worthley, D.L.; Muthupalani, S.; Wang, T.C. Mouse models of gastric cancer. Cancers 2013, 5, 92–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.C.; Goldenring, J.R.; Dangler, C.; Ito, S.; Mueller, A.; Jeon, W.K.; Koh, T.J.; Fox, J.G. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with helicobacter felis infection. Gastroenterology 1998, 114, 675–689. [Google Scholar] [CrossRef]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Roth, K.A.; Kapadia, S.B.; Martin, S.M.; Lorenz, R.G. Cellular immune responses are essential for the development of helicobacter felis-associated gastric pathology. J. Immunol. 1999, 163, 1490–1497. [Google Scholar] [PubMed]
- Lee, Y.; Urbanska, A.M.; Hayakawa, Y.; Wang, H.; Au, A.S.; Luna, A.M.; Chang, W.; Jin, G.; Bhagat, G.; Abrams, J.A.; et al. Gastrin stimulates a cholecystokinin-2-receptor-expressing cardia progenitor cell and promotes progression of Barrett’s-like esophagus. Oncotarget 2017, 8, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Tu, S.; Dubeykovskaya, Z.A.; Whary, M.T.; Muthupalani, S.; Rickman, B.H.; Rogers, A.B.; Lertkowit, N.; Varro, A.; Fox, J.G.; et al. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. Am. J. Pathol. 2009, 175, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Chang, W.; Jin, G.; Wang, T.C. Gastrin and upper GI cancers. Curr. Opin. Pharmacol. 2016, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Jin, G.; Wang, H.; Chen, X.; Westphalen, C.B.; Asfaha, S.; Renz, B.W.; Ariyama, H.; Dubeykovskaya, Z.A.; Takemoto, Y.; et al. CCK2R identifies and regulates gastric antral stem cell states and carcinogenesis. Gut 2015, 64, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Takaishi, S.; Menheniott, T.R.; Yang, X.; Shibata, W.; Jin, G.; Betz, K.S.; Kawakami, K.; Minamoto, T.; Tomasetto, C.; et al. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology 2011, 140, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Soutto, M.; Belkhiri, A.; Piazuelo, M.B.; Schneider, B.G.; Peng, D.; Jiang, A.; Washington, M.K.; Kokoye, Y.; Crowe, S.E.; Zaika, A.; et al. Loss of TFF1 is associated with activation of NF–κB-mediated inflammation and gastric neoplasia in mice and humans. J. Clin. Investig. 2011, 121, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; O’Rourke, J.; de Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Arnold, I.C.; Lee, J.Y.; Amieva, M.R.; Roers, A.; Flavell, R.A.; Sparwasser, T.; Muller, A. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 2011, 140, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, T.; Hirata, Y.; Hayakawa, Y.; Suzuki, N.; Sakitani, K.; Hikiba, Y.; Ihara, S.; Kinoshita, H.; Nakagawa, H.; Tateishi, K.; et al. Gastric metaplasia induced by Helicobacter pylori is associated with enhanced SOX9 expression via interleukin-1 signaling. Infect. Immun. 2015, 84, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Sayi, A.; Kohler, E.; Toller, I.M.; Flavell, R.A.; Müller, W.; Roers, A.; Müller, A. TLR-2-activated b cells suppress helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J. Immunol. 2011, 186, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Thamphiwatana, S.; Carmona, E.M.; Rickman, B.; Doran, K.S.; Obonyo, M. Deficiency of the myeloid differentiation primary response molecule MyD88 leads to an early and rapid development of helicobacter-induced gastric malignancy. Infect. Immun. 2014, 82, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, R.E.; Rose, S.; Westphalen, C.B.; Shibata, W.; Muthupalani, S.; Tailor, Y.; Friedman, R.A.; Han, W.; Fox, J.G.; Ferrante, A.W., Jr.; et al. Obesity accelerates helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut 2014, 63, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of commensal flora in Helicobacter pylori-infected INS—GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011, 140, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Syu, L.J.; El-Zaatari, M.; Eaton, K.A.; Liu, Z.; Tetarbe, M.; Keeley, T.M.; Pero, J.; Ferris, J.; Wilbert, D.; Kaatz, A.; et al. Transgenic expression of interferon-γ in mouse stomach leads to inflammation, metaplasia, and dysplasia. Am. J. Pathol. 