GH/IGF-1 Signaling and Current Knowledge of Epigenetics; a Review and Considerations on Possible Therapeutic Options

Abstract

{kind=link}
1. Introduction
2. Intracellular Signals Regulating Growth Hormone Actions
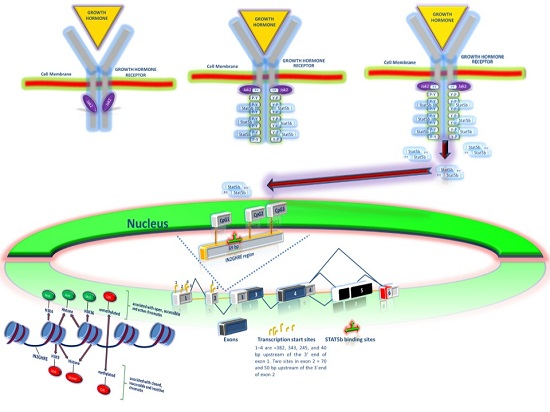
2.1. GH Signal Transduction Pathway
2.2. Intracellular Signals Regulating Growth Hormone Actions
2.3. Epigenetic Regulation of the GH-IGF-I Axis
2.3.1. Fetal Programming and Epigenetic Regulation at the IGF1 Locus
2.3.2. Epigenetic Alterations in Intrauterine Growth Retardation Open the Possibility for a New Pharmaceutical Approach in Short Statured Small for Gestational Age Subjects
3. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ChIP | Chromatin ImmunoPrecipitation |
| Cish | Cytokine Inducible SH2 Containing Protein gene [not Homo sapiens (human)] |
| DNMT | DNA Methyl Transferase |
| ECD | Extracellular Domain |
| EHMT2 | Euchromatic Histone Lysine N-Methyltransferase 2 |
| FDA | US Food and Drug Administration |
| GH | Growth Hormone |
| GHR | Growth Hormone Receptor |
| GHRE | Growth Hormone Response Element |
| H4Kac | lysine acetylation at histone 4 |
| H3K4 | Mono-methylation of lysine 4 on histone H3 |
| H3K9me3 | trimethylation (me3) of lysine 9 on histone H3 |
| H3K27me3 | trimethylation (me3) of lysine 27 on histone H3 |
| HDAC | Histone Deacetylase |
| ICD | Intracellular Domain |
| IGF1 | Insulin-like Growth Factor I gene [Homo sapiens (human)] |
| Igfals | Insulin Like Growth Factor Binding Protein Acid Labile Subunit gene [not Homo sapiens (human)] |
| IGF-I | Insulin-like Growth Factor-I (protein) |
| IN2GHRE | Intron 2 Growth Hormone Response Element |
| IUGR | Intrauterine Growth Retardation |
| JAK2 | Janus-Family Tyrosine Kinase-2 (JAK2) |
| miRNA | microRNA |
| PWS | Prader-Willi Syndrome |
| PWS-IC | Prader-Willi Syndrome-Imprinting Centre |
| rhGH | recombinant human Growth Hormone |
| SGA | Small for Gestational Age |
| SNRPN | Small Nuclear Ribonucleoprotein N Polypeptide gene (Homo sapiens) |
| Socs2 | Suppressor of cytokine signaling 2 gene (not Homo sapiens) |
| Spi2.1 | Transcription factor PU.1 gene (not Homo sapiens) |
| STAT | Signal Transducers and Activators of Transcription |
| T2DM | Type 2 Diabetes Mellitus |
| TCD | Transmembrane Domain |
References
- Dominici, F.P.; Argentino, D.P.; Muñoz, M.C.; Miquet, J.G.; Sotelo, A.I.; Turyn, D. Influence of the crosstalk between growth hormone and insulin signalling on the modulation of insulin sensitivity. Growth Horm. IGF Res. 2005, 15, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Fuh, G.; Cunningham, B.C.; Fukunaga, R.; Nagata, S.; Goeddel, D.V.; Wells, J.A. Rational Design of Potent Antagonists to the Human Growth Hormone Receptor. Science 1992, 256, 1677–1680. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J.; Hoang, H.N.; Fairlie, D.P.; Pelekanos, R.A.; Brown, R.J. New insights into growth hormone action. J. Mol. Endocrinol. 2006, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Adams, J.J.; Pelekanos, R.A.; Wan, Y.; McKinstry, W.J.; Palethorpe, K.; Seeber, R.M.; Monks, T.A.; Eidne, K.A.; Parker, M.W.; et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 2005, 12, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Behncken, S.N.; Billestrup, N.; Brown, R.; Amstrup, J.; Conway-Campbell, B.; Waters, M.J. Growth hormone (GH)-independent dimerization of GH receptor by a leucine zipper results in constitutive activation. J. Biol. Chem. 2000, 275, 17000–17007. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Schadt, E.E.; Wang, S.; Wang, H.; Arnold, A.P.; Ingram-Drake, L.; Drake, T.A.; Lusis, A.J. Tissue-specific expression and regulation of sexually dimorphic genes in mice\r10.1101/gr.5217506. Genome Res. 2006, 16, 995. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.A.; Hansen, L.H.; Wang, X.; Kopchick, J.J.; Gouilleux, F.; Groner, B.; Nielsen, J.H.; Møldrup, A.; Galsgaard, E.D.; Billestrup, N. The role of GH receptor tyrosine phosphorylation in Stat5 activation. J. Mol. Endocrinol. 1997, 18, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Smit, L.S.; Meyer, D.J.; Billestrup, N.; Schwartz, J.; Carter-su, C. The Role of the Growth Hormone ( GH ) Receptor and JAKI and JAK2 Kinases in the Activation of Stats 1, 3, and 5 by GH. Mol. Endocrinol. 1996, 10, 519–533. [Google Scholar] [PubMed]
