
Comparative Study of Ultrasonication-Induced and Naturally Self-Assembled Silk Fibroin-Wool Keratin Hydrogel Biomaterials

 ,
, 
Abstract
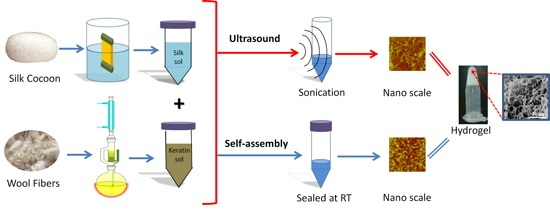
:
1. Introduction
2. Results and Discussion
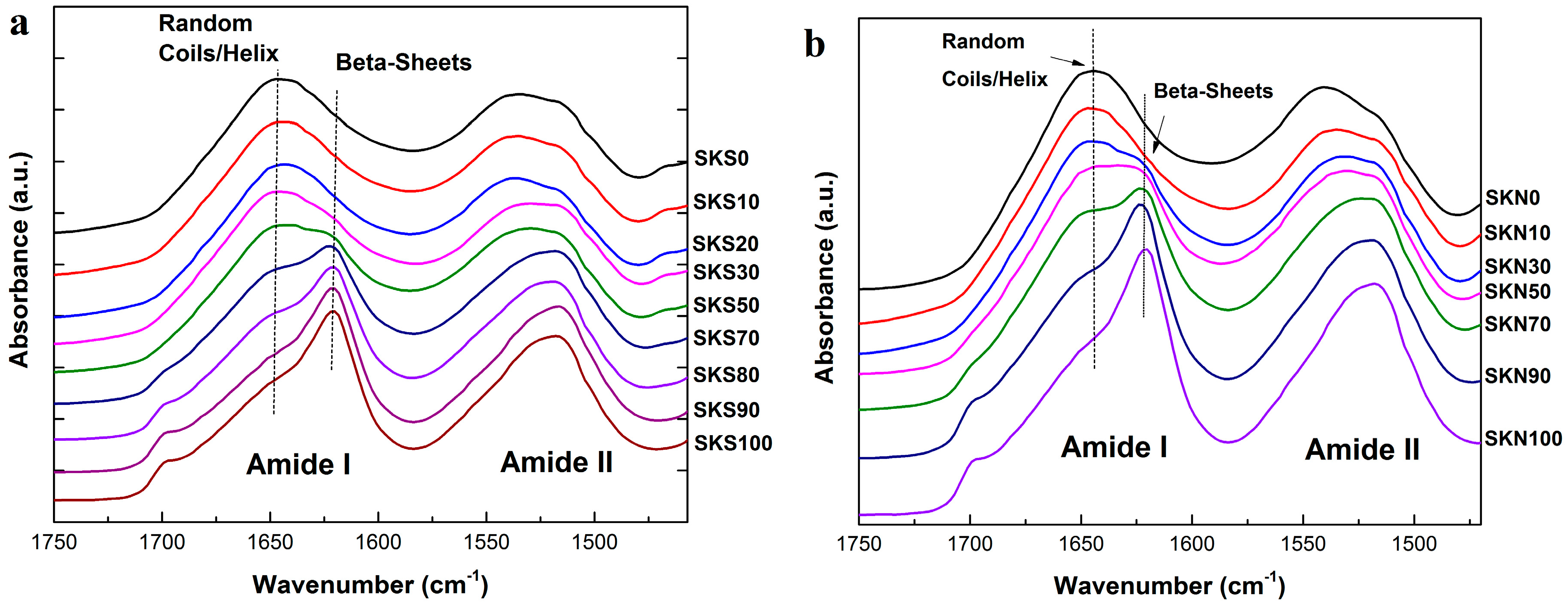
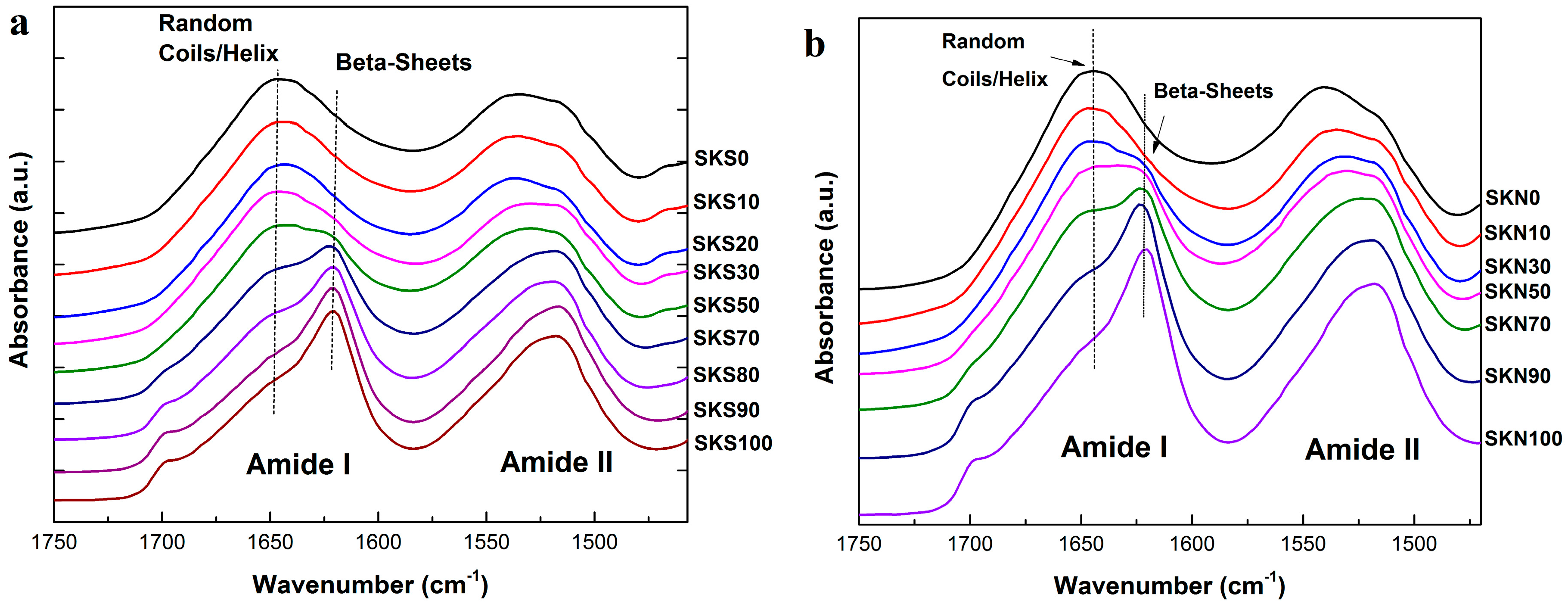
2.1. Structural Analysis
2.2. Morphology Analysis
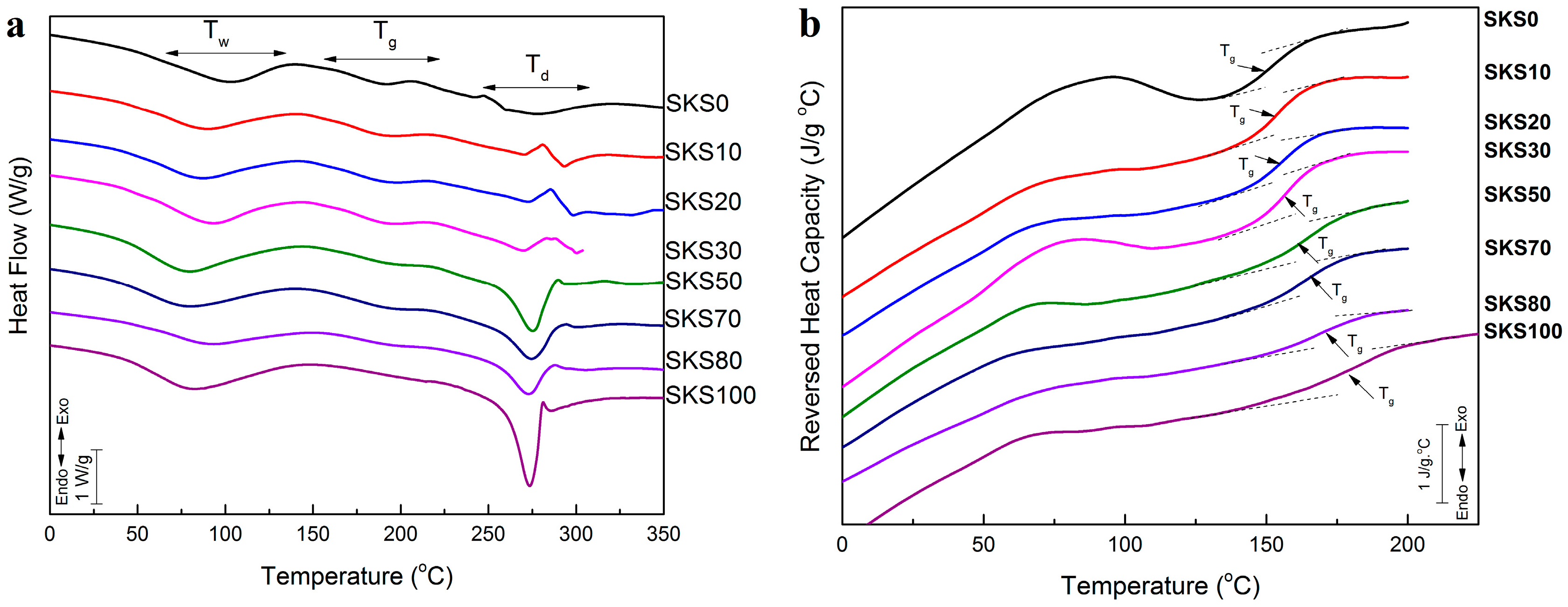
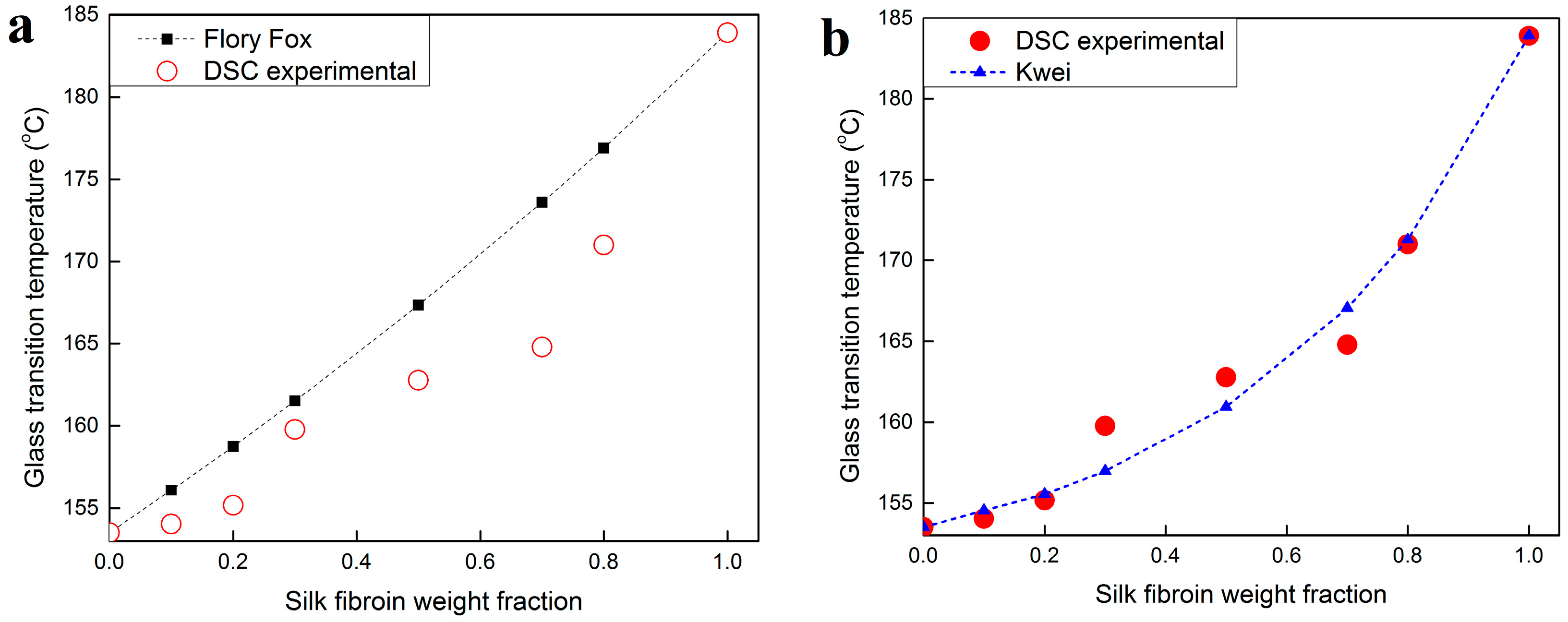
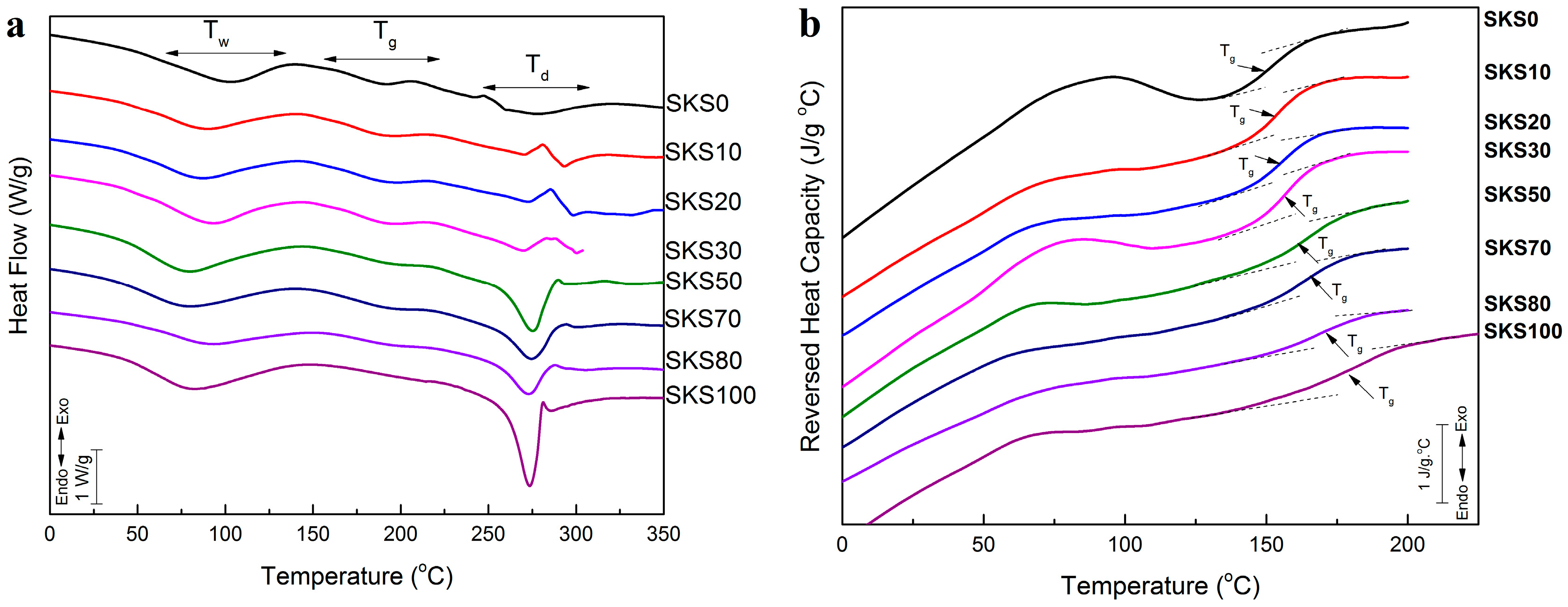
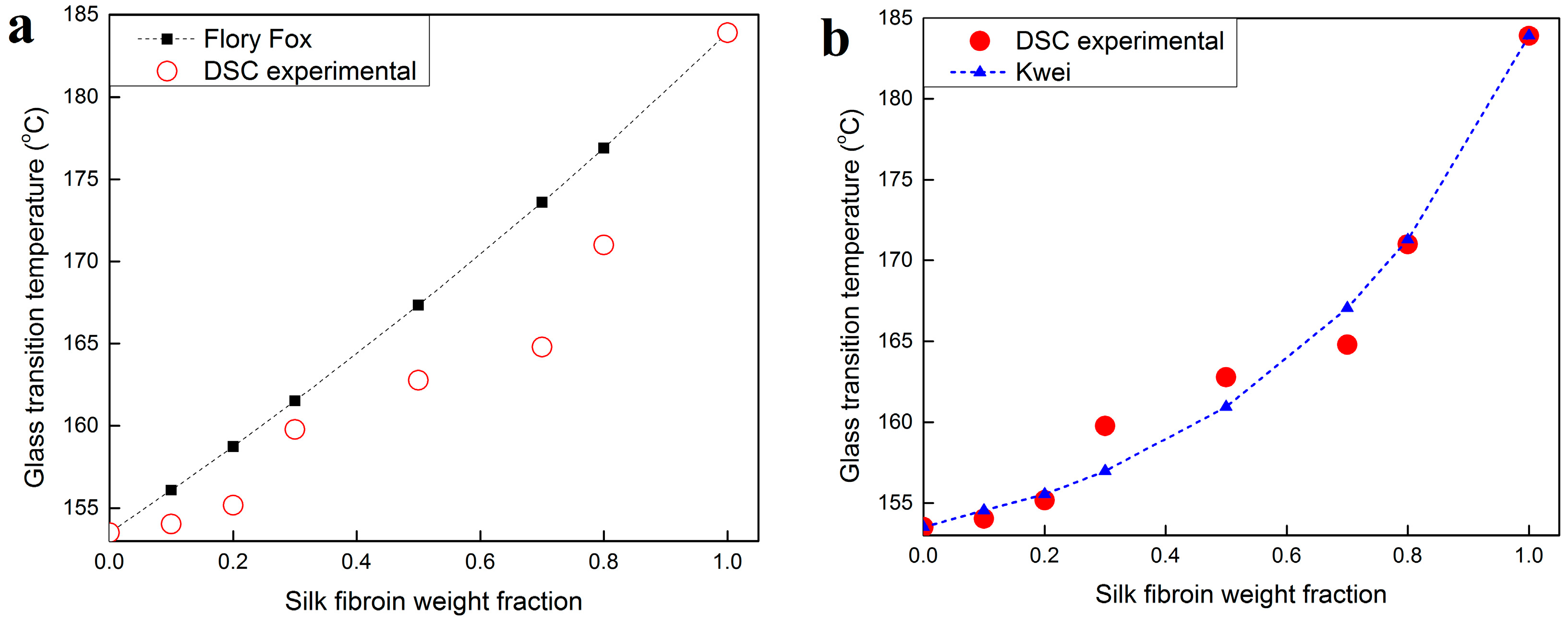
2.3. Thermal Analysis
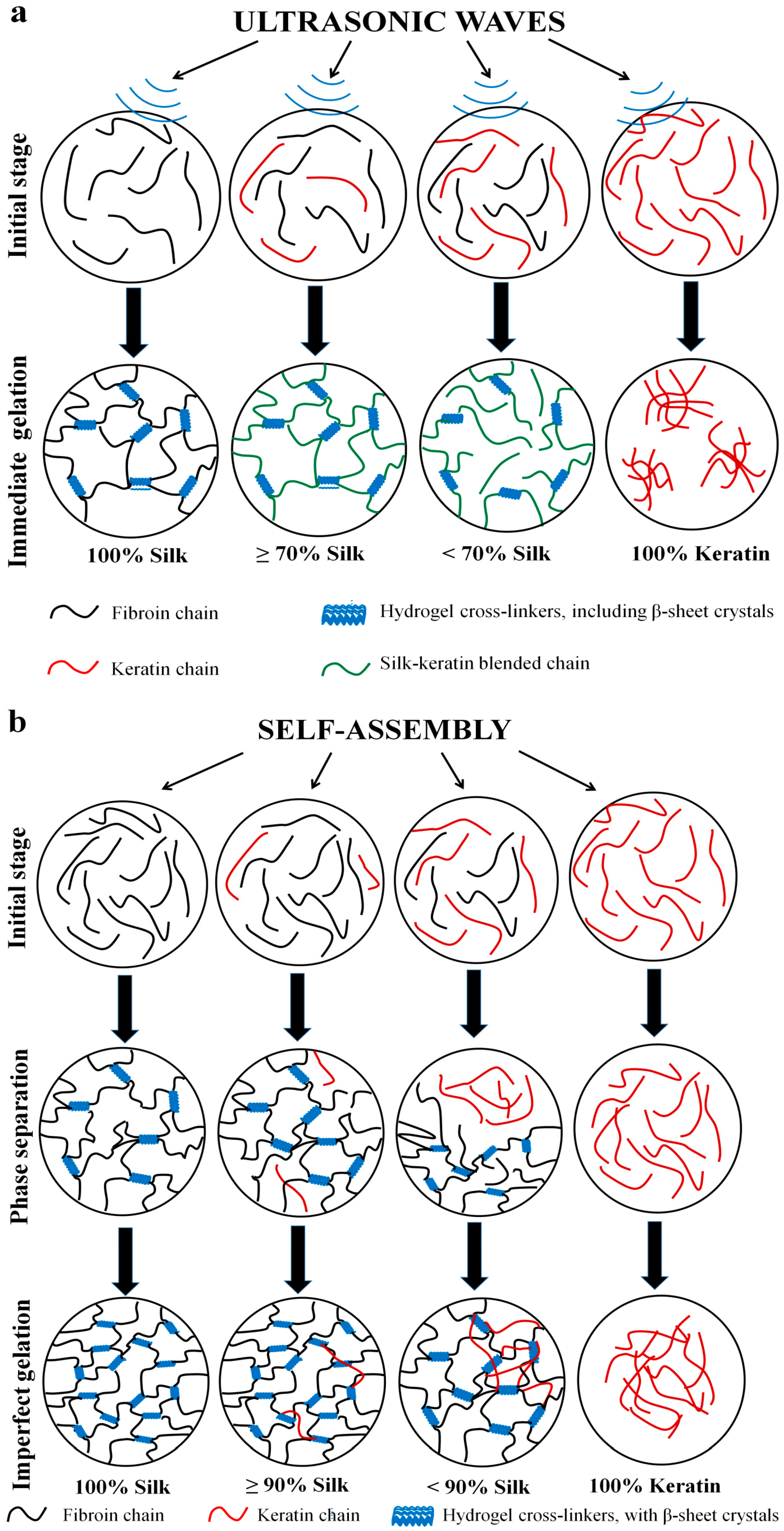
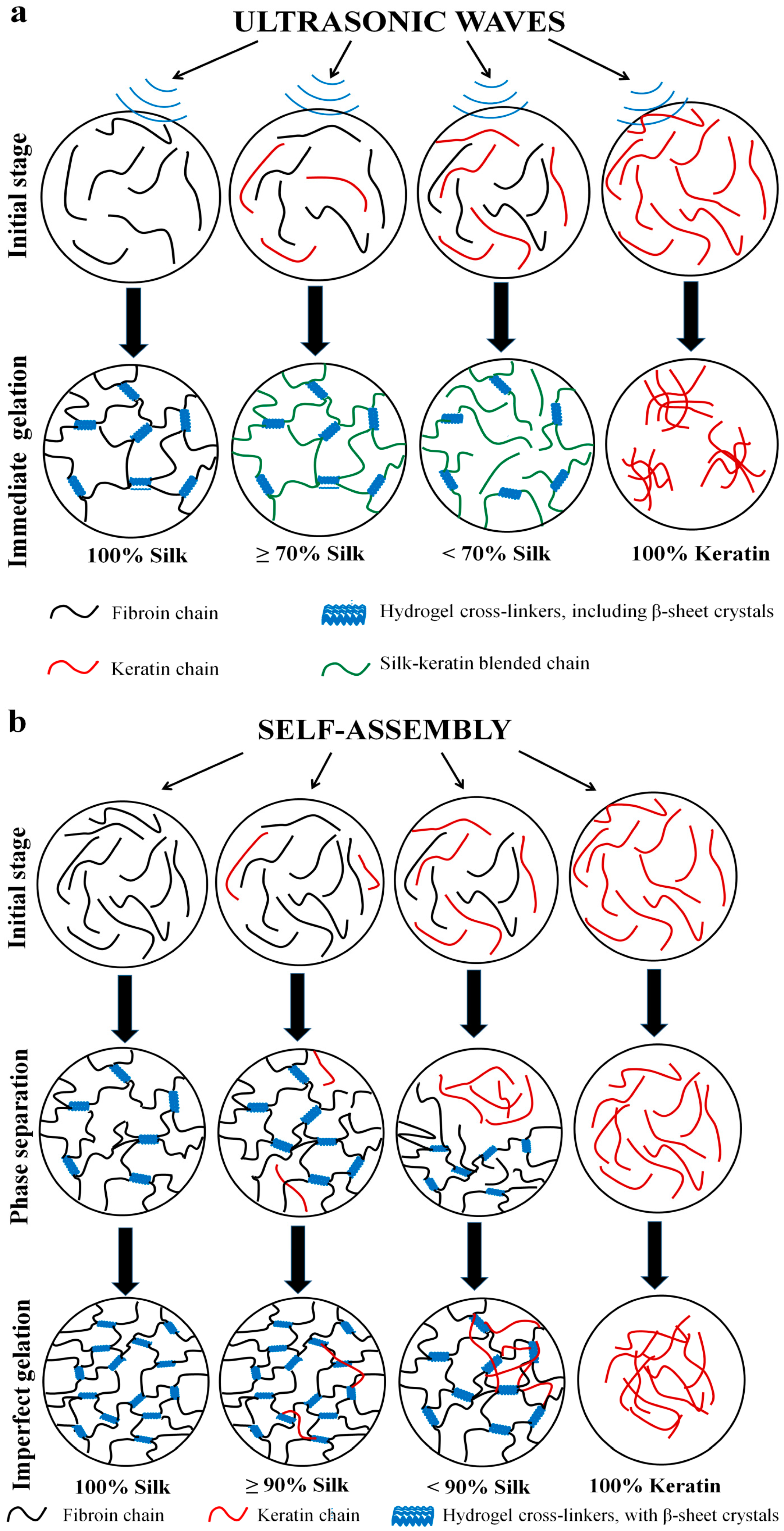
2.4. Gelation Mechanism and Condition
2.4.1. Nanoscale Level
2.4.2. Microscale Level
3. Experimental Section
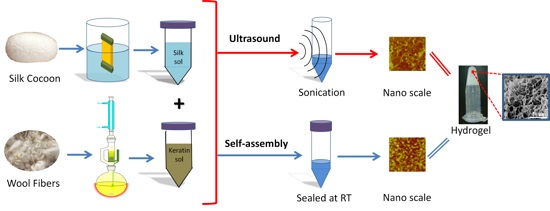
3.1. Material Preparation
3.1.1. Silk Solution
3.1.2. Wool Solution
