
Somatic Genetic Variation in Solid Pseudopapillary Tumor of the Pancreas by Whole Exome Sequencing

 ,
, 
Abstract
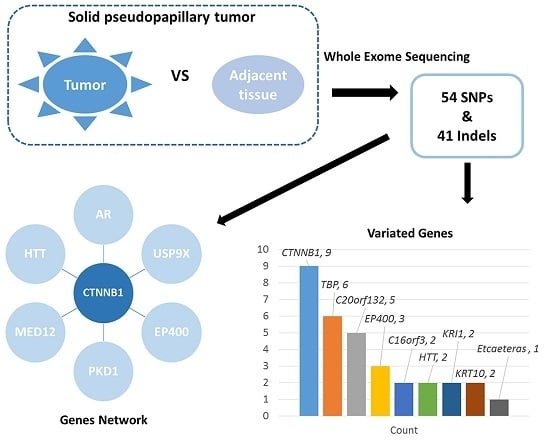
:
1. Introduction
2. Results
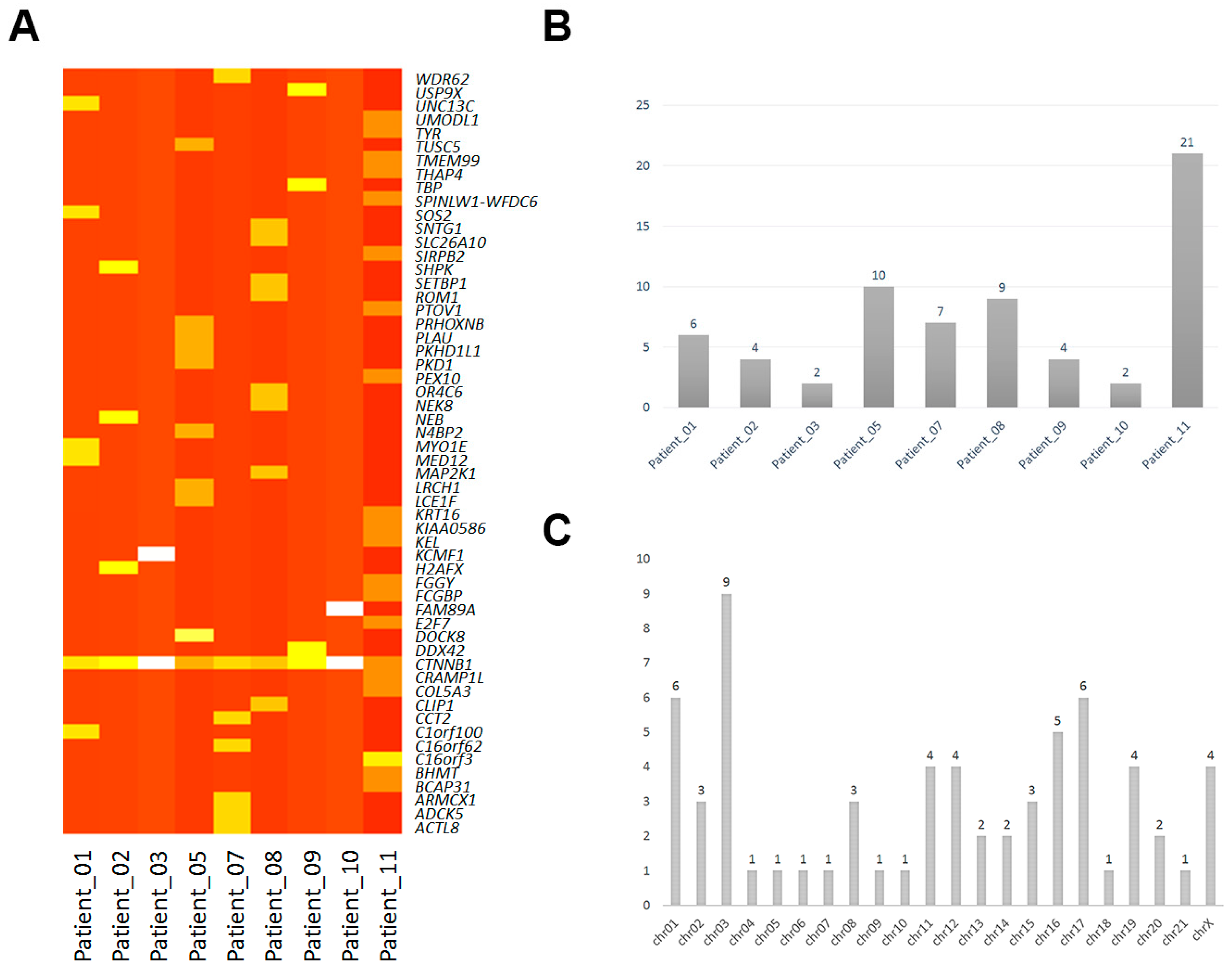
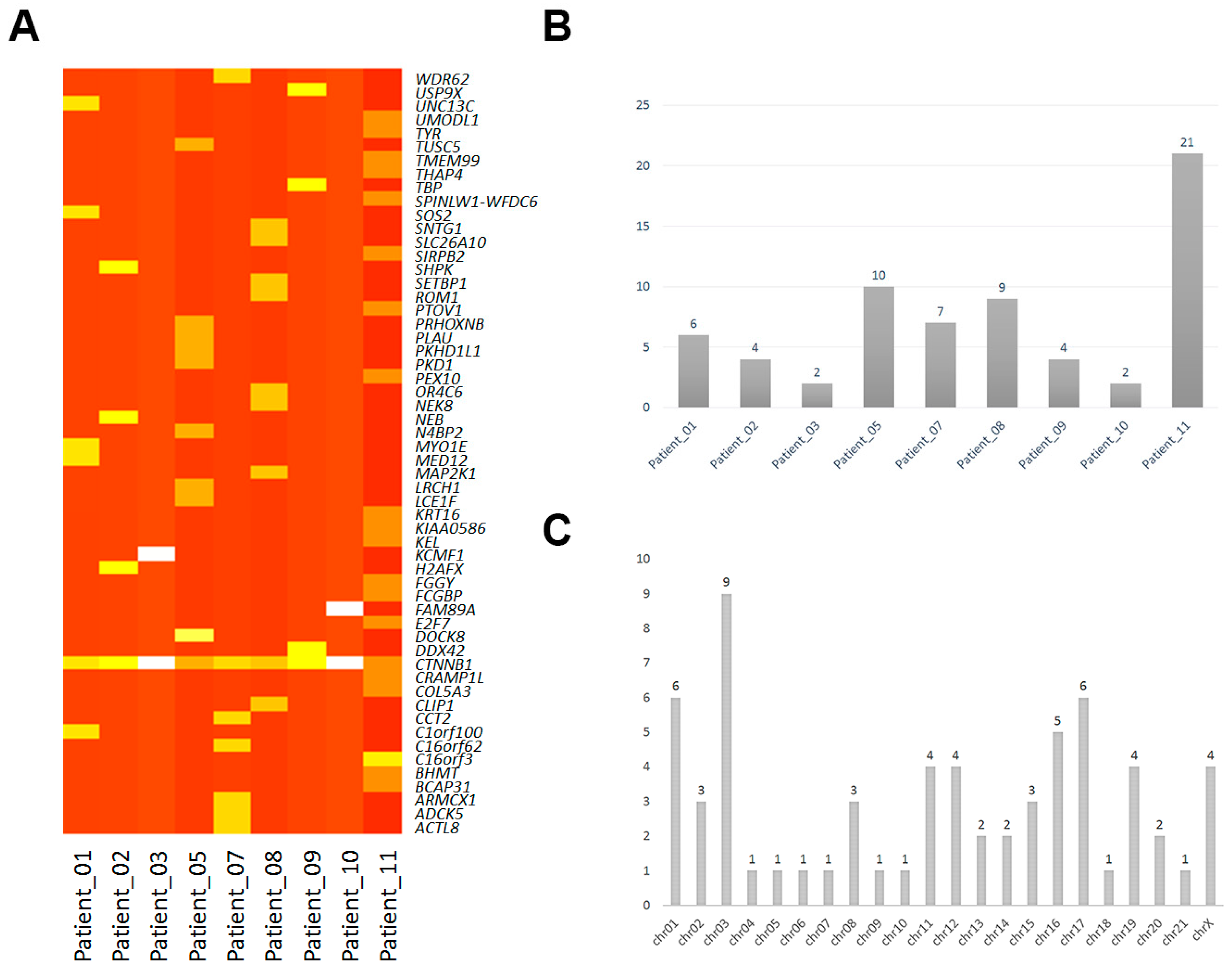
2.1. Mononucleotide Variation in Solid Pseudopapillary Tumor of the Pancreas (SPT)
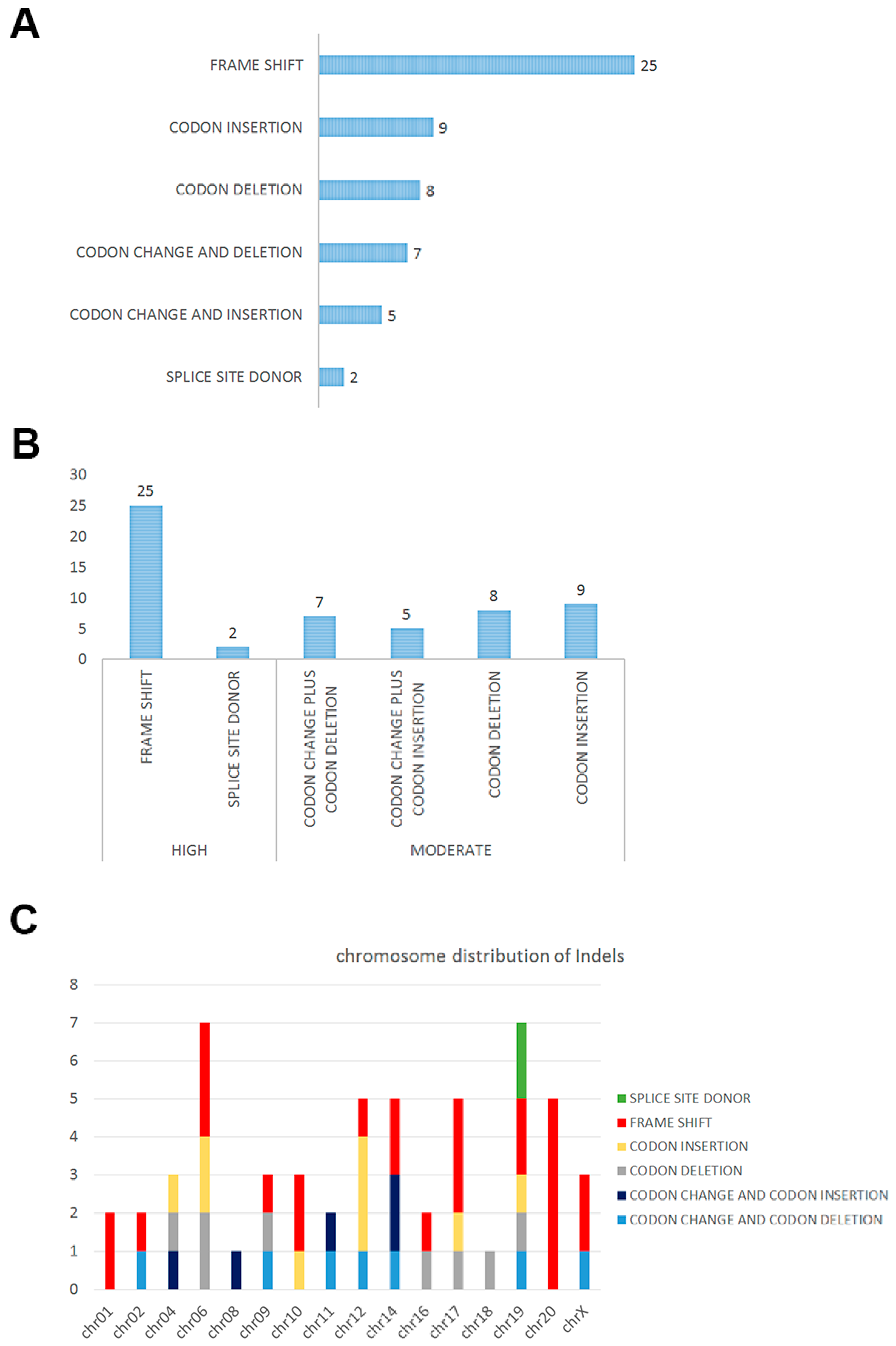
2.2. Insertions and Deletions in SPT
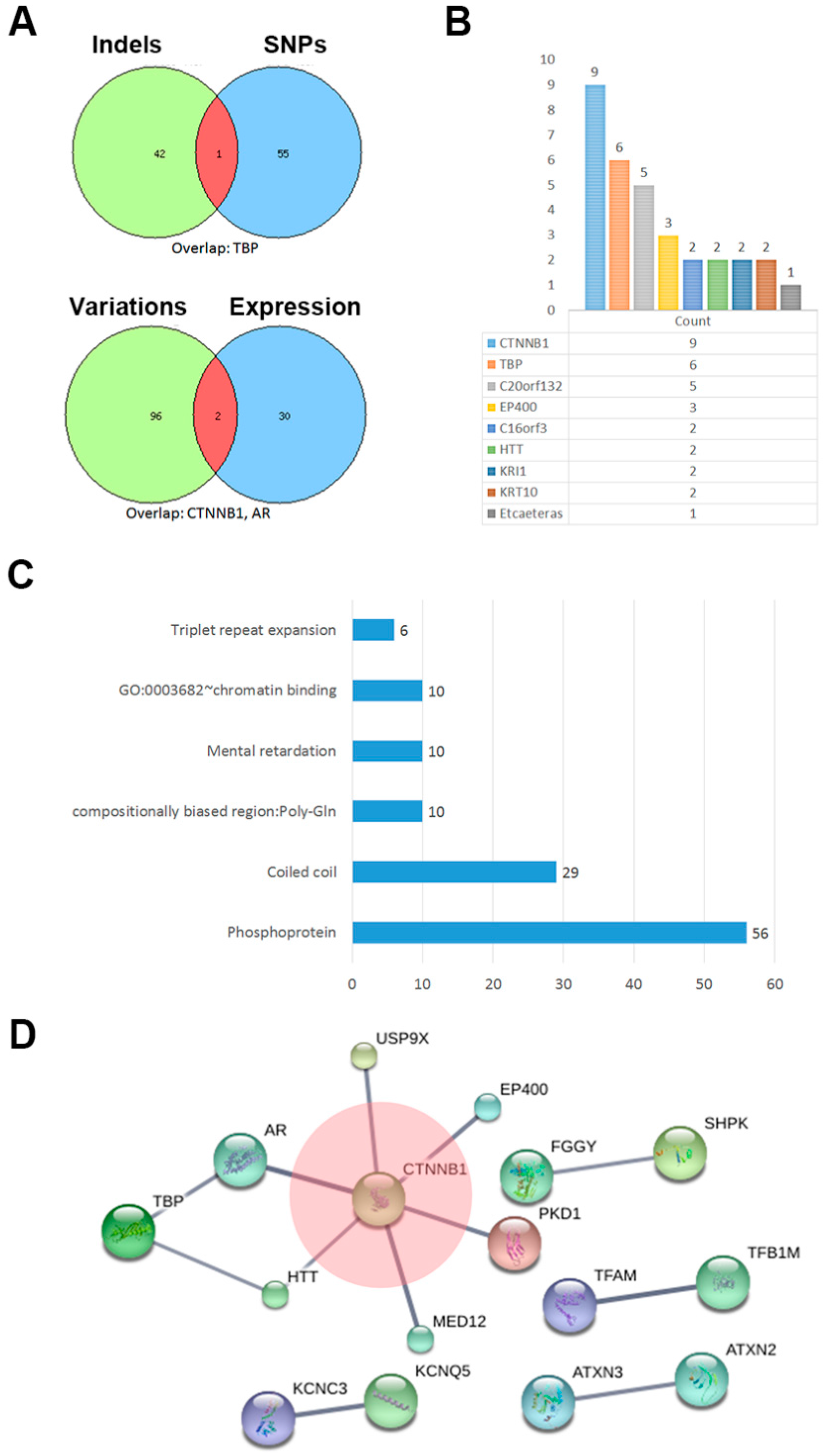
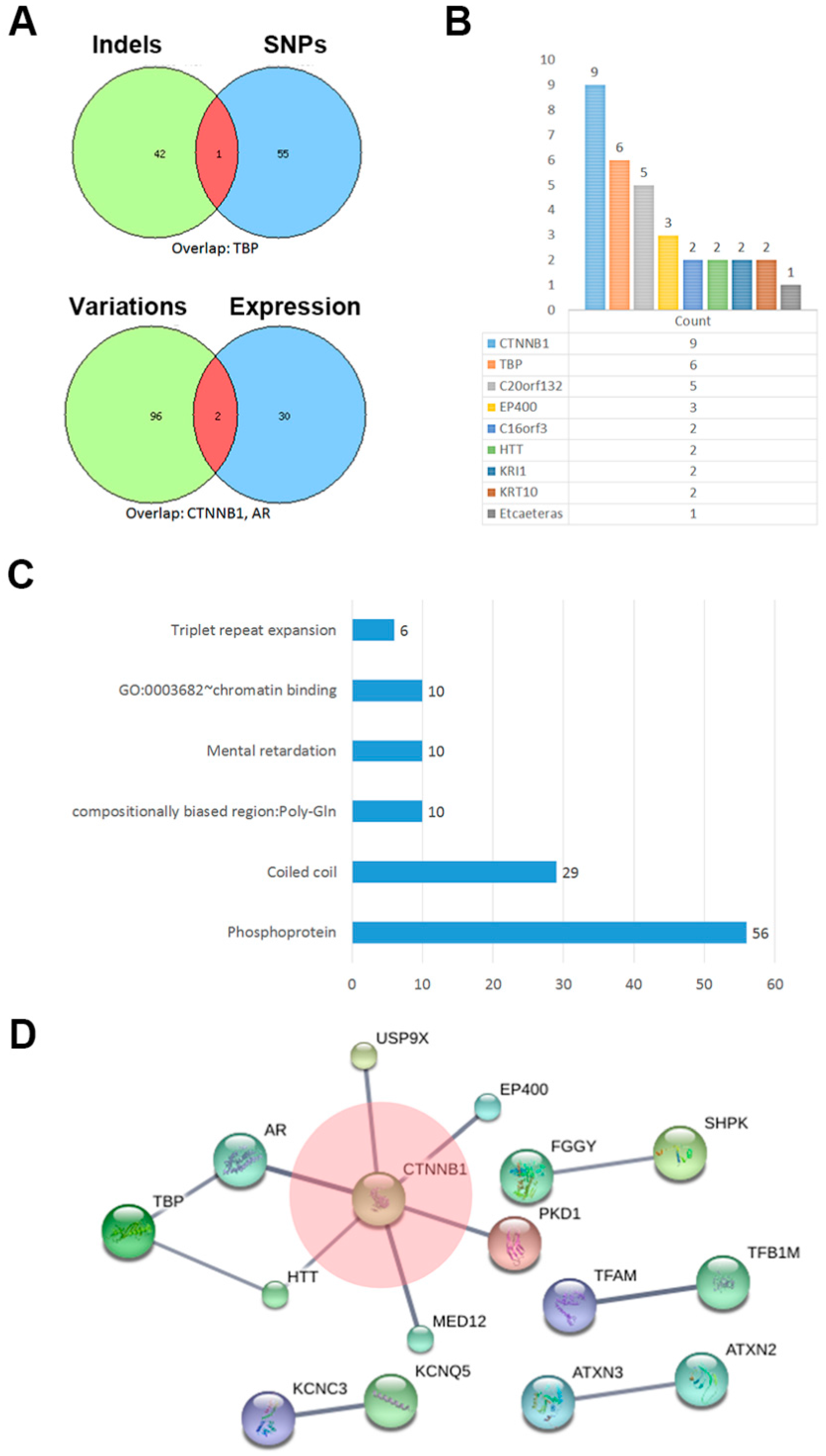
2.3. The Network of Indels and Single Nucleotide Polymorphisms (SNPs) Related Genes
3. Discussion
4. Materials and Methods
4.1. Patients and Tissues
4.2. DNA Extraction and Exome Sequencing
4.3. Read Mapping and Standard Bioinformatics Analysis
4.4. Exome Homozygosity Mapping
4.5. Cluster and Network
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, S.C.; Ng, K.F.; Yeh, T.S.; Chang, H.C.; Su, C.Y.; Chen, T.C. Clinicopathological analysis of β-catenin and Axin-1 in solid pseudopapillary neoplasms of the pancreas. Ann. Surg. Oncol. 2012, 19, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Kosmahl, M.; Seada, L.S.; Janig, U.; Harms, D.; Kloppel, G. Solid-pseudopapillary tumor of the pancreas: Its origin revisited. Virchows Arch. 2000, 436, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kato, K.; Notohara, K.; Hojo, H.; Ijiri, R.; Miyake, T.; Nagahara, N.; Sasaki, F.; Kitagawa, N.; Nakatani, Y.; Kobayashi, Y. Frequent β-catenin mutation and cytoplasmic/nuclear accumulation in pancreatic solid-pseudopapillary neoplasm. Cancer Res. 2001, 61, 8401–8404. [Google Scholar] [PubMed]
- Hallas, C.; Phillipp, J.; Domanowsky, L.; Kah, B.; Tiemann, K. BCL9L expression in pancreatic neoplasia with a focus on SPN: A possible explanation for the enigma of the benign neoplasia. BMC Cancer 2016, 16, 648. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kim, M.; Hwang, D.; Park, M.; Kim, W.K.; Kim, S.K.; Shin, J.; Park, E.S.; Kang, C.M.; Paik, Y.K.; et al. Characterization of gene expression and activated signaling pathways in solid-pseudopapillary neoplasm of pancreas. Mod. Pathol. 