- Bergad, P.L.; Schwarzenberg, S.J.; Humbert, J.T.; Amarasinghe, S.; Towle, H.C.; Berry, S.A.; Morrison, M.; Pearl, L. Inhibition of growth hormone action in models of inflammation. Am. J. Physiol. 2010, 55455, 1906–1917. [Google Scholar]
- Vidal, O.M.; Merino, R.; Rico-bautista, E.; Fernandez-perez, L.; Chia, D.J.; Woelfle, J.; Ono, M.; Lenhard, B.; Norstedt, G.; Rotwein, P. In Vivo Transcript Profiling and Phylogenetic Analysis Identifies Suppressor of Cytokine Signaling 2 as a Direct Signal Transducer and Activator of Transcription 5b Target in Liver. Mol. Endocrinol. 2007, 21, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Park, S.H.; Choi, H.K.; Waxman, D.J. Growth hormone activation of Stat 1, Stat 3, and Stat 5 in rat liver: Differential kinetics of hormone desensitization and growth hormone stimulation of both tyrosine phosphorylation and serine/threonine phosphorylation. J. Biol. Chem. 1996, 271, 5929–5940. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Day, N. Characterization and Cloning of STAT5 from IM-9 Cells and Its Activation by Growth Hormone. Mol. Endocrinol. 1996, 10, 508–518. [Google Scholar] [PubMed]
- Seidel, H.M.; Milocco, L.H.; Lamb, P.; Darnell, J.E.; Stein, R.B.; Rosen, J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc. Natl. Acad. Sci. USA 1995, 92, 3041–3045. [Google Scholar] [CrossRef] [PubMed]
- Gebert, C.A.; Park, S.H.; Waxman, D.J. Regulation of signal transducer and activator of transcription (STAT) 5b activation by the temporal pattern of growth hormone stimulation. Mol. Endocrinol. 1997, 11, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Gronowski, A.M.; Lestunff, C.R.P. Acute nuclear actions of growth hormone (GH): Cycloheximide inhibits inducible activator protein-1 activity, but does not block GH-regulated signal transducer and activator of transcription activation or gene expression. Endocrinology 1996, 137, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of Growth Hormone and Insulin-like Growth Factor 1 in Mouse Postnatal Growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Miquet, J.G.; Irene, P.E.; Díaz, M.E.; Rossi, S.P.; Sotelo, A.I.; Frungieri, M.B.; Hill, C.M.; Bartke, A.T.D. Attenuation of epidermal growth factor (EGF) signaling by growth hormone (GH). J. Endocrinol. 2017, 233, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Sodhi, A. Growth hormone-induced production of cytokines in murine peritoneal macrophages in vitro: Role of JAK/STAT, PI3K, PKC and MAP kinases. Immunobiology 2009, 214, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Chia, D.J.; Rotwein, P. Defining the epigenetic actions of growth hormone: Acute chromatin changes accompany GH-activated gene transcription. Mol. Endocrinol. 2010, 24, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Rotwein, P. Mapping the growth hormone-Stat5b-IGF-I transcriptional circuit. Trends Endocrinol. Metab. 2012, 23, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Oberbauer, A.M. The regulation of IGF-1 gene transcription and splicing during development and aging. Front. Endocrinol. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.I.; Stempien, M.M.; Fong, N.M.; Roll, L.B. Sequences of liver cDNAs encoding two different mouse insulin like growth factor I precursors. Nucleic Acids Res. 1986, 14, 7873–7882. [Google Scholar] [CrossRef] [PubMed]
- Lowe, W.L.; Lasky, S.R.; LeRoith, D.; Roberts, C.T. Distribution and regulation of rat insulin-like growth factor I messenger ribonucleic acids encoding alternative carboxyterminal E-peptides: Evidence for differential processing and regulation in liver. Mol. Endocrinol. 1988, 2, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Hepler, J.E.; Van Wyk, J.J.; Lund, P.K. Different half-lives of insulin-like growth factor I mRNAs that differ in length of 3' untranslated sequence. Endocrinology 1990, 127, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.R. The ABCs of IGF-I isoforms: Impact on muscle hypertrophy and implications for repair. Appl. Physiol. Nutr. Metab. 2006, 31, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Matheny, R.W.; Nindl, B.C.; Adamo, M.L. Minireview: Mechano-growth factor: A putative product of IGF-I gene expression involved in tissue repair and regeneration. Endocrinology 2010, 151, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Chia, D.J.; Varco-Merth, B.; Rotwein, P. Dispersed chromosomal Stat5b-binding elements mediate growth hormone-activated insulin-like growth factor-I gene transcription. J. Biol. Chem. 2010, 285, 17636–17647. [Google Scholar] [CrossRef] [PubMed]
