
The Epidermal Growth Factor Receptor (EGFR) Inhibitor Gefitinib Reduces but Does Not Prevent Tumorigenesis in Chemical and Hormonal Induced Hepatocarcinogenesis Rat Models

Abstract
:1. Introduction
2. Results
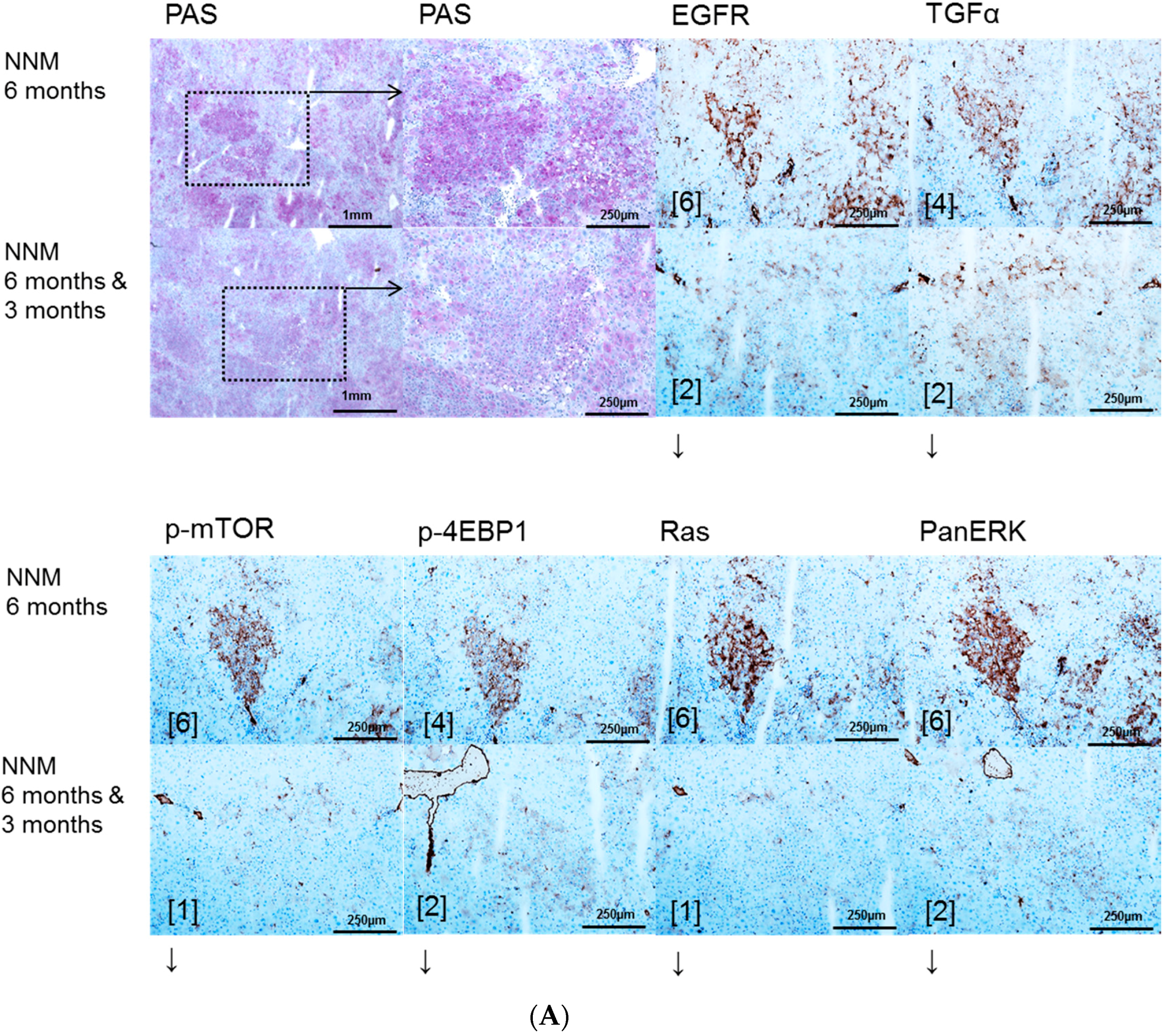
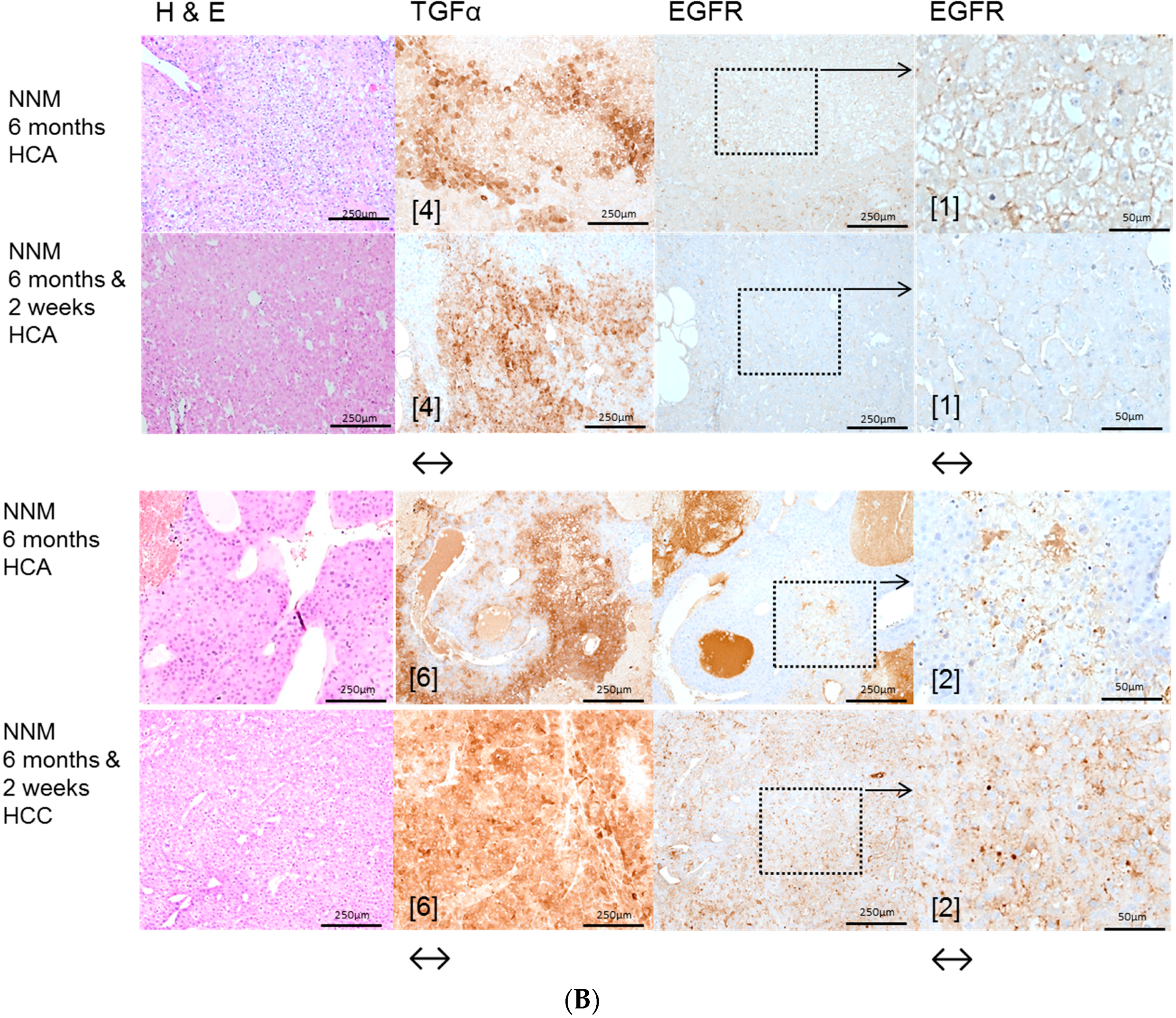
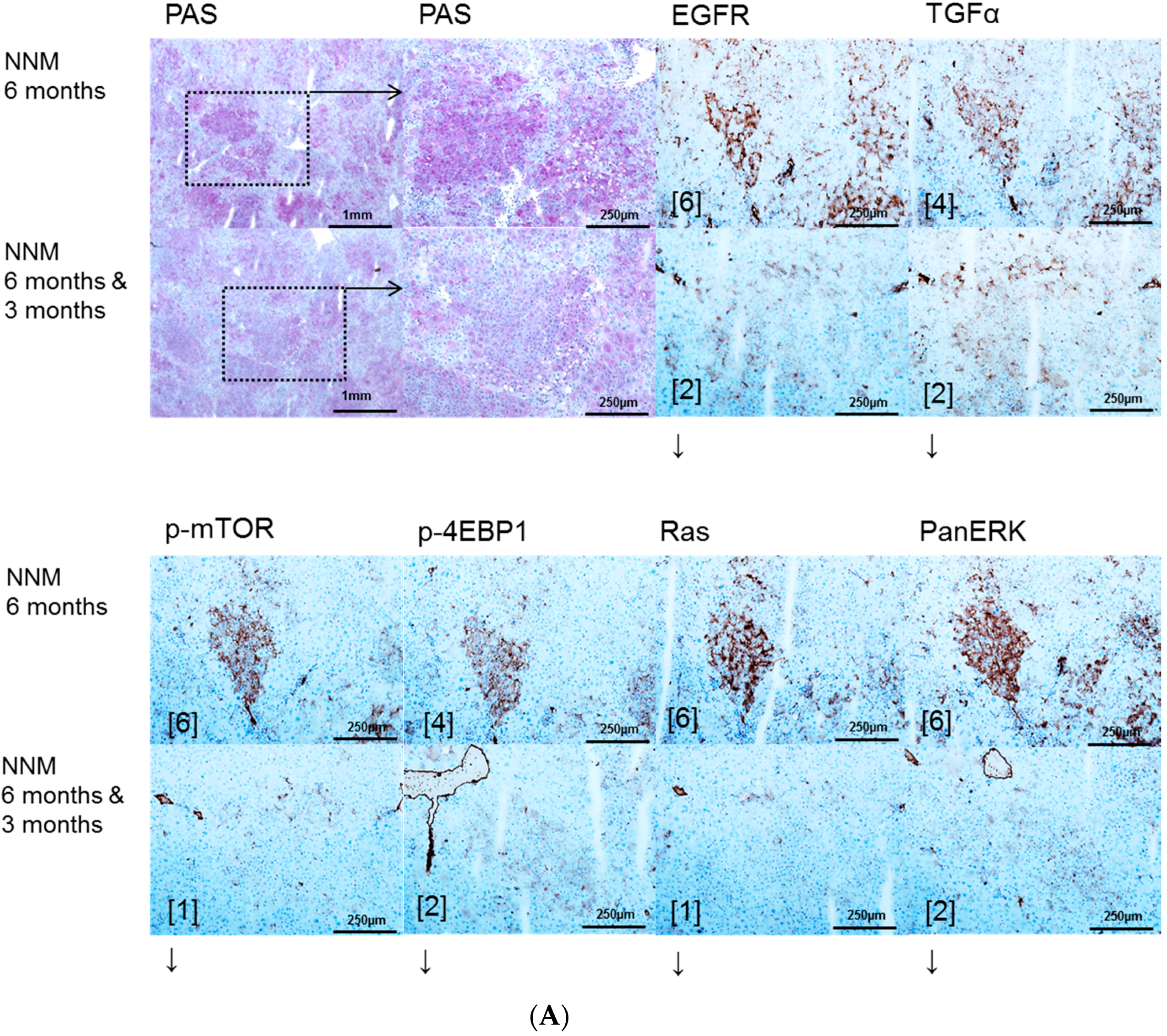
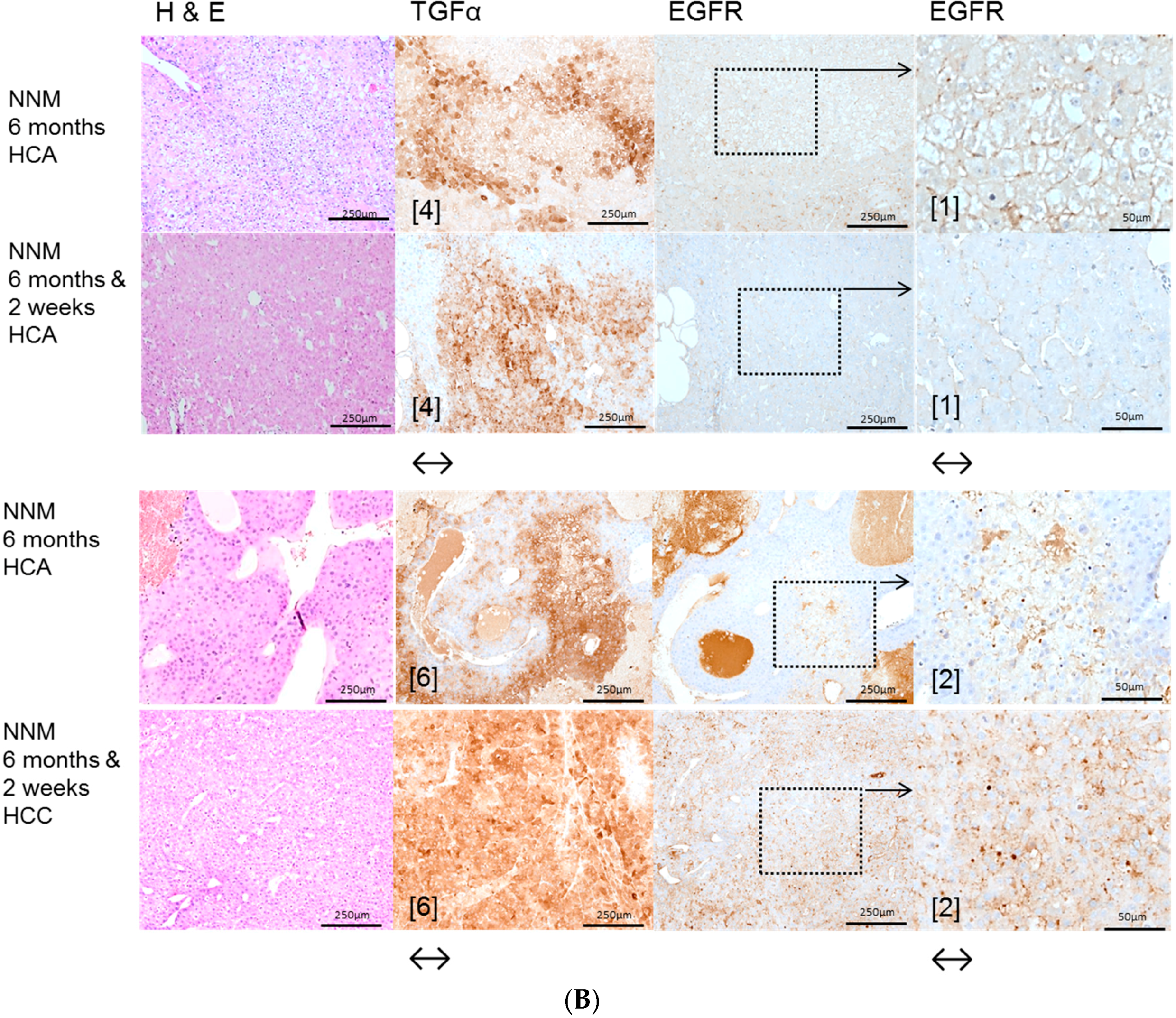
2.1. Hepatocellular Lesions after N-Nitrosomorpholine (NNM) Administration
2.1.1. Proliferative Activity
2.1.2. Volume Fraction of FAH and HCA
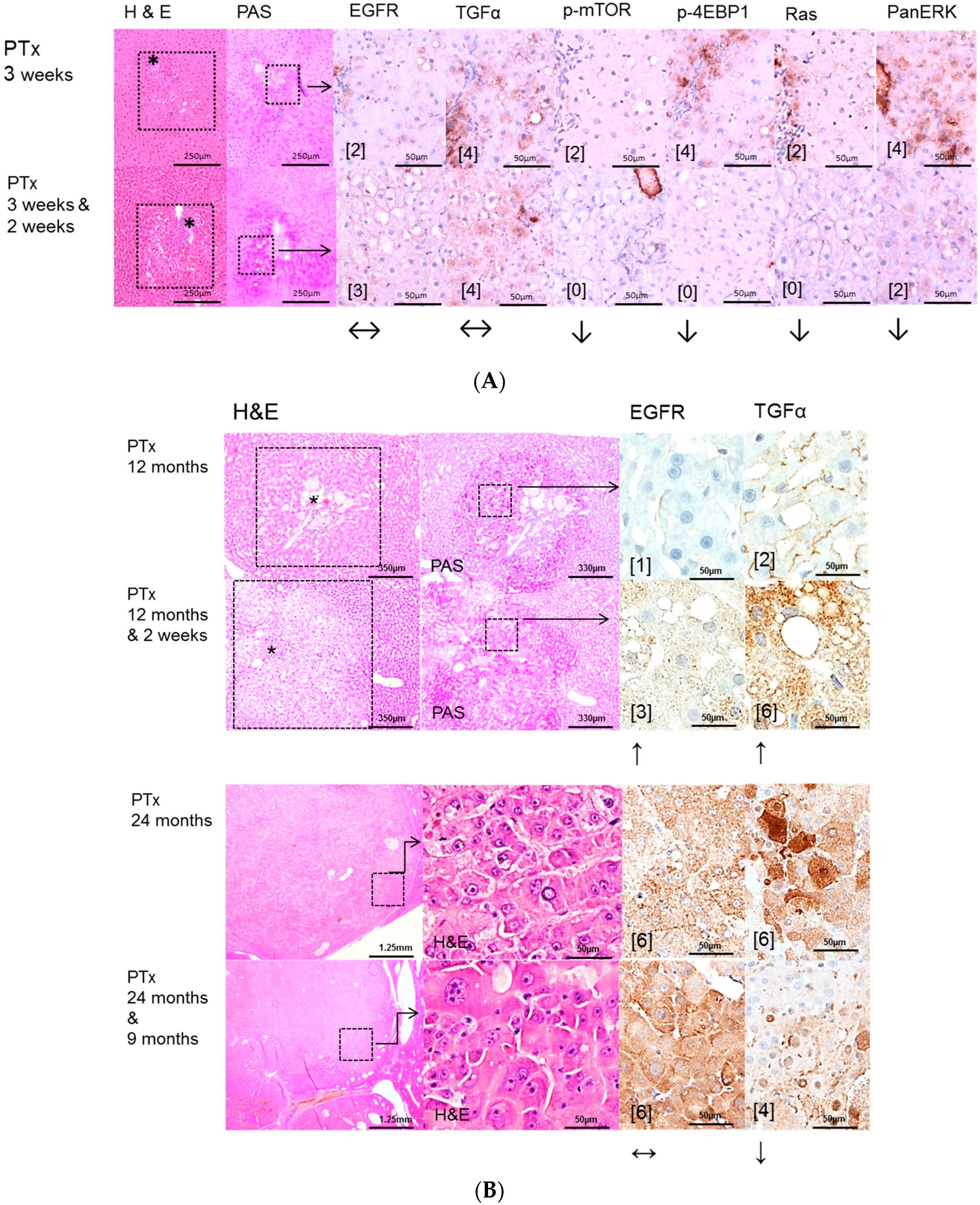