2014, 27, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Cavard, C.; Audebourg, A.; Letourneur, F.; Audard, V.; Beuvon, F.; Cagnard, N.; Radenen, B.; Varlet, P.; Vacher-Lavenu, M.C.; Perret, C.; et al. Gene expression profiling provides insights into the pathways involved in solid pseudopapillary neoplasm of the pancreas. J. Pathol. 2009, 218, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Terris, B.; Cavard, C. Diagnosis and molecular aspects of solid-pseudopapillary neoplasms of the pancreas. Semin. Diagn. Pathol. 2014, 31, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Stelter, A.A.; French, S.; Shen, S.; Qiu, S.; Venegas, R.; Wen, J.; Wang, H.Q.; Xie, J. Loss of cell-adhesion molecule complexes in solid pseudopapillary tumor of pancreas. Mod. Pathol. 2007, 20, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Lawrenson, K.; Iversen, E.S.; Tyrer, J.; Weber, R.P.; Concannon, P.; Hazelett, D.J.; Li, Q.; Marks, J.R.; Berchuck, A.; Lee, J.M.; et al. Common variants at the CHEK2 gene locus and risk of epithelial ovarian cancer. Carcinogenesis 2015, 36, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Sundram, V.; Ganju, A.; Hughes, J.E.; Khan, S.; Chauhan, S.C.; Jaggi, M. Protein kinase D1 attenuates tumorigenesis in colon cancer by modulating β-catenin/T cell factor activity. Oncotarget 2014, 5, 6867–6884. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Usp9X is required for lymphocyte activation and homeostasis through its control of ZAP70 ubiquitination and PKCβ kinase activity. J. Immunol. 2016, 196, 3438–3451. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.K.; Su, T.; Yen, L.; Jacquet, K.; Huang, C.; Cote, J.; Kurdistani, S.K.; Carey, M.F. EP400 deposits H3.3 into promoters and enhancers during gene activation. Mol. Cell 2016, 61, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; McGuire, J.R.; Sousa, C.M.; Fuhrmann, L.; Fitamant, J.; Leboucher, S.; Vacher, S.; du Montcel, S.T.; Bieche, I.; Bernet, A.; et al. Unraveling the role of huntingtin in breast cancer metastasis. J. Natl. Cancer Inst. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Xu, X.; Hecht, A.; Boyer, T.G. Mediator is a transducer of Wnt/β-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J.; Gustafsson, J. Interaction of the human androgen receptor transactivation function with the general transcription factor TFIIF. Proc. Natl. Acad. Sci. USA 1997, 94, 8485–8490. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zhang, S.; Yazdanparast, A.; Li, M.; Pawar, A.V.; Liu, Y.; Inavolu, S.M.; Cheng, L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genom. 2016, 17, 525. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Oda, T.; Hashimoto, S.; Akashi, Y.; Miyamoto, R.; Enomoto, T.; Satomi, K.; Morishita, Y.; Ohkohchi, N. Pancreatic neuroendocrine tumor and solid-pseudopapillary neoplasm: Key immunohistochemical profiles for differential diagnosis. World J. Gastroenterol. 2016, 22, 8596–8604. [Google Scholar] [CrossRef] [PubMed]
- Kominami, A.; Fujino, M.; Murakami, H.; Ito, M. β-catenin mutation in ovarian solid pseudopapillary neoplasm. Pathol. Int. 2014, 64, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhang, X.; Feng, X.; Fan, X.; Jin, Z. The crosstalk between microRNAs and the Wnt/β-catenin signaling pathway in cancer. Oncotarget 2016. [Google Scholar] [CrossRef]
- Rocha, P.P.; Scholze, M.; Bleiss, W.; Schrewe, H. Med12 is essential for early mouse development and for canonical Wnt and Wnt/PCP signaling. Development 2010, 137, 2723–2731. [Google Scholar] [CrossRef] [PubMed]
- Izrailit, J.; Jaiswal, A.; Zheng, W.; Moran, M.F.; Reedijk, M. Cellular stress induces TRB3/USP9x-dependent Notch activation in cancer. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Papavramidis, T.; Papavramidis, S. Solid pseudopapillary tumors of the pancreas: Review of 718 patients reported in English literature. J. Am. Coll. Surg. 2005, 200, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Kloppel, G.; Hruban, R.H.; Klimstra, D.S.; Maitra, A.; Morohoshi, T.; Notohara, K.; Shimizu, M.; Terris, B. Solid-pseudopapillary neoplasm of the pancreas. In WHO Classification of Tumours of the Digestive System; Bosman, F.T., Carneiro, F., Hruban, R.H., Theise, N.D., Eds.; IARC: Lyon, France, 2010; pp. 327–330. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Gender | Age (Years) | Size (mm) | TNM Stage | Location | Distant Metastasis (Yes/No) | CA19-9 Value | Surgical Procedures |
|---|---|---|---|---|---|---|---|---|
| 1 | male | 35 | 18 | I | head | No | no abnormal | distal pancreatectomy |