2012, 181, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Ariyama, H.; Westphalen, C.B.; Worthley, D.L.; Muthupalani, S.; Asfaha, S.; Dubeykovskaya, Z.; Quante, M.; Fox, J.G.; Wang, T.C. Stromal cell-derived factor-1 overexpression induces gastric dysplasia through expansion of stromal myofibroblasts and epithelial progenitors. Gut 2013, 62, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, S.; Dubeykovskiy, A.N.; Tomita, H.; Yang, X.; Stokes, S.; Shibata, W.; Friedman, R.A.; Ariyama, H.; Dubeykovskaya, Z.A.; Muthupalani, S.; et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology 2013, 144, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Buzzelli, J.N.; Chalinor, H.V.; Pavlic, D.I.; Sutton, P.; Menheniott, T.R.; Giraud, A.S.; Judd, L.M. IL33 is a stomach alarmin that initiates a skewed TH2 response to injury and infection. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Ohtani, M.; Jones, E.K.; Wang, T.C. Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2−/− C57BL6 × Sv129 Helicobacter pylori-infected mice. Am. J. Pathol. 2007, 171, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Fox, J.G.; Wang, T.C. Isthmus stem cells are the origins of metaplasia in the gastric corpus. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.H.; Lee, J.R.; Joshi, V.; Playford, R.J.; Poulsom, R.; Wright, N.A.; Goldenring, J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab. Investig. 1999, 79, 639–646. [Google Scholar] [PubMed]
- Goldenring, J.R.; Nam, K.T.; Wang, T.C.; Mills, J.C.; Wright, N.A. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: Time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology 2010, 138, 2207–2210. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.J.; Menheniott, T.R.; O’Connor, L.; Walduck, A.K.; Fox, J.G.; Kawakami, K.; Minamoto, T.; Ong, E.K.; Wang, T.C.; Judd, L.M.; et al. Helicobacter pylori infection promotes methylation and silencing of trefoil factor 2, leading to gastric tumor development in mice and humans. Gastroenterology 2010, 139, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Silberg, D.G.; Sullivan, J.; Kang, E.; Swain, G.P.; Moffett, J.; Sund, N.J.; Sackett, S.D.; Kaestner, K.H. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology 2002, 122, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, H.; Hakamata, Y.; Sato, K.; Eda, A.; Yanaka, I.; Honda, S.; Osawa, H.; Kaneko, Y.; Sugano, K. Conversion of gastric mucosa to intestinal metaplasia in cdx2-expressing transgenic mice. Biochem. Biophys. Res. Commun. 2002, 294, 470–479. [Google Scholar] [CrossRef]
- Mutoh, H.; Sakurai, S.; Satoh, K.; Osawa, H.; Hakamata, Y.; Takeuchi, T.; Sugano, K. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: Comparative study with Cdx2 transgenic mice. Gut 2004, 53, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, H.; Sakurai, S.; Satoh, K.; Tamada, K.; Kita, H.; Osawa, H.; Tomiyama, T.; Sato, Y.; Yamamoto, H.; Isoda, N.; et al. Development of gastric carcinoma from intestinal metaplasia in Cdx2-transgenic mice. Cancer Res. 2004, 64, 7740–7747. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Ericksen, R.E.; Takaishi, S.; Wang, S.S.; Dubeykovskiy, Z.; Shibata, W.; Betz, K.S.; Muthupalani, S.; Rogers, A.B.; Fox, J.G.; et al. K-ras mutation targeted to gastric tissue progenitor cells results in chronic inflammation, an altered microenvironment, and progression to intraepithelial neoplasia. Cancer Res. 2010, 70, 8435–8445. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.C.; Bell, K.M.; Yan, J.; Gu, G.; Chung, C.H.; Washington, M.K.; Means, A.L. Epithelial tissues have varying degrees of susceptibility to krasG12D-initiated tumorigenesis in a mouse model. PLoS ONE 2011, 6, e16786. [Google Scholar] [CrossRef] [PubMed]