- Woelfle, J.; Chia, D.J.; Rotwein, P. Mechanisms of Growth Hormone (GH) Action prior to the onset of transcription from both major and. Biochemistry 2003, 278, 51261–51266. [Google Scholar]
- Wang, Y.; Jiang, H. Identification of a Distal STAT5-binding DNA region that may mediate growth hormone regulation of insulin-like growth factor-I gene expression *. J. Biol. Chem. 2005, 280, 10955–10963. [Google Scholar] [CrossRef] [PubMed]
- Eleswarapu, S.; Gu, Z.; Jiang, H. Growth hormone regulation of insulin-like growth factor-I gene expression may be mediated by multiple distal signal transducer and activator of transcription 5 binding sites. Endocrinology 2008, 149, 2230–2240. [Google Scholar] [CrossRef] [PubMed]
- Conaway, R.C.; Sato, S.; Tomomori-Sato, C.; Yao, T.; Conaway, J.W. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem. Sci. 2005, 30, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Bilenky, M.; Tam, A.; Zhao, Y.; Zeng, T.; Thiessen, N.; Cezard, T.; Fejes, A.P.; Wederell, E.D.; Cullum, R. Others Genome-wide relationship between histone H3 lysine 4 mono-and tri-methylation and transcription factor binding. Genome Res. 2008, 18, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J. Capturing Chromosome Conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.D.; Laz, E.V.; Su, T.; Waxman, D.J. Male-Specific Hepatic Bcl6: Growth Hormone-Induced Block of Transcription Elongation in Females and Binding to Target Genes Inversely Coordinated with STAT5. Mol. Endocrinol. 2009, 23, 1914–1926. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, G.; Huo, J.S.; Barney, D.; Wang, Z.; Livshiz, T.; States, D.J.; Qin, Z.S.; Schwartz, J. Computational and functional analysis of growth hormone (GH)-regulated genes identifies the transcriptional repressor B-cell lymphoma 6 (Bc16) as a participant in GH-regulated transcription. Endocrinology 2009, 150, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.W.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG Islands Identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Bmre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are trimethylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.J.; Schneider, R.; Bauer, U.-M.; Bannister, A.J.; Morrison, A.; O’Carroll, D.; Firestein, R.; Cleary, M.; Jenuwein, T.; Herrera, R.E.; et al. Rb targets histone H3 methylation and HP1 to promoters. Nature 2001, 412, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Altucci, L.; Rots, M.G. Epigenetic drugs: From chemistry via biology to medicine and back. Clin. Epigenet. 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Rando, O.J. Combinatorial complexity in chromatin structure and function: Revisiting the histone code. Curr. Opin. Genet. Dev. 2012, 22, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; McKnight, R.A.; Callaway, C.W.; Yu, X.; Lane, R.H.; Majnik, A.V. Intrauterine growth restriction disrupts developmental epigenetics around distal growth hormone response elements on the rat hepatic IGF-1 gene. FASEB J. 2015, 29, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Shimatsu, A.; Rotwein, P. Mosaic evolution of the insulin-like growth factors. Organization, sequence, and expression of the rat insulin-like growth factor I gene. J. Biol. Chem. 1987, 262, 7894–7900. [Google Scholar] [PubMed]
- Kikuchi, K.; Bichell, D.P.; Rotwein, P. Chromatin changes accompany the developmental activation of insulin-like growth factor I gene transcription. J. Biol. Chem. 1992, 267, 21505–21511. [Google Scholar] [PubMed]