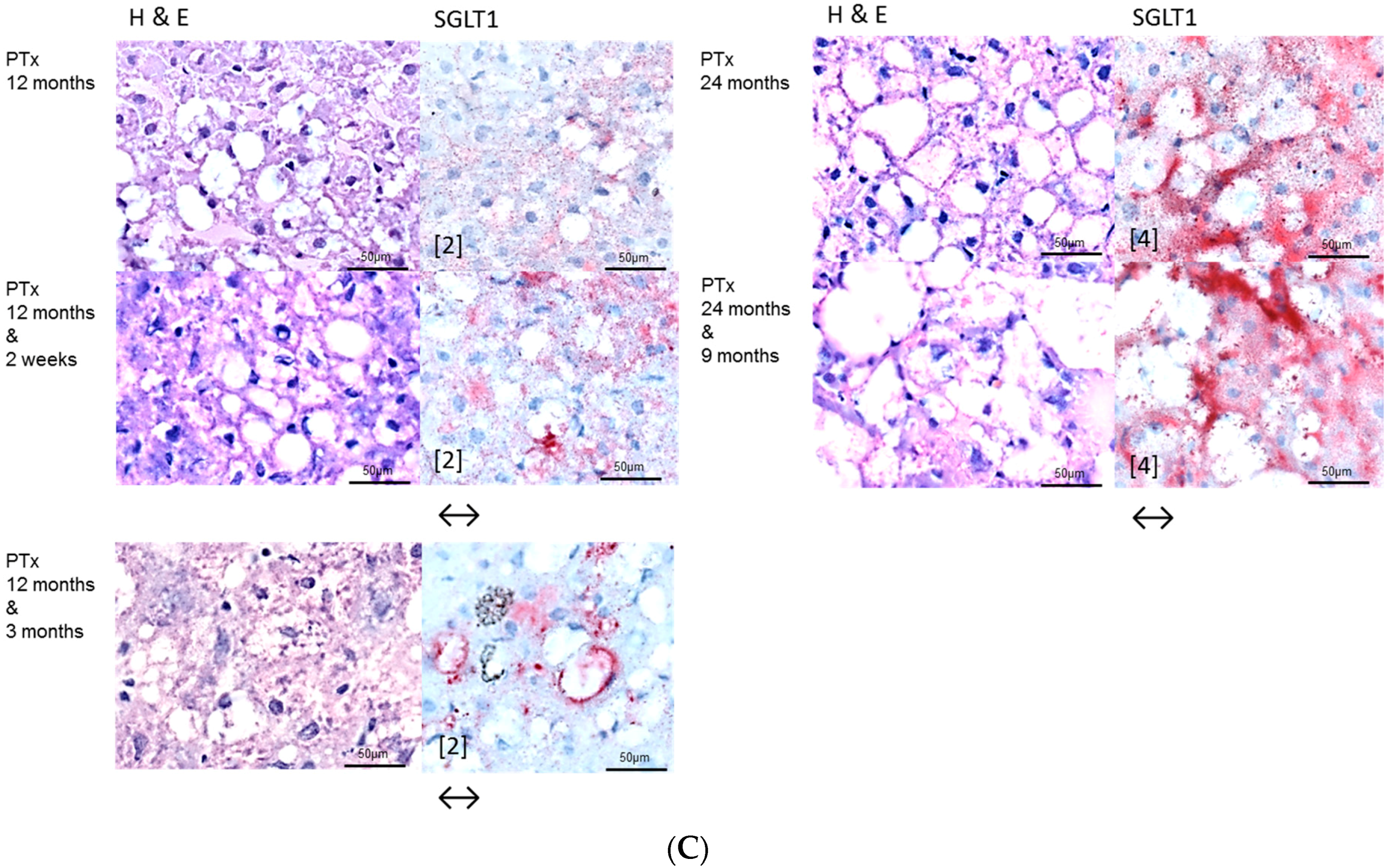
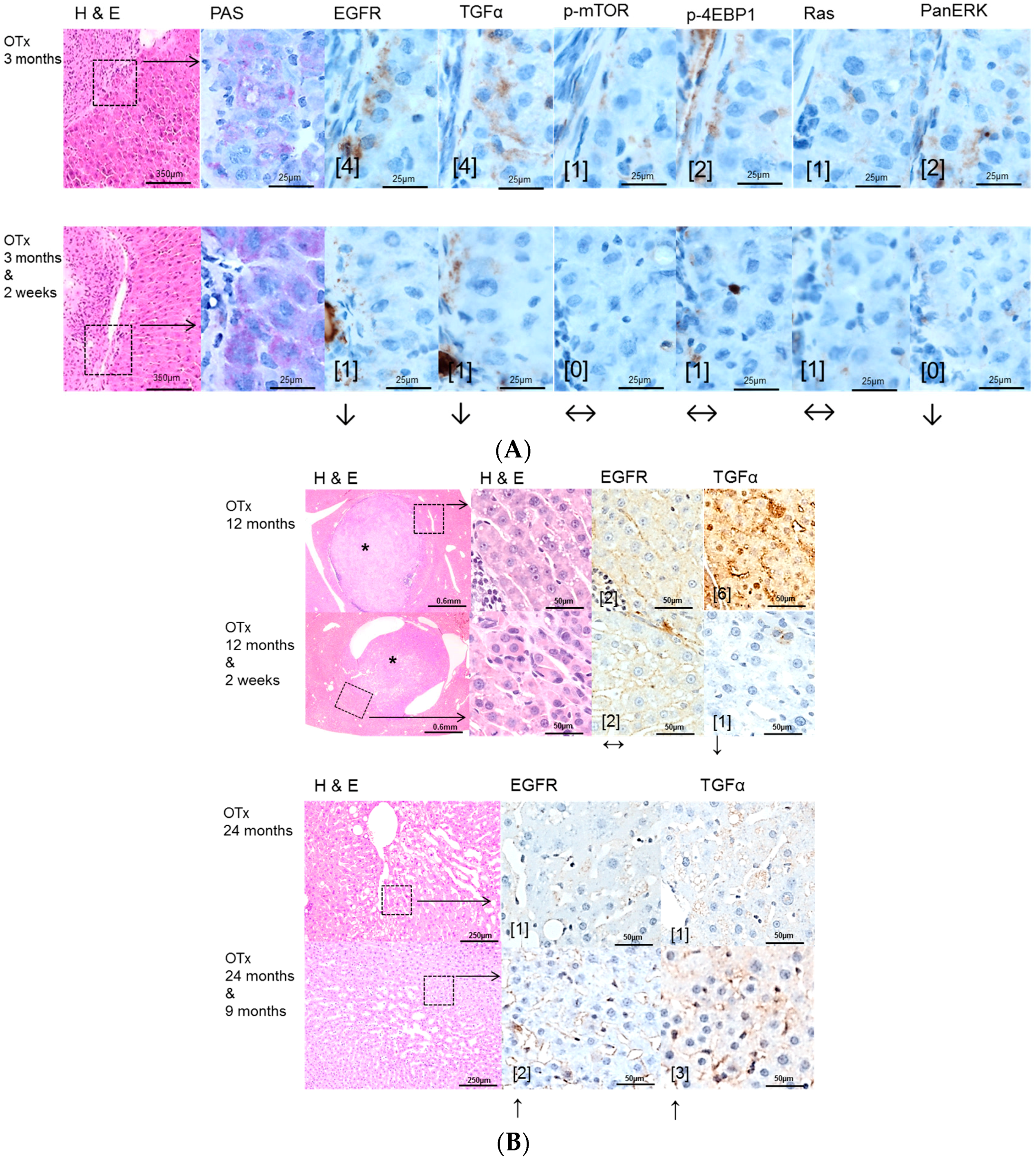
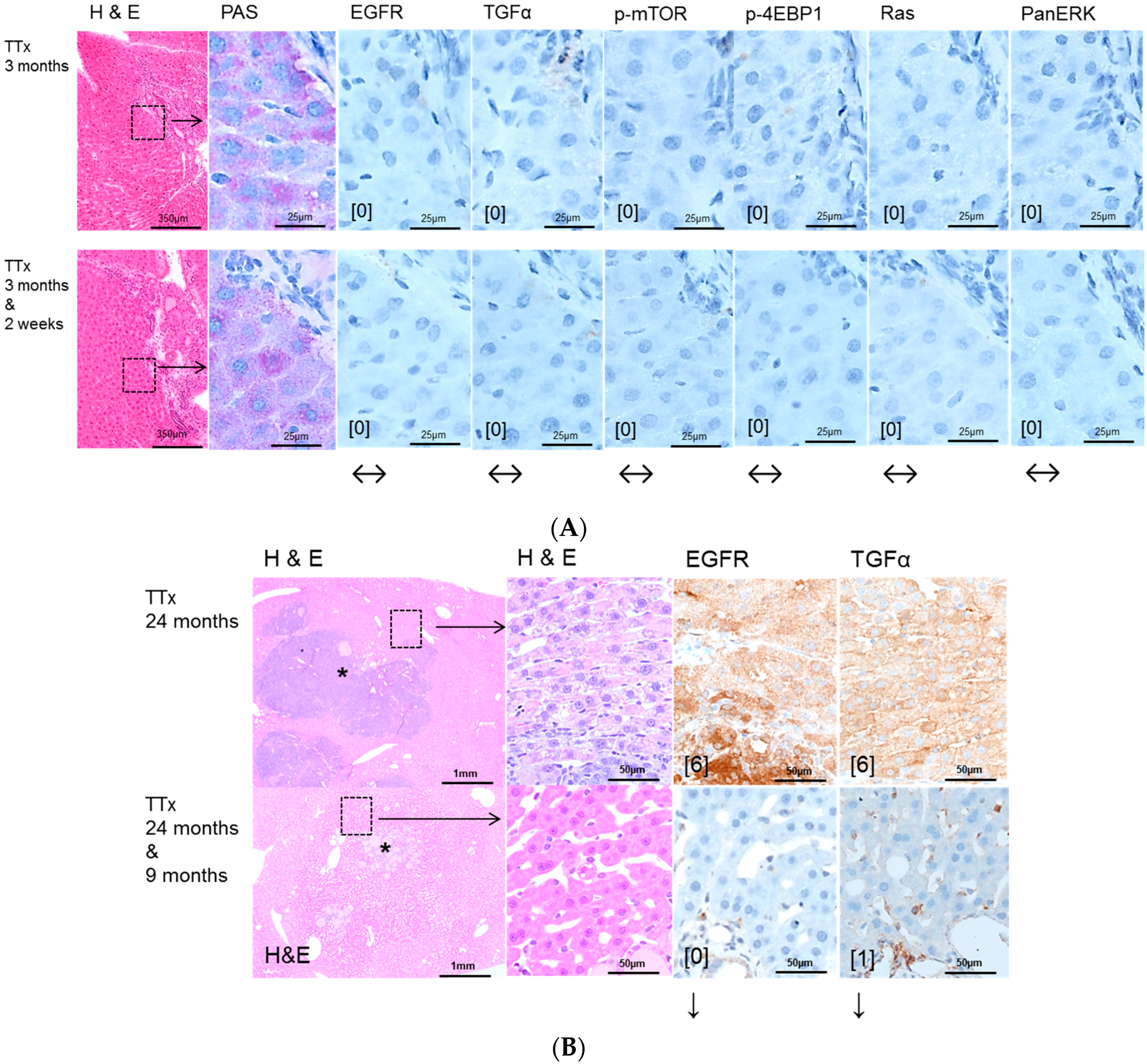
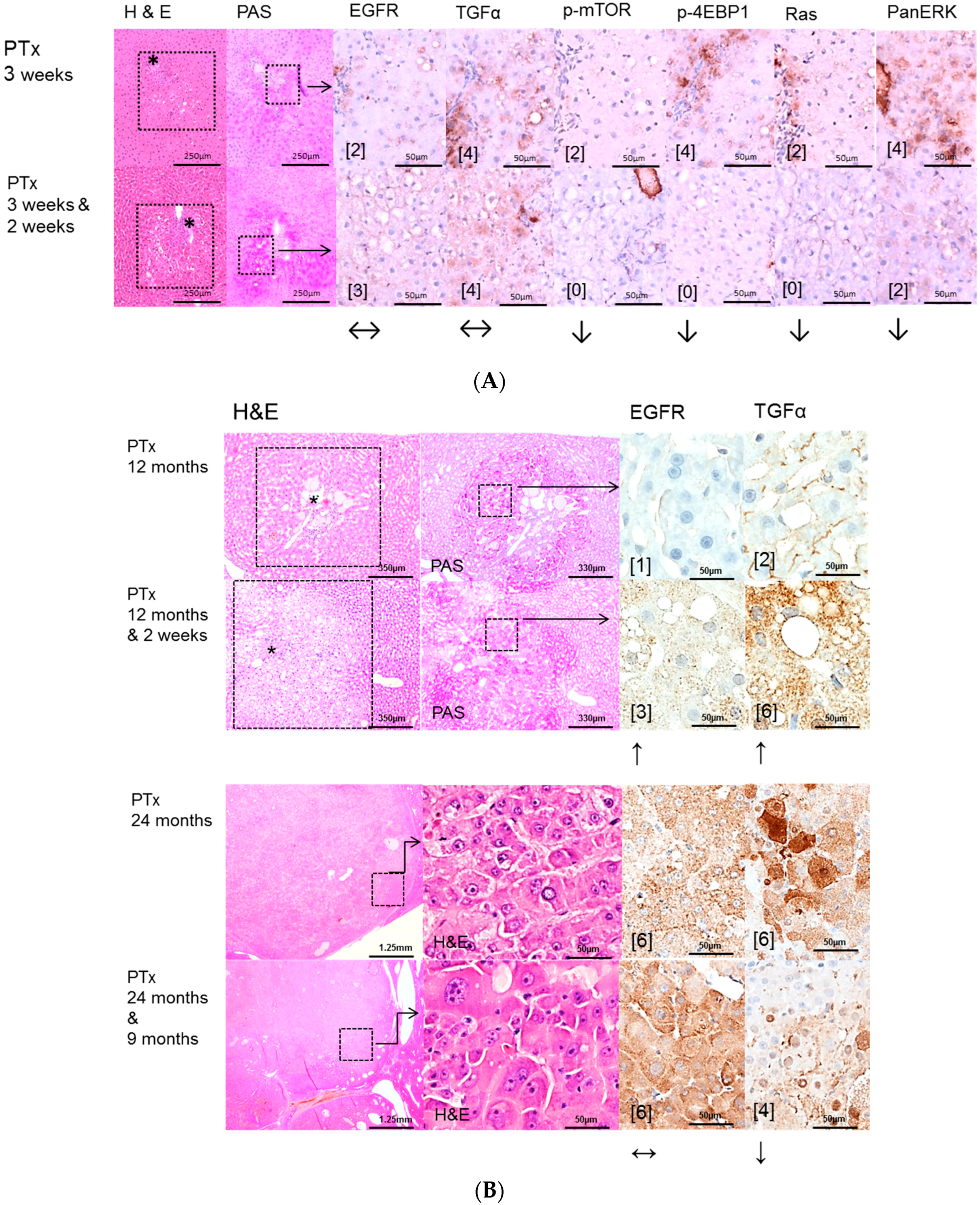
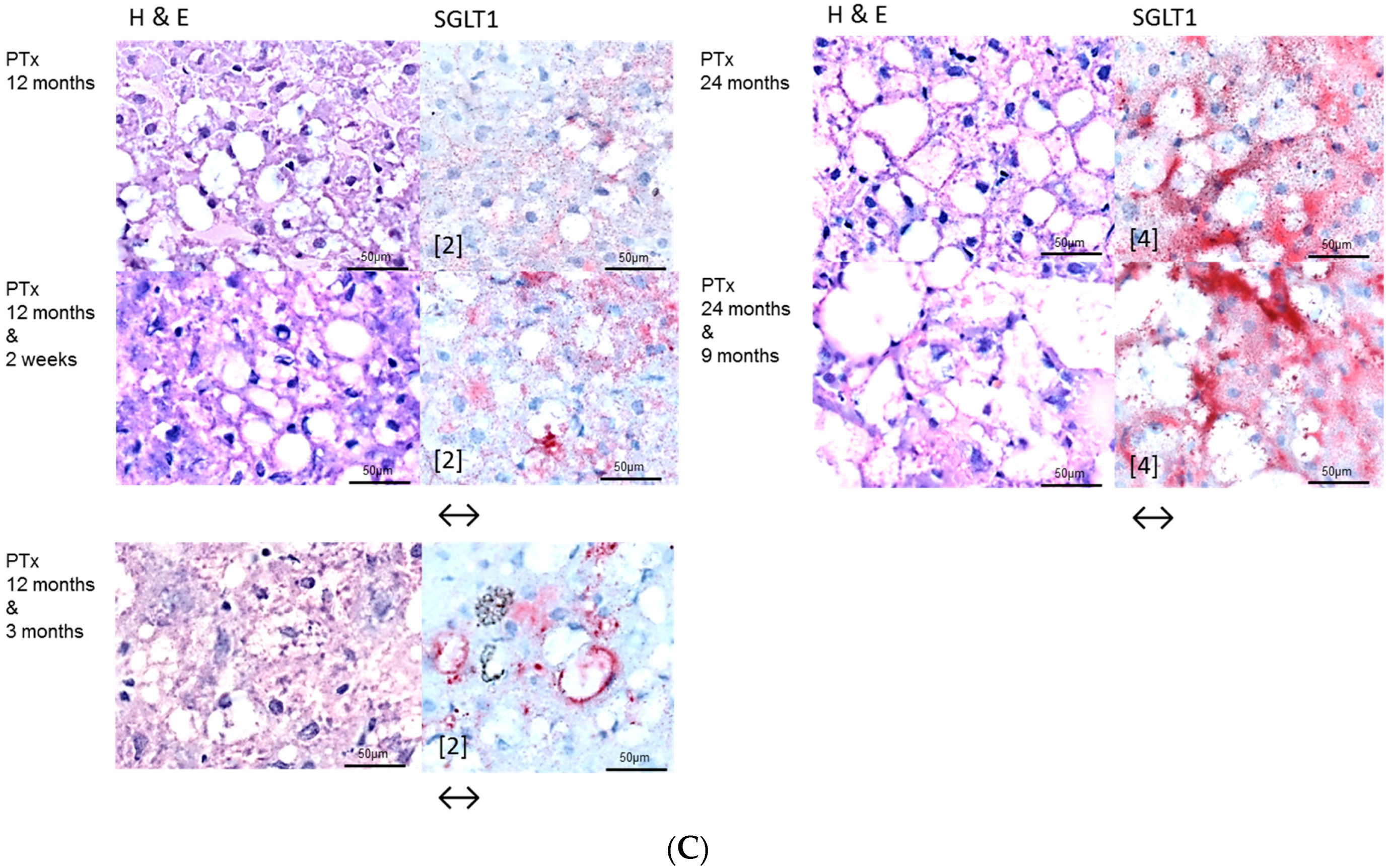
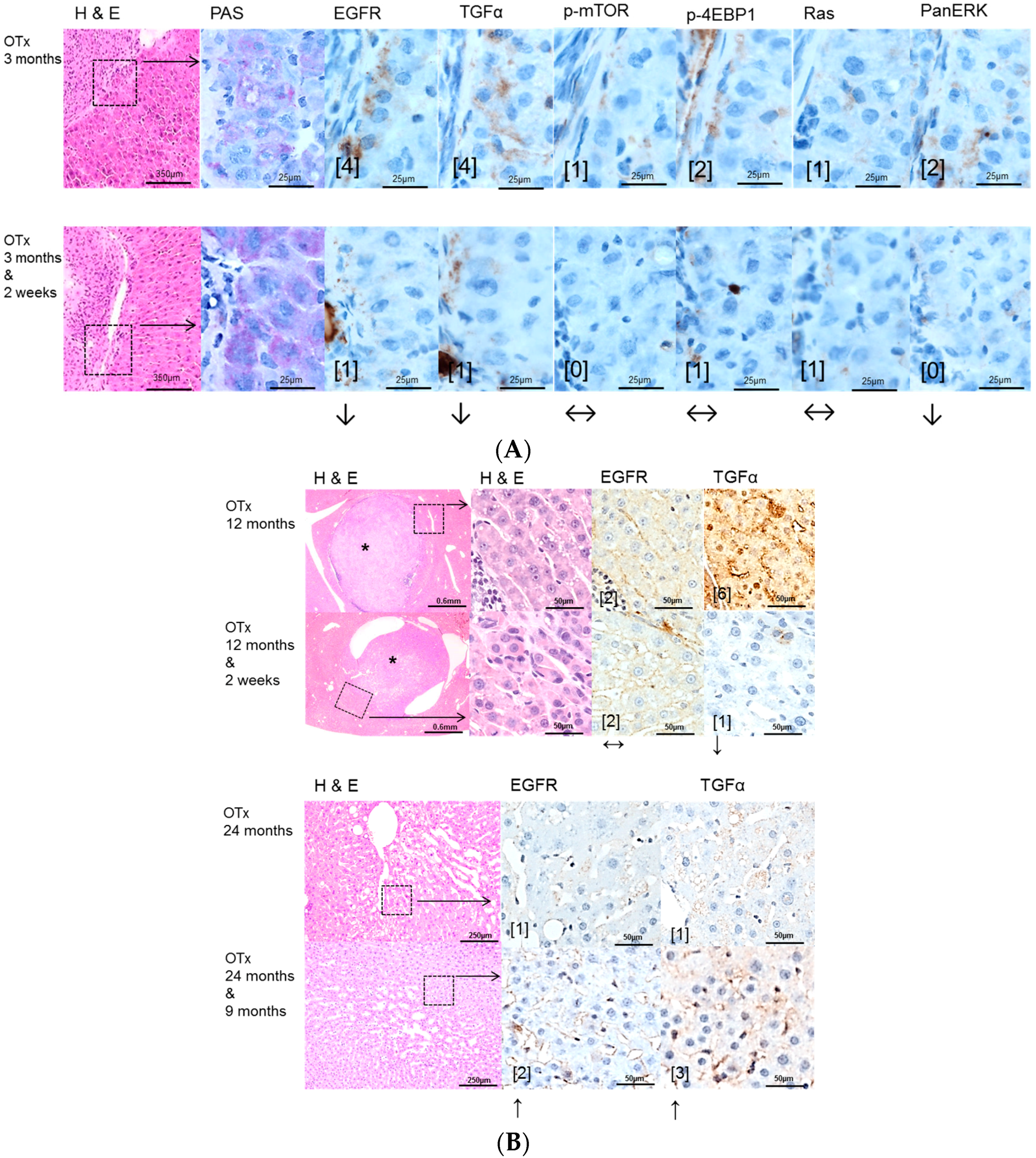
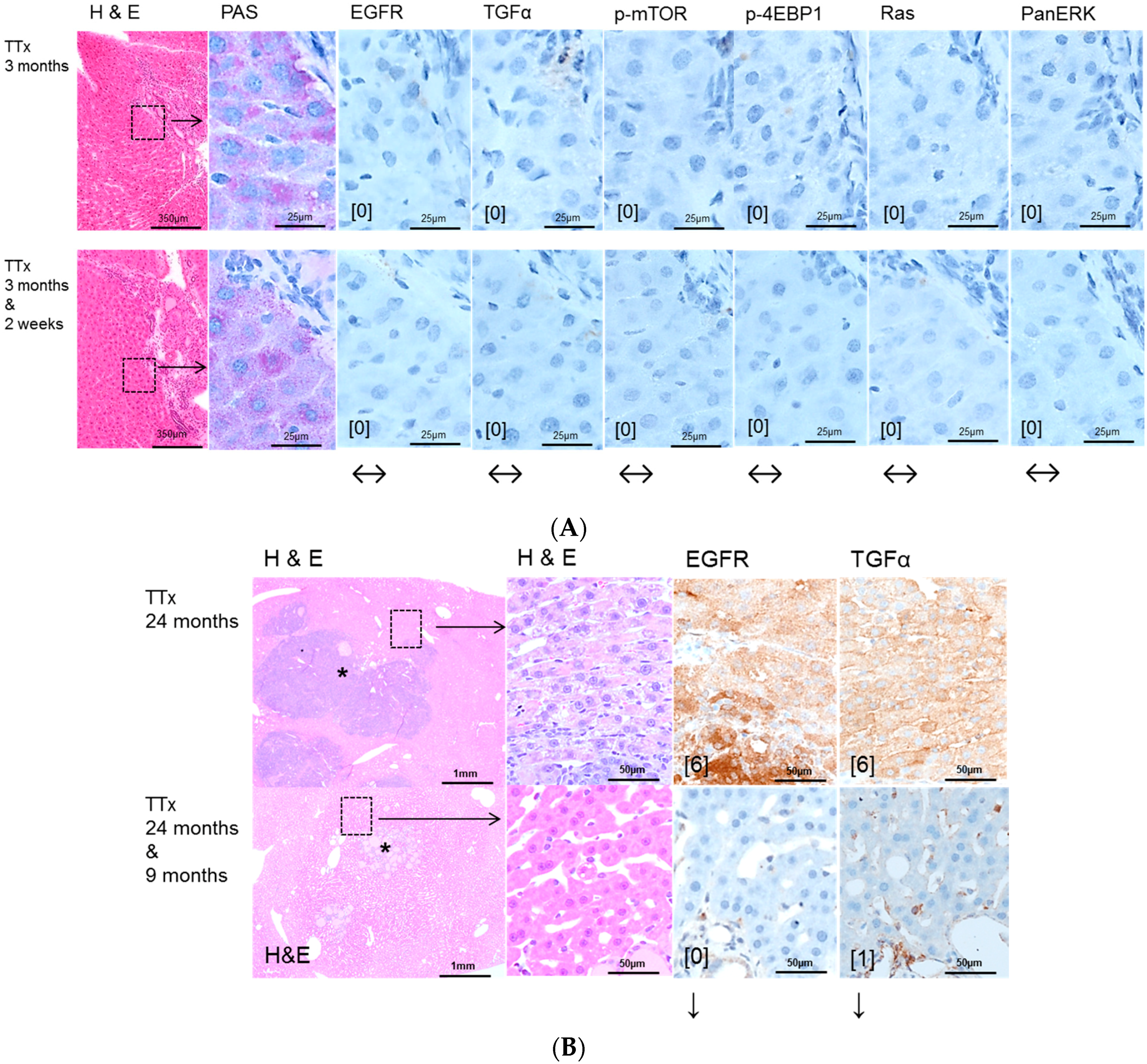
2.2. Hepatocellular Lesions in Transplantation Models
2.2.1. Short Term Experiments
2.2.2. Long Term Experiments
2.3. Effects of Gefitinib on Expression Levels of EGFR, TGFα and Downstream Effectors in Different Hepatocarcinogenesis Models
3. Discussion
4. Experimental Section (for Details Refer to Supplementary Materials and Methods)