3.1.3. Ultrasonication-Induced Hydrogel
3.1.4. Naturally Assembled Hydrogel
3.2. Fourier Transform Infrared Spectroscopy (FTIR)
3.3. Differential Scanning Calorimetry (DSC)
3.4. Scanning Electron Microscopy (SEM)
3.5. Atomic Force Microscope (AFM)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Advert. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Barbucci, R. Hydrogels: Biological Properties and Applications; Springer Science & Business Media: New York, NY, USA, 2009. [Google Scholar]
- Hu, X.; Lu, Q.; Sun, L.; Cebe, P.; Wang, X.; Zhang, X.; Kaplan, D.L. Biomaterials from ultrasonication-induced silk fibroin-hyaluronic acid hydrogels. Biomacromolecules 2010, 11, 3178–3188. [Google Scholar] [CrossRef] [PubMed]
- Suslick, K.S. The chemical effects of ultrasound. Sci. Am. 1989, 260, 62–68. [Google Scholar] [CrossRef]
- Min, B.M.; Lee, G.; Kim, S.H.; Nam, Y.S.; Lee, T.S.; Park, W.H. Electrospinning of silk fibroin nanofibers and its effect on the adhesion and spreading of normal human keratinocytes and fibroblasts in vitro. Biomaterials 2004, 25, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Torculas, M.; Medina, J.; Xue, W.; Hu, X. Protein-based bioelectronics. ACS Biomater. Sci. Eng. 2016, 2, 1211–1223. [Google Scholar] [CrossRef]
- Talukdar, S.; Mandal, M.; Hutmacher, D.W.; Russell, P.J.; Soekmadji, C.; Kundu, S.C. Engineered silk fibroin protein 3D matrices for in vitro tumor mode. Biomaterials 2011, 32, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, O.J.; Lee, M.C.; Moon, B.M.; Ju, H.W.; Lee, J.M.; Kim, J.H.; Kim, D.W.; Park, C.H. Fabrication of 3D porous silk scaffolds by particulate (salt/sucrose) leaching for bone tissue reconstruction. Int. J. Biol. Macromol. 