- Matkar, S.S.; Durham, A.; Brice, A.; Wang, T.C.; Rustgi, A.K.; Hua, X. Systemic activation of K-ras rapidly induces gastric hyperplasia and metaplasia in mice. Am. J. Cancer Res. 2011, 1, 432–445. [Google Scholar] [PubMed]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Hendley, A.M.; Bailey, J.M.; Leach, S.D.; Goldenring, J.R. Expression of activated ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology 2016, 150, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Thiem, S.; Eissmann, M.F.; Elzer, J.; Jonas, A.; Putoczki, T.L.; Poh, A.; Nguyen, P.; Preaudet, A.; Flanagan, D.; Waring, P.; et al. Stomach-specific activation of oncogenic kRAS and STAT3-dependent inflammation cooperatively promote gastric tumorigenesis in a preclinical model. Cancer Res. 2016, 76, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, J.; Kimura, S.; Yamamura, A.; Koh, C.P.; Hossain, M.Z.; Heng, D.L.; Kohu, K.; Chih-Cheng Voon, D.; Hiai, H.; Unno, M.; et al. Identification of stem cells in the epithelium of the stomach corpus and antrum of mice. Gastroenterology 2016, 152, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Leushacke, M.; Tan, S.H.; Wong, A.; Swathi, Y.; Hajamohideen, A.; Tan, L.T.; Goh, J.; Wong, E.; Denil, S.; Murakami, K.; et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat. Cell. Biol. 2017, 19, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R.; Ray, G.S.; Soroka, C.J.; Smith, J.; Modlin, I.M.; Meise, K.S.; Coffey, R.J., Jr. Overexpression of transforming growth factor-α alters differentiation of gastric cell lineages. Dig. Dis. Sci. 1996, 41, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Fukusato, T.; Kawaharada, U.; Kuboyama, S.; Merlino, G.; Tsutsumi, Y. Histochemical analysis of hyperplastic stomach of TGF-α transgenic mice. Dig. Dis. Sci. 1997, 42, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; Lee, H.J.; Mok, H.; Romero-Gallo, J.; Crowe, J.E., Jr.; Peek, R.M., Jr.; Goldenring, J.R. Amphiregulin-deficient mice develop spasmolytic polypeptide expressing metaplasia and intestinal metaplasia. Gastroenterology 2009, 136, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; O’Neal, R.; Lee, Y.S.; Lee, Y.C.; Coffey, R.J.; Goldenring, J.R. Gastric tumor development in Smad3-deficient mice initiates from forestomach/glandular transition zone along the lesser curvature. Lab. Investig. 2012, 92, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Mao, M.; Keeley, T.M.; El-Zaatari, M.; Lee, H.J.; Eaton, K.A.; Samuelson, L.C.; Merchant, J.L.; Goldenring, J.R.; Todisco, A. Bone morphogenetic protein signaling regulates gastric epithelial cell development and proliferation in mice. Gastroenterology 2010, 139, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, J.; van den Brink, G.R. Morphogens and the parietal cell: Shaping up acid secretion. Gastroenterology 2010, 139, 1830–1833. [Google Scholar] [CrossRef] [PubMed]
- Bleuming, S.A.; He, X.C.; Kodach, L.L.; Hardwick, J.C.; Koopman, F.A.; Ten Kate, F.J.; van Deventer, S.J.; Hommes, D.W.; Peppelenbosch, M.P.; Offerhaus, G.J.; et al. Bone morphogenetic protein signaling suppresses tumorigenesis at gastric epithelial transition zones in mice. Cancer Res. 2007, 67, 8149–8155. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Yamaguchi, H.; Ogawa, M.; Wang, T.C.; Lee, J.R.; Goldenring, J.R. Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild-type and gastrin-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G362–375. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; Lee, H.J.; Sousa, J.F.; Weis, V.G.; O’Neal, R.L.; Finke, P.E.; Romero-Gallo, J.; Shi, G.; Mills, J.C.; Peek, R.M., Jr.; et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010, 139, 2028–2037 e2029. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.J.; Khurana, S.S.; Geahlen, J.H.; Kohli, K.; Waller, R.A.; Mills, J.C. Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 2012, 142, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Burclaff, J.; Osaki, L.H.; Liu, D.; Goldenring, J.R.; Mills, J.C. Targeted apoptosis of parietal cells is insufficient to induce metaplasia in stomach. Gastroenterology 2016, 152, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.P.; Weis, V.G.; Nam, K.T.; Sousa, J.F.; Fingleton, B.; Goldenring, J.R. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology 2014, 146, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.P.; Meyer, A.R.; DeSalvo, C.; Choi, E.; Schlegel, C.; Petersen, A.; Engevik, A.C.; Prasad, N.; Levy, S.E.; Peebles, R.S.; et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Lantz, T.L.; Vlacich, G.; Keeley, T.M.; Samuelson, L.C.; Coffey, R.J.; Goldenring, J.R.; Powell, A.E. Lrig1+ gastric isthmal progenitor cells restore normal gastric lineage cells during damage recovery in adult mouse stomach. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Menheniott, T.R.; O’Connor, L.; Chionh, Y.T.; Dabritz, J.; Scurr, M.; Rollo, B.N.; Ng, G.Z.; Jacobs, S.; Catubig, A.; Kurklu, B.; et al. Loss of gastrokine-2 drives premalignant gastric inflammation and tumor progression. J. Clin. Investig. 2016, 126, 1383–1400. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Wang, T.C. Isthmus progenitors, not chief cells, are the likely origin of metaplasia in eR1–CreERT; LSL-KrasG12D mice. Gastroenterology 2017, 152, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Fox, J.G.; Wang, T.C. The origins of gastric cancer from gastric stem cells: Lessons from mouse models. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.C.; Goldenring, J.R. Metaplasia in the stomach arises from gastric chief cells. CMGH 2017, 4, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J.; Merchant, J.L.; Wang, T.C.; Chandrasoma, P.; Fox, J.G.; Genta, R.M.; Goldenring, J.R.; Hayakawa, Y.; Kuipers, E.J.; Lund, P.K.; et al. A summary of the 2016 james W. Freston conference of the american gastroenterological association: Intestinal metaplasia in the esophagus and stomach: Origins, differences, similarities and significance. Gastroenterology 2017, 153, e6–e13. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; O’Neal, R.L.; Coffey, R.J.; Finke, P.E.; Barker, N.; Goldenring, J.R. Spasmolytic polypeptide-expressing metaplasia (SPEM) in the gastric oxyntic mucosa does not arise from Lgr5-expressing cells. Gut 2012, 61, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Investig. 2014, 124, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Willet, S.G.; Bankaitis, E.D.; Xu, Y.; Wright, C.V.; Gu, G. Non-parallel recombination limits cre-loxP-based reporters as precise indicators of conditional genetic manipulation. Genesis 2013, 51, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Naumann, M. What a disorder: Proinflammatory signaling pathways induced by Helicobacter pylori. Trends Microbiol. 2010, 18, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Clyne, M.; Tegtmeyer, N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell. Commun. Signal. 2011, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell. Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Sue, S.; Shibata, W.; Maeda, S. Helicobacter pylori-induced signaling pathways contribute to intestinal metaplasia and gastric carcinogenesis. Biomed. Res. Int. 2015, 2015, 737621. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing CagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar] [PubMed]