4.1. Animal Treatments
4.2. Administration of NNM
4.3. Transplantation Models
4.3.1. Diabetes Induction and Intraportal Pancreatic Islet Transplantation (PTx)
4.3.2. Thyroidectomy and Intraportal Transplantation of Thyroid Follicles (TTx)
4.3.3. Ovariectomy and Intraportal Transplantation of Ovarian Fragments (OTx)
4.3.4. Control Groups
4.3.5. Gefitinib Treatment
4.3.6. Application of the Nucleoside Analog 5-Bromo-2′-deoxyuridine (BrdU)
4.3.7. Animal Sacrifices and Tissue Processing
4.3.8. Immunohistochemistry
4.3.9. Morphologic Investigations
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, A.I.; Khan, S.A.; Toledano, M.B.; Waked, I.; Taylor-Robinson, S.D. Hepatocellular carcinoma: Epidemiology, risk factors and pathogenesis. World J. Gastroenterol. 2008, 14, 4300–4308. [Google Scholar] [CrossRef] [PubMed]
- Evert, M.; Dombrowski, F. Hepatocellular carcinoma in the non-cirrhotic liver. Der Pathol. 2008, 29, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Sood, G.K. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1901. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Wells, A. EGF receptor. Int. J. Biochem. Cell Biol. 1999, 31, 637–643. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [PubMed]
- Höpfner, M.; Schuppan, D.; Scherübl, H. Growth factor receptors and related signalling pathways as targets for novel treatment strategies of hepatocellular cancer. World J. Gastroenterol. 2008, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Carr, B.I.; Nalesnik, M.A. Concomitant and isolated expression of TGF-α and EGF-R in human hepatoma cells supports the hypothesis of autocrine, paracrine, and endocrine growth of human hepatoma. J. Surg. Oncol. 1995, 58, 240–245. [Google Scholar] [PubMed]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Pomatico, G.; de Placido, S.; Bianco, A.R.; Tortora, G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin. Cancer Res. 2000, 6, 2053–2063. [Google Scholar] [PubMed]
- Lopes, G.; Ho, C.; Liau, K.; Chung, A.; Cheow, P.; Chang, A.; Consortium, S.H.C. Adjuvant gefitinib in hepatocellular carcinoma (HCC): An ongoing pilot study by the Singapore Hepatocellular Carcinoma Consortium. In Proceedings of the 2009 Gastrointestinal Cancers Symposium, San Francisco, CA, USA, 15–17 January 2009.