2015, 78, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Kaplan, D.; Cebe, P. Determining β-sheet crystallinity in fibrous proteins by thermal analysis and infrared spectroscopy. Macromolecules 2006, 39, 6161–6170. [Google Scholar] [CrossRef]
- Sah, M.K.; Pramanik, K. Regenerated silk fibroin from B. mori silk cocoon for tissue engineering applications. Int. J. Environ. Sci. Dev. 2010, 1, 404–408. [Google Scholar] [CrossRef]
- Kasoju, N.; Naresh, K.; Utpal, B. Corrigendum: silk fibroin based biomimetic artificial extracellular matrix for hepatic tissue engineering applications. Biomed. Mater. 2013, 8, 490–501. [Google Scholar] [CrossRef]
- Phetnin, R.; Rattanachan, S.T. Bio-hybrid composite scaffold from silk fibroin/chitosan/mesoporous bioactive glass microspheres for tissue engineering applications. Adv. Mater. Res. 2015, 1131, 79–83. [Google Scholar] [CrossRef]
- Anonymous. Encyclopedic Dictionary of Polymers; Gooch, J.W., Ed.; Springer: New York, NY, USA, 2011; p. 410. [Google Scholar]
- Vasconcelos, A.; Freddi, G.; Cavaco-Paulo, A. Biodegradable materials based on silk fibroin and keratin. Biomacromolecules 2008, 9, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Çalamak, S.; Erdogdu, C.; Ozalp, M.; Ulubayram, K. Silk fibroin based antibacterial bionanotextiles as wound dressing materials. Mater. Sci. Eng. C 2014, 43, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Fabry, B.; Boccaccini, A.R. Fibrous protein-based hydrogels for cell encapsulation. Biomaterials 2014, 35, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Shariati, S.R.P.; Seyedsina, M.; Esmaiel, J. Hydrogels for cell encapsulation and bioprinting. Stem Cell Biol. Regen. Med. 2015, 89–108. [Google Scholar]