| 2 | male | 33 | 50 | II | body and tail | No | no abnormal | distal pancreatectomy |
| 3 | male | 26 | 70 | II | body and tail | No | no abnormal | distal pancreatectomy |
| 5 | female | 43 | 108 | II | head | Yes | no abnormal | total pancreatectomy |
| 7 | female | 30 | 45 | II | body and tail | No | no abnormal | distal pancreatectomy |
| 8 | female | 31 | 45 | II | head | No | no abnormal | pancreaticoduodenectomy |
| 9 | female | 25 | 50 | II | body and tail | No | no abnormal | distal pancreatectomy |
| 10 | female | 25 | NA | II | head | No | no abnormal | pancreaticoduodenectomy |
| 11 | male | 51 | 138 | IV | body and tail | Yes | no abnormal | distal pancreatectomy |
| Samples | Gene | Biotype | Transcript | Codon | Chromosome | Alleles |
|---|---|---|---|---|---|---|
| Patient_01 | C1orf100 | Missense | NM_001012970:p.Tyr78Cys | tAt/tGt | chr01 | het |
| CTNNB1 | Missense | NM_001098209:p.Asp32Tyr | Gac/Tac | chr03 | het | |
| MED12 | Missense | NM_005120:p.Arg1295Cys | Cgt/Tgt | chrX | hom | |
| MYO1E | Missense | NM_004998:p.Ser179Arg | agT/agG | chr15 | het | |
| SOS2 | Missense | NM_006939:p.Leu793Ile | Ctt/Att | chr14 | het | |
| UNC13C | Missense | NM_001080534:p.Lys1395Met | aAg/aTg | chr15 | het | |
| Patient_02 | CTNNB1 | Missense | NM_001098209:p.Asp32Gly | gAc/gGc | chr03 | het |
| H2AFX | Missense | NM_002105:p.Leu98Arg | cTg/cGg | chr11 | het | |
| NEB | Missense | NM_001164507:p.Asp5797Asn | Gat/Aat | chr02 | het | |
| SHPK | Missense | NM_013276:p.Glu477Asp | gaA/gaC | chr17 | het | |
| Patient_03 | CTNNB1 | Missense | NM_001098209:p.Gly34Arg | Gga/Aga | chr03 | het |
| KCMF1 | Missense | NM_020122:p.Arg257His | cGt/cAt | chr02 | het | |
| Patient_05 | CTNNB1 | Missense | NM_001098209:p.Ser37Pro | Tct/Cct | chr03 | het |
| DOCK8 | Missense | NM_203447:p.Val245Met | Gtg/Atg | chr09 | het | |
| LCE1F | Missense | NM_178354:p.Arg83His | cGt/cAt | chr01 | het | |
| LRCH1 | Missense | NM_001164211:p.His745Arg | cAt/cGt | chr13 | het | |
| N4BP2 | Missense | NM_018177:p.Thr92Ile | aCc/aTc | chr04 | het | |
| PKD1 | Missense | NM_001009944:p.Arg4249Cys | Cgc/Tgc | chr16 | het | |
| PKHD1L1 | Missense | NM_177531:p.Ile2532Ser | aTt/aGt | chr08 | het | |