- Ogura, K.; Maeda, S.; Nakao, M.; Watanabe, T.; Tada, M.; Kyutoku, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Virulence factors of Helicobacter pylori responsible for gastric diseases in mongolian gerbil. J. Exp. Med. 2000, 192, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. cag, A pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Ziska, E.; Brinkmann, V.; Zimny-Arndt, U.; Fauconnier, A.; Jungblut, P.R.; Naumann, M.; Meyer, T.F. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type iv secretion apparatus. Cell. Microbiol 2000, 2, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Puls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Maeda, S.; Mitsuno, Y.; Tateishi, K.; Yanai, A.; Akanuma, M.; Yoshida, H.; Kawabe, T.; Shiratori, Y.; Omata, M. Helicobacter pylori CagA protein activates serum response element-driven transcription independently of tyrosine phosphorylation. Gastroenterology 2002, 123, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Mitsuno, Y.; Yoshida, H.; Maeda, S.; Ogura, K.; Hirata, Y.; Kawabe, T.; Shiratori, Y.; Omata, M. Helicobacter pylori induced transactivation of SRE and AP-1 through the ERK signalling pathway in gastric cancer cells. Gut 2001, 49, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Murata-Kamiya, N.; Hirayama, T.; Ohba, Y.; Hatakeyama, M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J. Exp. Med. 2010, 207, 2157–2174. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Maeda, S.; Mitsuno, Y.; Akanuma, M.; Yamaji, Y.; Ogura, K.; Yoshida, H.; Shiratori, Y.; Omata, M. Helicobacter pylori activates the cyclin D1 gene through mitogen-activated protein kinase pathway in gastric cancer cells. Infect. Immun. 2001, 69, 3965–3971. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Pathak, S.K.; Chatterjee, G.; Pathak, S.; Basu, J.; Kundu, M. Helicobacter pylori protein HP0175 transactivates epidermal growth factor receptor through TLR4 in gastric epithelial cells. J. Biol. Chem. 2008, 283, 32369–32376. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.C.; Asim, M.; Verriere, T.G.; Piazuelo, M.B.; Suarez, G.; Romero-Gallo, J.; Delgado, A.G.; Wroblewski, L.E.; Barry, D.P.; Peek, R.M., Jr.; et al. Epidermal growth factor receptor inhibition downregulates Helicobacter pylori-induced epithelial inflammatory responses, DNA damage and gastric carcinogenesis. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Israel, D.A.; Washington, M.K.; Krishna, U.; Fox, J.G.; Rogers, A.B.; Neish, A.S.; Collier-Hyams, L.; Perez-Perez, G.I.; Hatakeyama, M.; et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2005, 102, 10646–10651. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M., Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Bronte-Tinkew, D.M.; Terebiznik, M.; Franco, A.; Ang, M.; Ahn, D.; Mimuro, H.; Sasakawa, C.; Ropeleski, M.J.; Peek, R.M., Jr.; Jones, N.L. Helicobacter pylori cytotoxin-associated gene a activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res. 2009, 69, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.O.; Kim, J.H.; Choi, Y.J.; Pillinger, M.H.; Kim, S.Y.; Blaser, M.J.; Lee, Y.C. Helicobacter pylori CagA phosphorylation status determines the GP130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J. Biol. Chem. 2010, 285, 16042–16050. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Hirata, Y.; Kinoshita, H.; Sakitani, K.; Nakagawa, H.; Nakata, W.; Takahashi, R.; Sakamoto, K.; Maeda, S.; Koike, K. Differential roles of ASK1 and TAK1 in Helicobacter pylori-induced cellular responses. Infect. Immun. 2013, 81, 4551–4560. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.D.; Cha, J.; Lo, J.; Falkow, S.; Tompkins, L.S. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1999, 96, 14559–14564. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Ohmae, T.; Shibata, W.; Maeda, S.; Ogura, K.; Yoshida, H.; Kawabe, T.; Omata, M. MyD88 and tnf receptor-associated factor 6 are critical signal transducers in Helicobacter pylori-infected human epithelial cells. J. Immunol 2006, 176, 3796–3803. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Hirata, Y.; Nakagawa, H.; Sakamoto, K.; Hikiba, Y.; Kinoshita, H.; Nakata, W.; Takahashi, R.; Tateishi, K.; Tada, M.; et al. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Hirata, Y.; Sakitani, K.; Nakagawa, H.; Nakata, W.; Kinoshita, H.; Takahashi, R.; Takeda, K.; Ichijo, H.; Maeda, S.; et al. Apoptosis signal-regulating kinase-1 inhibitor as a potent therapeutic drug for the treatment of gastric cancer. Cancer Sci 2012, 103, 2181–2185. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Maeda, S.; Hikiba, Y.; Yanai, A.; Sakamoto, K.; Nakagawa, H.; Ogura, K.; Karin, M.; Omata, M. C-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008, 68, 5031–5039. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Teshima, S.; Kuwano, Y.; Oka, A.; Kishi, K.; Rokutan, K. Helicobacter pylori lipopolysaccharide induces apoptosis of cultured guinea pig gastric mucosal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G726–734. [Google Scholar] [PubMed]