- Weber, E.; Bannasch, P. Dose and time dependence of the cellular phenotype in rat hepatic preneoplasia and neoplasia induced by continuous oral exposure to N-Nitrosomorpholine. Carcinogenesis 1994, 15, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, F.; Lehringer-Polzin, M.; Pfeifer, U. Hyperproliferative liver acini after intraportal islet transplantation in streptozotocin-induced diabetic rats. Lab. Investig. 1994, 71, 688–699. [Google Scholar] [PubMed]
- Dombrowski, F.; Flaschka, C.; Klotz, L.; von Netzer, B.; Schulz, C.; Lehnert, H.; Evert, M. Hepatocellular neoplasms after intrahepatic transplantation of ovarian fragments into ovariectomized rats. Hepatology 2006, 43, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, F.; Klotz, L.; Hacker, H.J.; Li, Y.; Klingmüller, D.; Brix, K.; Herzog, V.; Bannasch, P. Hyperproliferative hepatocellular alterations after intraportal transplantation of thyroid follicles. Am. J. Pathol. 2000, 156, 99–113. [Google Scholar] [CrossRef]
- Dombrowski, F.; Mathieu, C.; Evert, M. Hepatocellular neoplasms induced by low-number pancreatic islet transplants in autoimmune diabetic BB/PFD rats. Cancer Res. 2006, 66, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, A.; Carr, B.I.; Azzarone, A.; Polimeno, L.; Wang, Z.; van Thiel, D.H.; Subbotin, V.; Prelich, J.G.; Starzl, T.E. Hepatocyte proliferation and gene expression induced by triiodothyronine in vivo and in vitro. Hepatology 1994, 20, 1237–1241. [Google Scholar] [CrossRef]
- Hufnagl, K.; Parzefall, W.; Marian, B.; Käfer, M.; Bukowska, K.; Schulte-Hermann, R.; Grasl-Kraupp, B. Role of transforming growth factor alpha and prostaglandins in preferential growth of preneoplastic rat hepatocytes. Carcinogenesis 2001, 22, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of ERB-B receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Sahin, F.; Kannangai, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef] [PubMed]
- Takami, T.; Kaposi-Novak, P.; Uchida, K.; Gomez-Quiroz, L.E.; Conner, E.A.; Factor, V.M.; Thorgeirsson, S.S. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. 2007, 67, 9844–9851. [Google Scholar] [CrossRef] [PubMed]
- Breuhahn, K.; Longerich, T.; Schirmacher, P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006, 25, 3787–3800. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Burgart, L.J.; Sahai, V.; Kakar, S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am. J. Clin. Pathol. 2008, 129, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Matsumoto, K.; Nagahara, T.; Murawaki, Y. Gefitinib and the modulation of the signaling pathways downstream of epidermal growth factor receptor in human liver cancer cells. J. Gastroenterol. 2006, 41, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Höpfner, M.; Sutter, A.P.; Huether, A.; Schuppan, D.; Zeitz, M.; Scherübl, H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J. Hepatol. 2004, 41, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, E.; Housset, C.; Cacheux, W.; Wendum, D.; Desbois-Mouthon, C.; Rey, C.; Clergue, F.; Poupon, R.; Barbu, V.; Rosmorduc, O. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005, 41, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Solorzano, C.C.; Harbison, M.T.; Ozawa, S.; Tsan, R.; Fan, D.; Abbruzzese, J.; Traxler, P.; Buchdunger, E.; Radinsky, R.; et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000, 60, 2926–2935. [Google Scholar] [PubMed]
- Evert, M.; Sun, J.; Pichler, S.; Slavova, N.; Schneider-Stock, R.; Dombrowski, F. Insulin receptor, insulin receptor substrate-1, Raf-1, and Mek-1 during hormonal hepatocarcinogenesis by intrahepatic pancreatic islet transplantation in diabetic rats. Cancer Res. 2004, 64, 8093–8100. [Google Scholar] [CrossRef] [PubMed]
- Evert, M.; Calvisi, D.F.; Evert, K.; de Murtas, V.; Gasparetti, G.; Mattu, S.; Destefanis, G.; Ladu, S.; Zimmermann, A.; Delogu, S.; et al. AKT/mTOR Activation Induces a Module of Metabolic Changes Contributing to Growth in Insulin-induced Hepatocarcinogenesis. Hepatology 2012, 55, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Ribback, S.; Calvisi, D.F.; Cigliano, A.; Sailer, V.; Peters, M.; Rausch, J.; Heidecke, C.D.; Birth, M.; Dombrowski, F. Molecular and metabolic changes in human liver clear cell foci resemble the alterations occurring in rat hepatocarcinogenesis. J. Hepatol. 2013, 58, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.S.H.; Kobayashi, S.; Costa, D.B. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin. Lung Cancer 2009, 10, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Merlino, G.; Fausto, N. Development of liver tumors in transforming growth factor alpha transgenic mice. Cancer Res. 1992, 52, 5162–5170. [Google Scholar] [PubMed]
- Steinway, S.N.; Dang, H.; You, H.; Rountree, C.B.; Ding, W. The EGFR/ErbB3 Pathway acts as a compensatory survival mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0128159. [Google Scholar] [CrossRef] [PubMed]
- Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Wendum, D.; Clapéron, A.; Chrétien, Y.; Rey, C.; Scatton, O.; Soubrane, O.; Conti, F.; et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013, 73, 3974–3986. [Google Scholar] [CrossRef] [PubMed]
- Bodzin, A.S.; Wei, Z.; Hurtt, R.; Gu, T.; Doria, C. Gefitinib resistance in HCC mahlavu cells: Upregulation of CD133 expression, activation of IGF-1R signaling pathway, and enhancement of IGF-1R nuclear translocation. J. Cell. Physiol. 2012, 227, 2947–2952. [Google Scholar] [CrossRef] [PubMed]
- Desbois-Mouthon, C.; Baron, A.; Blivet-Van Eggelpoël, M.J.; Fartoux, L.; Venot, C.; Bladt, F.; Housset, C.; Rosmorduc, O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: Rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 5445–5456. [Google Scholar] [PubMed]
- Scharf, J.G.; Braulke, T. The role of the IGF axis in hepatocarcinogenesis. Horm. Metab. Res. 2003, 35, 685–693. [Google Scholar] [PubMed]
- Huether, A.; Höpfner, M.; Sutter, A.P.; Baradari, V.; Schuppan, D.; Scherübl, H. Signaling pathways involved in the inhibition of epidermal growth factor receptor by erlotinib in hepatocellular cancer. World J. Gastroenterol. 2006, 12, 5160–5167. [Google Scholar] [PubMed]
- Bannasch, P.; Zerban, H. Predictive value of hepatic preneoplastic lesions as early indicators ofcarcinogenic response. In Mechanisms of Carcinogenesis in Risk Identification; Vainio, H., Magee, P.N., McGregor, D.B., McMichael, A.J., Eds.; International Agency for Research on Cancer: Lyon, France, 1992; pp. 389–427. [Google Scholar]
- Dombrowski, F.; Bannasch, P.; Pfeifer, U. Hepatocellular neoplasms induced by low-number pancreatic islet transplants in streptozotocin diabetic rats. Am. J. Pathol. 1997, 150, 1071–1087. [Google Scholar] [PubMed]
- Weibel, E.R. Stereological principles for morphometry in electron microscopic cytology. Int. Rev. Cytol. 1969, 26, 235–302. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Duration of NNM Administration | Gefitinib Administration | N | BrdU-LI FAH, % | BrdU-LI Extrafocal Liver Tissue, % |
|---|---|---|---|---|