| PLAU | Missense | NM_002658:p.His224Gln | caC/caG | chr10 | het | |
| PRHOXNB | Missense | NM_001105577:p.Gly116Arg | Ggt/Cgt | chr13 | het | |
| TUSC5 | Missense | NM_172367:p.Ser93Thr | Tcc/Acc | chr17 | het | |
| Patient_07 | ACTL8 | Missense | NM_030812:p.Arg48His | cGt/cAt | chr01 | het |
| ADCK5 | Missense | NM_174922:p.Arg449His | cGc/cAc | chr08 | het | |
| ARMCX1 | Missense | NM_016608:p.Cys144Tyr | tGc/tAc | chrX | het | |
| C16orf62 | Missense | NM_020314:p.Ala53Glu | gCg/gAg | chr16 | het | |
| CCT2 | Missense | NM_006431:p.Gly98Asp | gGc/gAc | chr12 | het | |
| CTNNB1 | Missense | NM_001098209:p.Ser33Cys | tCt/tGt | chr03 | het | |
| WDR62 | Missense | NM_001083961:p.Val407Ile | Gtt/Att | chr19 | het | |
| Patient_08 | CLIP1 | Missense | NM_001247997:p.Ile450Val | Att/Gtt | chr12 | het |
| CTNNB1 | Missense | NM_001098209:p.Ser37Phe | tCt/tTt | chr03 | het | |
| MAP2K1 | Missense | NM_002755:p.Leu42His | cTt/cAt | chr15 | het | |
| NEK8 | Missense | NM_178170:p.Asp530Asn | Gac/Aac | chr17 | het | |
| OR4C6 | Missense | NM_001004704:p.Phe104Ser | tTc/tCc | chr11 | het | |
| ROM1 | Missense | NM_000327:p.Ala265Glu | gCa/gAa | chr11 | het | |
| SETBP1 | Missense | NM_015559:p.Tyr1327Cys | tAt/tGt | chr18 | het | |
| SLC26A10 | Missense | NM_133489:p.Val488Met | Gtg/Atg | chr12 | het | |
| SNTG1 | Missense | NM_018967:p.Arg202Gln | cGa/cAa | chr08 | het | |
| Patient_09 | CTNNB1 | Missense | NM_001098209:p.Ser37Phe | tCt/tTt | chr03 | het |
| DDX42 | Missense | NM_007372:p.Thr581Ala | Acc/Gcc | chr17 | het | |
| TBP | Missense | NM_003194:p.Thr106Ala | Acg/Gcg | chr06 | het | |
| USP9X | Missense | NM_001039590:p.Asn2098Ser | aAt/aGt | chrX | het | |
| Patient_10 | CTNNB1 | Missense | NM_001098209:p.Ser33Phe | tCt/tTt | chr03 | het |
| FAM89A | Missense | NM_198552:p.Ser175Cys | tCc/tGc | chr01 | het | |
| Patient_11 | BCAP31 | Missense | NM_001139457:p.Ile190Val | Att/Gtt | chrX | het |
| BHMT | Missense | NM_001713:p.Asp105Asn | Gac/Aac | chr05 | het | |
| C16orf3 | Missense | NM_001214:p.Val60Ile | Gta/Ata | chr16 | het | |
| C16orf3 | Missense | NM_001214:p.Ser57Gly | Agc/Ggc | chr16 | het | |
| COL5A3 | Missense | NM_015719:p.Gly533Val | gGa/gTa | chr19 | het | |
| CRAMP1L | Missense | NM_020825:p.Pro818Thr | Ccc/Acc | chr16 | het | |