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF–κ B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Akanuma, M.; Mitsuno, Y.; Hirata, Y.; Ogura, K.; Yoshida, H.; Shiratori, Y.; Omata, M. Distinct mechanism of Helicobacter pylori-mediated NF–κ B activation between gastric cancer cells and monocytic cells. J. Biol. Chem. 2001, 276, 44856–44864. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Kwok, T.; Hartig, R.; Konig, W.; Backert, S. Nf-kappab activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.; Yang, X.D.; Tsang, Y.H.; Li, J.D.; Higashi, H.; Hatakeyama, M.; Peek, R.M.; Blanke, S.R.; Chen, L.F. Helicobacter pylori CagA activates NF–κB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009, 10, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Nakata, W.; Hayakawa, Y.; Nakagawa, H.; Sakamoto, K.; Kinoshita, H.; Takahashi, R.; Hirata, Y.; Maeda, S.; Koike, K. Anti-tumor activity of the proteasome inhibitor bortezomib in gastric cancer. Int. J. Oncol. 2011, 39, 1529–1536. [Google Scholar] [PubMed]
- Sakitani, K.; Hirata, Y.; Hayakawa, Y.; Serizawa, T.; Nakata, W.; Takahashi, R.; Kinoshita, H.; Sakamoto, K.; Nakagawa, H.; Akanuma, M.; et al. Role of interleukin-32 in Helicobacter pylori-induced gastric inflammation. Infect. Immun. 2012, 80, 3795–3803. [Google Scholar] [CrossRef] [PubMed]
- Varro, A.; Noble, P.J.; Pritchard, D.M.; Kennedy, S.; Hart, C.A.; Dimaline, R.; Dockray, G.J. Helicobacter pylori induces plasminogen activator inhibitor 2 in gastric epithelial cells through nuclear factor-κB and RhoA: Implications for invasion and apoptosis. Cancer Res. 2004, 64, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Zhou, J.; Hayakawa, Y.; Wang, T.C.; Bass, A.J. RhoA mutations identified in diffuse gastric cancer. Cancer Cell 2014, 26, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RhoA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Maeda, S.; Ohmae, T.; Shibata, W.; Yanai, A.; Ogura, K.; Yoshida, H.; Kawabe, T.; Omata, M. Helicobacter pylori induces IκB kinase α nuclear translocation and chemokine production in gastric epithelial cells. Infect. Immun. 2006, 74, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Yoshida, H.; Ogura, K.; Mitsuno, Y.; Hirata, Y.; Yamaji, Y.; Akanuma, M.; Shiratori, Y.; Omata, M. H. Pylori activates through a signaling pathway involving ikappab kinases, NF–κB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000, 119, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Hirata, Y.; Maeda, S.; Ogura, K.; Ohmae, T.; Yanai, A.; Mitsuno, Y.; Yamaji, Y.; Okamoto, M.; Yoshida, H.; et al. Caga protein secreted by the intact type IV secretion system leads to gastric epithelial inflammation in the mongolian gerbil model. J. Pathol. 2006, 210, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Torok, A.M.; Bouton, A.H.; Goldberg, J.B. Helicobacter pylori induces interleukin-8 secretion by toll-like receptor 2- and toll-like receptor 5-dependent and -independent pathways. Infect. Immun. 