- Davey, H.W.; Xie, T.; McLachlan, M.J.; Wilkins, R.J.; Waxman, D.J.; Grattan, D.R. STAT5b is required for GH-induced liver Igf-I gene expression. Endocrinology 2001, 142, 3836–3841. [Google Scholar] [CrossRef] [PubMed]
- Papers, J.B.C.; Doi, M.; Woelfle, J.; Billiard, J.; Rotwein, P. Acute Control of Insulin-like Growth Factor-I Gene Transcription by Growth Hormone through Stat5b *. J. Biol. Chem. 2003, 278, 22696–22702. [Google Scholar]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.G.; Beall, M.H. Adult Sequelae of Intrauterine Growth Restriction. Semin. Perinatol. 2008, 32, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Varvarigou, A.A. Intrauterine Growth Restriction as a Potential Risk Factor for Disease Onset in Adulthood. J. Pediatr. Endocrinol. Metab. 2010, 23, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Yu, X.; Callaway, C.W.; Lane, R.H.; McKnight, R.A. Epigenetics: Intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. FASEB J. 2009, 23, 2438–2449. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.U.N.; Sun, H.; Danila, D.C.; Johnson, S.R.; Zhou, Y.; Swearingen, B.; Klibanski, A. Loss of Expression of GADD45γ, a Growth Inhibitory Gene, in Human Pituitary Adenomas: Implications for Tumorigenesis. J. Clin. Endocrinol. Metab. 2002, 87, 1262–1267. [Google Scholar] [PubMed]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non – Small Cell Lung Cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Gius, D.; Cui, H.; Bradbury, C.M.; Cook, J.; Smart, D.D.K.; Zhao, S.; Young, L.; Brandenburg, S.A.; Hu, Y.; Bisht, K.S.; et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell 2004, 6, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, S.; Buiting, K.; Rogan, P.K.; Buxton, J.L.; Driscoll, D.J.; Arnemann, J.; König, R.; Malcolm, S.; Horsthemke, B.; Nicholls, R.D. Minimal definition of the imprinting center and fixation of chromosome 15q11-q13 epigenotype by imprinting mutations. Proc. Natl. Acad. Sci. USA 1996, 93, 7811–7815. [Google Scholar] [CrossRef] [PubMed]
- Fulmer-Smentek, S.B.; Francke, U. Association of acetylated histones with paternally expressed genes in the Prader--Willi deletion region. Hum. Mol. Genet. 2001, 10, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Allis, C.D.; Wagstaff, J. Parent-Specific Complementary Patterns of Histone H3 Lysine 9 and H3 Lysine 4 Methylation at the Prader-Willi Syndrome Imprinting Center. AJHG 2001, 69, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, H.-M.; Xiong, Y.; Sciaky, N.; Hulbert, S.W.; Cao, X.; Everitt, J.I.; Jin, J.; Roth, B.L.; Jiang, Y. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader–Willi syndrome. Nat. Med. 2016, 23, 213. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, E.M.; Williamson, K.E.; Reyon, D.; Zou, J.Y.; Ram, O.; Joung, J.K.; Bernstein, B.E. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013, 31, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Cano-Rodriguez, D.; Rots, M.G. Epigenetic Editing: On the Verge of Reprogramming Gene Expression at Will. Curr. Genet. Med. Rep. 2016, 4, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Nery, E.D.; Ebner, D.; Montoya, M.C.; Östling, P.; Pietiäinen, V.; Price, L.S.; et al. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Nava, F.; Lanes, R. GH/IGF-1 Signaling and Current Knowledge of Epigenetics; a Review and Considerations on Possible Therapeutic Options. Int. J. Mol. Sci. 2017, 18, 1624. https://doi.org/10.3390/ijms18101624
Álvarez-Nava F, Lanes R. GH/IGF-1 Signaling and Current Knowledge of Epigenetics; a Review and Considerations on Possible Therapeutic Options. International Journal of Molecular Sciences. 2017; 18(10):1624. https://doi.org/10.3390/ijms18101624
Chicago/Turabian StyleÁlvarez-Nava, Francisco, and Roberto Lanes. 2017. "GH/IGF-1 Signaling and Current Knowledge of Epigenetics; a Review and Considerations on Possible Therapeutic Options" International Journal of Molecular Sciences 18, no. 10: 1624. https://doi.org/10.3390/ijms18101624
APA StyleÁlvarez-Nava, F., & Lanes, R. (2017). GH/IGF-1 Signaling and Current Knowledge of Epigenetics; a Review and Considerations on Possible Therapeutic Options. International Journal of Molecular Sciences, 18(10), 1624. https://doi.org/10.3390/ijms18101624