| CTNNB1 | Missense | NM_001098209:p.Asp32Tyr | Gac/Tac | chr03 | het | |
| E2F7 | Missense | NM_203394:p.Phe873Val | Ttt/Gtt | chr12 | het | |
| FCGBP | Missense | NM_003890:p.Gly4778Asp | gGc/gAc | chr19 | het | |
| FGGY | Missense | NM_001113411:p.Ser21Asn | aGt/aAt | chr01 | het | |
| KEL | Missense | NM_000420:p.Arg393Gln | cGg/cAg | chr07 | het | |
| KIAA0586 | Missense | NM_001244189:p.Lys953Ile | aAa/aTa | chr14 | het | |
| KRT16 | Missense | NM_005557:p.Gly69Cys | Ggc/Tgc | chr17 | het | |
| PEX10 | Missense | NM_153818:p.Leu221His | cTc/cAc | chr01 | het | |
| PTOV1 | Missense | NM_017432:p.Lys212Met | aAg/aTg | chr19 | het | |
| SIRPB2 | Missense | NM_001122962:p.Gly94Arg | Ggg/Agg | chr20 | het | |
| SPINLW1-WFDC6 | Missense | NM_001198986:p.Glu141Lys | Gaa/Aaa | chr20 | het | |
| THAP4 | Missense | NM_015963:p.Gly111Ser | Ggt/Agt | chr02 | het | |
| TMEM99 | Missense | NM_001195386:p.Asp195Asn | Gac/Aac | chr17 | het | |
| TYR | Missense | NM_000372:p.Thr292Met | aCg/aTg | chr11 | het | |
| UMODL1 | Missense | NM_173568:p.Asp814Glu | gaC/gaG | chr21 | het |
| Impact | Function | Chr | Gene | Reference | Observation | Alleles |
|---|---|---|---|---|---|---|
| High | FS | chr01 | AHDC1 | T | TG | het |
| High | FS | chr01 | LRRIQ3 | G | GT | het |
| High | FS | chr10 | NOC3L | AT | A | het |
| High | FS | chr10 | TFAM | CA | C | het |
| High | FS | chr12 | TDG | G | GA | het |
| High | FS | chr14 | CCNK | G | GC | het |
| High | FS | chr14 | PAPOLA | TG | T | het |
| High | FS | chr16 | IRX5 | AGG | A | het |
| High | FS | chr17 | ACSF2 | T | TAA | het |
| High | FS | chr17 | KRT10 | CCGCCG | C | het |
| High | FS | chr17 | KRT10 | TG | T | het |
| High | FS | chr19 | CAPN12 | G | GC | het |
| High | FS | chr19 | KCNC3 | C | CG | het |
| High | FS | chr02 | SNED1 | A | AC | het |
| High | FS | chr20 | C20orf132 | GACCT | G | het |
| High | FS | chr20 | C20orf132 | GC | G | het |
| High | FS | chr20 | C20orf132 | GAGGAGTT | G | het |
| High | FS | chr20 | C20orf132 | CG | C | het |
| High | FS | chr20 | C20orf132 | TGG | T | het |
| High | FS | chr06 | TBP | AGC | A | het |
| High | FS | chr06 | TBP | AG | A | het |
| High | FS | chr06 | TFB1M | CAA | C | het |