2005, 73, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.F., Jr.; Mitchell, A.; Li, G.; Ding, S.; Fitzmaurice, A.M.; Ryan, K.; Crowe, S.; Goldberg, J.B. Toll-like receptor (TLR) 2 and TLR 5, but not TLR 4, are required for Helicobacter pylori-induced NF–κB activation and chemokine expression by epithelial cells. J. Biol. Chem. 2003, 278, 32552–32560. [Google Scholar] [CrossRef] [PubMed]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Memet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol 2004, 5, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IκB-kinase-β accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 2010, 138, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hikiba, Y.; Nakagawa, H.; Hayakawa, Y.; Yanai, A.; Akanuma, M.; Ogura, K.; Hirata, Y.; Kaestner, K.H.; Omata, M.; et al. Inhibitor of κB kinase β regulates gastric carcinogenesis via interleukin-1α expression. Gastroenterology 2010, 139, 226–238 e226. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Tsukamoto, T.; Toyoda, T.; Mori, A.; Tanaka, H.; Maekita, T.; Ichinose, M.; Tatematsu, M.; Ushijima, T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010, 70, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, A.; Colleran, A.; Ryan, A.; Mann, J.; Egan, L.J. Regulation of NF–κB responses by epigenetic suppression of Iκα expression in HCT116 intestinal epithelial cells. Am. J. Physiol Gastrointest Liver Physiol 2010, 299, G96–G105. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric cancer originating from bone marrow-derived cells. Science 2004, 306, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rabben, H.L.; Zhao, C.M.; Hayakawa, Y.; Wang, T.C.; Chen, D. Vagotomy and gastric tumorigenesis. Curr. Neuropharmacol. 2016, 14, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Type of Metaplasia | Histological Characteristics | Representative Markers |
|---|---|---|
| Intestinal metaplasia (IM) | Intestine-like gland structures Intestine-specific cell lineages | CDX1, CDX2 MUC2, TFF3, Villin Alcian blue staining at pH 2.5 |
| Complete IM | Small intestinal phenotype Enterocytes with brush borders Well-defined goblet cells Paneth cells | Loss of gastric mucins (MUC1, 5AC, 6) |
| Incomplete IM | Colonic gland-like structures Irregular mucin droplets | Brown staining by high iron diamine (HID) Both gastric and intestinal mucins may be present |
| Spasmolytic polypeptide-expressing metaplasia (SPEM) | Deep antral gland-like structure Expansion of a mucous neck cell-like lineage | TFF2, MUC6, GSII Alcian blue staining at pH 2.5 |
| Type of Metaplasia | Histological Characteristics | Representative Markers |
|---|---|---|
| SPEM in Helicobacter infection or after transgenic activation of Ras–MAPK signaling | Accompanied by atrophy, foveolar hyperplasia, and inflammation Dysplasia at later time points No development of IM Generally irreversible | TFF2, MUC6, GSII CD44v9, SOX9 Alcian blue staining at pH 2.5 |
| IM in transgenic Cdx1/2 mouse models | Complete IM Enterocytes with brush borders Well-defined goblet cells Paneth cells Irreversible | CDX1 or CDX2 MUC2, TFF3 Alcian blue staining at pH 2.5 Brown staining by HID |
| Acute drug-induced injury (DMP-777, L-635, high-dose tamoxifen) | Acute loss of parietal cells and mature chief cells with inflammatory infiltration Reversible after discontinuation of drug treatment | TFF2, MUC6, GSII Low expression of gastric intrinsic factor CD44v9 (DMP-777 and L-635) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinoshita, H.; Hayakawa, Y.; Koike, K. Metaplasia in the Stomach—Precursor of Gastric Cancer? Int. J. Mol. Sci. 2017, 18, 2063. https://doi.org/10.3390/ijms18102063
Kinoshita H, Hayakawa Y, Koike K. Metaplasia in the Stomach—Precursor of Gastric Cancer? International Journal of Molecular Sciences. 2017; 18(10):2063. https://doi.org/10.3390/ijms18102063
Chicago/Turabian StyleKinoshita, Hiroto, Yoku Hayakawa, and Kazuhiko Koike. 2017. "Metaplasia in the Stomach—Precursor of Gastric Cancer?" International Journal of Molecular Sciences 18, no. 10: 2063. https://doi.org/10.3390/ijms18102063
APA StyleKinoshita, H., Hayakawa, Y., & Koike, K. (2017). Metaplasia in the Stomach—Precursor of Gastric Cancer? International Journal of Molecular Sciences, 18(10), 2063. https://doi.org/10.3390/ijms18102063