| 3 months | 7 | 68.26 ± 6.82 § | 18.07 ± 2.72 | |
| 3 months | 2 weeks HD | 7 | 60.80 ± 2.80 § | 18.33 ± 2.88 |
| 3 months | 3 months LD | 7 | 55.49 ± 3.00 § | 12.53 ± 1.29 |
| Duration of NNM Administration | Gefitinib Administration | N | BrdU-LI FAH % | BrdU-LI HCA % | BrdU-LI HCC % |
|---|---|---|---|---|---|
| 6 months | 6 | 85.50 ± 1.90 | 89.73 ± 1.13 | 97.14 ± 0.32 | |
| 6 months | 2 weeks HD | 7 | 73.89 ± 3.17 | 81.03 ± 2.70 | 95.49 ± 0.82 |
| 6 months | 3 months LD | 7 | 70.36 ± 1.75 # | 76.03 ± 1.75 | 92.67 ± 1.58 |
| Duration of NNM Administration | Gefitinib Administration | N | Volume Fraction FAH % | Volume Fraction HCA % | Number of HCC per Animal |
|---|---|---|---|---|---|
| 3 months | 14 | 7.25 ± 0.90 | - | - | |
| 3 months | 2 weeks HD | 14 | 4.92 ± 0.87 # | - | - |
| 3 months | 3 months LD | 15 | 1.58 ± 0.28 # | - | - |
| 6 months | 12 | 85.86 ± 0.67 | 10.36 ± 0.89 | 7.93 ± 0.92 | |
| 6 months | 2 weeks HD | 15 | 84.82 ± 0.83 | 10.51 ± 0.56 | 6.53 ± 1.61 |
| 6 months | 3 months LD | 15 | 88.01 ± 0.57 # | 5.43 ± 0.57 # | 4.87 ± 0.77 # |
| Treatment Procedure | Time after Tx | Gefitinib Administration | N | BrdU-LI FAH % | BrdU-LI Extrafocal Liver Tissue, % |
|---|---|---|---|---|---|
| PTx | 3 weeks | 4 | 10.7 ± 0.99 § | 3.48 ± 0.77 | |
| 3 weeks | 2 weeks HD | 4 | 5.74 ± 0.49 § | 1.25 ± 0.26 | |
| 6 months | 5 | 6.06 ± 0.44 § | 1.17 ± 0.08 | ||
| 6 months | 2 weeks HD | 6 | 2.27 ± 0.13 §,# | 0.60 ± 0.03 # | |
| 6 months | 3 months LD | 6 | 3.55 ± 0.22 § | 0.74 ± 0.04 | |
| 12 months | 5 | 2.43 ± 1.39 | 0.35 ± 0.27 | ||
| 12 months | 2 weeks HD | 9 | 1.26 ± 0.44 § | 0.09 ± 0.05 | |
| 12 months | 3 months LD | 7 | 1.55 ± 0.51 | 0.49 ± 0.20 | |
| 24 months | 4 | 10.50 ± 6.94 | 6.69 ± 2.80 | ||
| 24 months | 9 months LD | 3 | 19.20 ± 5.82 | 5.33 ± 3.78 | |
| OTx | 3 months | 7 | 29.79 ± 2.68 § | 2.66 ± 0.39 | |
| 3 months | 2 weeks HD | 6 | 12.67 ± 2.64 §,# | 2.67 ± 1.59 | |
| 12 months | 6 | 16.52 ± 2.47 § | 5.68 ± 0.10 | ||
| 12 months | 2 weeks HD | 7 | 8.73 ± 0.88 §,# | 5.12 ± 0.94 | |
| 12 months | 3 months LD | 4 | 13.55 ± 2.50 | 6.29 ± 0.49 | |
| 24 months | 6 | 6.50 ± 1.56 | 2.26 ± 0.58 | ||
| 24 months | 9 months LD | 5 | 11.10 ± 1.77 | 6.35 ± 5.22 | |
| TTx | 3 months | 7 | 12.25 ± 1.33 § | 2.84 ± 0.53 | |
| 3 months | 2 weeks HD | 8 | 7.69 ± 1.07 # | 6.61 ± 1.63 # | |
| 12 months | 8 | 11.04 ± 1.45 § | 1.07 ± 0.18 | ||
| 12 months | 2 weeks HD | 6 | 11.36 ± 1.55 § | 0.69 ± 0.28 | |
| 12 months | 3 months LD | 6 | 10.62 ± 1.91 § | 2.31 ± 1.08 | |
| 24 months | 7 | 9.33 ± 2.67 § | 1.58 ± 0.90 | ||
| 24 months | 9 months LD | 9 | 6.34 ± 1.23 § | 1.13 ± 0.33 |
| Treatment Procedure | Time after Tx | Gefitinib Administration | Rats with HCA | Rats with HCC | Rats with Transplant Hyperplasia or Tumors |
|---|---|---|---|---|---|
| PTx | 12 months | 16 (34) | 0 (34) | 0 | |
| 12 months | 2 weeks HD | 8 (17) | 0 (17) | 0 | |
| 12 months | 3 months LD | 11 (17) | 0 (17) | 0 | |
| 24 months | 5 (19) | 1 (19) | 0 | ||
| 24 months | 9 months LD | 3 (10) | 1 (10) | 0 | |
| OTx | 12 months | 0 (15) | 0 (15) | 3 (15) | |
| 12 months | 2 weeks HD | 1 (16) | 0 (16) | 6 (16) | |
| 12 months | 3 months LD | 1 (16) | 0 (16) | 7 (16) | |
| 24 months | 9 (20) | 1 (20) | 10 (20) | ||
| 24 months | 9 months LD | 9 (21) | 1 (21) | 12 (21) | |
| TTx | 12 months | 1 (15) | 0 (15) | 0 (15) | |
| 12 months | 2 weeks HD | 2 (14) | 0 (14) | 1 (14) | |
| 12 months | 3 months LD | 2 (13) | 0 (13) | 1 (13) | |
| 24 months | 3 (15) | 0 (15) | 2 (15) | ||
| 24 months | 9 months LD | 8 (15) | 0 (15) | 2 (15) |
| Treatment Procedure | Sacrifice Time Point | Gefitinib Administration | N | Sacrifice Time Point | Gefitinib Administration | N | |
|---|---|---|---|---|---|---|---|
| Experimental groups | Control groups | ||||||
| PTx (male) | 3 weeks | - | 8 | male | 3 weeks | - | 10 |
| 3 weeks | 2 weeks HD | 12 | 3 weeks | 2 weeks HD | 10 | ||
| 6 months | - | 10 | 3 months | - | 15 | ||
| 6 months | 2 weeks HD | 14 | 3 months | 2 weeks HD | 15 | ||
| 6 months | 3 months LD | 12 | 3 months | 3 months LD | 15 | ||
| 12 months | - | 34 | 6 months | - | 26 | ||
| 12 months | 2 weeks HD | 17 | 6 months | 2 weeks HD | 25 | ||
| 12 months | 3 months LD | 17 | 6 months | 3 months LD | 25 | ||
| 24 months | - | 19 | 12 months | - | 11 | ||
| 24 months | 9 months LD | 10 | 12 months | 2 weeks HD | 10 | ||
| 12 months | 3 months LD | 10 | |||||
| TTx (male) | 3 months | - | 15 | 24 months | - | 19 | |
| 3 months | 2 weeks HD | 15 | 24 months | 9 months LD | 24 | ||
| 12 months | - | 15 | |||||
| 12 months | 2 weeks HD | 14 | female | 3 months | - | 10 | |
| 12 months | 3 months LD | 13 | 3 months | 2 weeks HD | 10 | ||
| 24 months | - | 15 | 12 months | - | 14 | ||
| 24 months | 9 months LD | 15 | 12 months | 2 weeks HD | 9 | ||
| 12 months | 3 months LD | 9 | |||||
| OTx (female) | 3 months | - | 15 | 24 months | - | 21 | |
| 3 months | 2 weeks HD | 15 | 24 months | 9 months LD | 17 | ||
| 12 months | - | 15 | |||||
| 12 months | 2 weeks HD | 16 | |||||
| 12 months | 3 months LD | 16 | |||||
| 24 months | - | 20 | |||||
| 24 months | 9 months LD | 21 | |||||
| NNM | 3 months | - | 15 | ||||
| 3 months | 2 weeks HD | 15 | |||||
| 3 months | 3 months LD | 15 | |||||
| 6 months | - | 15 | |||||
| 6 months | 2 weeks HD | 15 | |||||
| 6 months | 3 months LD | 15 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribback, S.; Sailer, V.; Böhning, E.; Günther, J.; Merz, J.; Steinmüller, F.; Utpatel, K.; Cigliano, A.; Peters, K.; Pilo, M.G.; et al. The Epidermal Growth Factor Receptor (EGFR) Inhibitor Gefitinib Reduces but Does Not Prevent Tumorigenesis in Chemical and Hormonal Induced Hepatocarcinogenesis Rat Models. Int. J. Mol. Sci. 2016, 17, 1618. https://doi.org/10.3390/ijms17101618
Ribback S, Sailer V, Böhning E, Günther J, Merz J, Steinmüller F, Utpatel K, Cigliano A, Peters K, Pilo MG, et al. The Epidermal Growth Factor Receptor (EGFR) Inhibitor Gefitinib Reduces but Does Not Prevent Tumorigenesis in Chemical and Hormonal Induced Hepatocarcinogenesis Rat Models. International Journal of Molecular Sciences. 2016; 17(10):1618. https://doi.org/10.3390/ijms17101618
Chicago/Turabian StyleRibback, Silvia, Verena Sailer, Enrico Böhning, Julia Günther, Jaqueline Merz, Frauke Steinmüller, Kirsten Utpatel, Antonio Cigliano, Kristin Peters, Maria G. Pilo, and et al. 2016. "The Epidermal Growth Factor Receptor (EGFR) Inhibitor Gefitinib Reduces but Does Not Prevent Tumorigenesis in Chemical and Hormonal Induced Hepatocarcinogenesis Rat Models" International Journal of Molecular Sciences 17, no. 10: 1618. https://doi.org/10.3390/ijms17101618
APA StyleRibback, S., Sailer, V., Böhning, E., Günther, J., Merz, J., Steinmüller, F., Utpatel, K., Cigliano, A., Peters, K., Pilo, M. G., Evert, M., Calvisi, D. F., & Dombrowski, F. (2016). The Epidermal Growth Factor Receptor (EGFR) Inhibitor Gefitinib Reduces but Does Not Prevent Tumorigenesis in Chemical and Hormonal Induced Hepatocarcinogenesis Rat Models. International Journal of Molecular Sciences, 17(10), 1618. https://doi.org/10.3390/ijms17101618