| High | FS | chr09 | PHF2 | A | AG | het |
| High | FS | chrX | PLXNA3 | T | TG | het |
| High | FS | chrX | RBM10 | CA | C | het |
| High | SSD | chr19 | KRI1 | CCATCA | C | het |
| High | SSD | chr19 | KRI1 | CCATCA | C | het |
| Moderate | C & D | chr11 | SCUBE2 | GGCA | G | het |
| Moderate | C & D | chr12 | ATXN2 | GGCT | G | het |
| Moderate | C & D | chr14 | MAP3K9 | GCCT | G | het |
| Moderate | C & D | chr19 | SAFB2 | GTAC | G | het |
| Moderate | C & D | chr02 | GIGYF2 | CACA | C | het |
| Moderate | C & D | chr09 | TPRN | TTCC | T | het |
| Moderate | C & D | chrX | AR | AAGAGACTAGCCCCAG | A | het |
| Moderate | C & I | chr11 | KRTAP5-8 | T | TCCG | het |
| Moderate | C & I | chr14 | ATXN3 | C | CCTG | het |
| Moderate | C & I | chr14 | IRF2BPL | C | CTGCTGT | het |
| Moderate | C & I | chr04 | HTT | A | AACAGCC | het |
| Moderate | C & I | chr08 | ATAD2 | A | ATCG | het |
| Moderate | CD | chr16 | APOBR | TGGGACAGCCTCAGGAGGGGAGGAGGCC | T | het |
| Moderate | CD | chr17 | KDM6B | TCAC | T | het |
| Moderate | CD | chr18 | MBD2 | CGCA | C | het |
| Moderate | CD | chr19 | ARID3A | GGGA | G | het |
| Moderate | CD | chr04 | ADAM29 | GTGACACCCTCCCAGAGGCAACCTCAGT | G | het |
| Moderate | CD | chr06 | KCNQ5 | AGCG | A | het |
| Moderate | CD | chr06 | TBP | GCAA | G | het |
| Moderate | CD | chr09 | RNF20 | TGTTGACTCTGAAGACTCA | T | het |
| Moderate | CI | chr10 | C10orf140 | C | CCCTCCT | het |
| Moderate | CI | chr12 | EP400 | A | ACAG | het |
| Moderate | CI | chr12 | EP400 | A | ACAG | het |
| Moderate | CI | chr12 | EP400 | G | GCAA | het |
| Moderate | CI | chr17 | KRTAP4-5 | T | TGGCAGCAGCTGGGGC | het |
| Moderate | CI | chr19 | ZNF814 | C | CATA | het |
| Moderate | CI | chr04 | HTT | A | ACCGCCGCCG | het |
| Moderate | CI | chr06 | TBP | A | ACAG | het |
| Moderate | CI | chr06 | TBP | A | ACAG | het |
| Category | Term | Count | Genes | Benjamin | FDR |
|---|---|---|---|---|---|
| Up keywords | Phosphoprotein | 56 | PLXNA3, KCNC3, TUSC5, E2F7, CCT2, CTNNB1, KCNQ5, MAP3K9, H2AFX, RBM10, AR, CCNK, C16ORF62, MED12, KRT10, KIAA0586, MBD2, LRCH1, KRT16, BHMT, NEK8, CLIP1, UNC13C, RNF20, GIGYF2, EP400, KDM6B, PTOV1, IRX5, THAP4, KEL, USP9X, NOC3L, N4BP2, TFAM, APOBR, PKD1, KRI1, DDX42, MAP2K1, HTT, MYO1E, ARID3A, ATAD2, DOCK8, SAFB2, ATXN2, ATXN3, PAPOLA, PHF2, KCMF1, WDR62, IRF2BPL, PLAU, TPRN, AHDC1 | 0.004376 | 0.086018 |
| Up keywords | Coiled coil | 29 | LRRIQ3, THAP4, NOC3L, TBP, N4BP2, MAP3K9, PKD1, KRI1, DDX42, AR, MAP2K1, HTT, ATAD2, KRT10, KIAA0586, BCAP31, ATXN2, ATXN3, PHF2, KCMF1, LRCH1, IRF2BPL, KRT16, CLIP1, UNC13C, RNF20, GIGYF2, EP400, TPRN | 0.003907 | 0.102373 |
| Up seqfeature | Compositionally biased region: Poly-Gln | 10 | ATXN2, CCNK, AR, ATXN3, KCNC3, IRF2BPL, HTT, KIAA0586, TBP, EP400 | 1.95 × 10−5 | 4.58 × 10−5 |
| Up keywords | Mental retardation | 10 | IRX5, MAP2K1, WDR62, USP9X, SETBP1, MED12, DOCK8, CTNNB1, AHDC1, BCAP31 | 6.05 × 10−4 | 0.007916 |
| Goterm mf direct | GO:0003682~chromatin binding | 10 | TFAM, AR, NOC3L, ARID3A, MED12, ATAD2, MBD2, RNF20, EP400, KDM6B | 0.022881 | 0.147425 |
| Up keywords | Triplet repeat expansion | 6 | ATXN2, AR, ATXN3, IRF2BPL, HTT, TBP | 3.76 × 10−5 | 2.46 × 10−4 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.; Luo, G.; Jin, K.; Long, J.; Cheng, H.; Lu, Y.; Wang, Z.; Yang, C.; Xu, J.; Ni, Q.; et al. Somatic Genetic Variation in Solid Pseudopapillary Tumor of the Pancreas by Whole Exome Sequencing. Int. J. Mol. Sci. 2017, 18, 81. https://doi.org/10.3390/ijms18010081
Guo M, Luo G, Jin K, Long J, Cheng H, Lu Y, Wang Z, Yang C, Xu J, Ni Q, et al. Somatic Genetic Variation in Solid Pseudopapillary Tumor of the Pancreas by Whole Exome Sequencing. International Journal of Molecular Sciences. 2017; 18(1):81. https://doi.org/10.3390/ijms18010081
Chicago/Turabian StyleGuo, Meng, Guopei Luo, Kaizhou Jin, Jiang Long, He Cheng, Yu Lu, Zhengshi Wang, Chao Yang, Jin Xu, Quanxing Ni, and et al. 2017. "Somatic Genetic Variation in Solid Pseudopapillary Tumor of the Pancreas by Whole Exome Sequencing" International Journal of Molecular Sciences 18, no. 1: 81. https://doi.org/10.3390/ijms18010081
APA StyleGuo, M., Luo, G., Jin, K., Long, J., Cheng, H., Lu, Y., Wang, Z., Yang, C., Xu, J., Ni, Q., Yu, X., & Liu, C. (2017). Somatic Genetic Variation in Solid Pseudopapillary Tumor of the Pancreas by Whole Exome Sequencing. International Journal of Molecular Sciences, 18(1), 81. https://doi.org/10.3390/ijms18010081