- McKittrick, J.; Chen, P.Y.; Bodde, S.G.; Yang, W.; Novitskaya, E.E.; Meyers, M.A. The structure, functions, and mechanical properties of keratin. Jom 2012, 64, 449–468. [Google Scholar] [CrossRef]
- Rattanavises, W.; Oonkhanond, B. The gelation study of silk fibroin for biomedical application. Adv. Mater. Res. 2012, 506, 385–388. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Sow, W.T.; Devi, D.; Ng, K.W.; Mandal, B.B.; Cho, N.J. Correction: Silk fibroin-keratin based 3D scaffolds as a dermal substitute for skin tissue engineering. Integr. Biol. 2015, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: New York, NY, USA, 1953; pp. 595–602. [Google Scholar]
- Peppas, N.A.; Huang, Y.; Torres-Lugo, M.; Ward, J.H.; Zhang, J. Physicochemical foundations and structural design of hydrogels in medicine and biology. Annu. Rev. Biomed. Eng. 2000, 2, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Rouse, J.G.; Dyke, M.E.V. A review of keratin-based biomaterials for biomedical applications. Materials 2010, 3, 999–1014. [Google Scholar] [CrossRef]
- Hu, X.; Lu, Q.; Kaplan, D.L.; Cebe, P. Microphase separation controlled β-sheet crystallization kinetics in fibrous proteins. Macromolecules 2009, 42, 2079–2087. [Google Scholar] [CrossRef]
- Cebe, P.; Partlow, B.P.; Kaplan, D.L.; Andreas, W.; Evgeny, Z.; Christoph, S. Using flash DSC for determining the liquid state heat capacity of silk fibroin. Thermochim. Acta 2015, 615, 8–14. [Google Scholar] [CrossRef]
- Pyda, M.; Hu, X.; Cebe, P. Heat capacity of silk fibroin based on the vibrational motion of poly(amino acid)s in the presence and absence of water. Macromolecules 2008, 41, 4786–4798. [Google Scholar] [CrossRef]
- Jin, H.J.; Kaplan, D.L. Mechanism of silk processing in insects and spiders. Nature 2003, 424, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Guo, Y.Q.; Gu, L.Z.; Ding, E.Y. Crystalline-amorphous phase transition of poly(ethylene glycol)/cellulose blend. Macromolecules 1995, 28, 6551–6555. [Google Scholar] [CrossRef]
- Elashmawi, I.S.; Hakeem, N.A.; Abdelrazek, E.M. Spectroscopic and thermal studies of PS/PVAc blends. Phys. B Condens. Matter 2008, 403, 3547–3552. [Google Scholar] [CrossRef]
- Weng, L.; Vijayaraghavan, R.; MacFarlane, D.R.; Elliott, G.D. Application of the Kwei equation to model the Tg behavior of binary blends of sugars and salts. Crybiology 2014, 68, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Kwei, T.K.; Pearce, E.M.; Pennacchia, J.R.; Charton, M. Correlation between the glass transition temperatures of polymer mixtures and intermolecular force parameters. Macromolecules 1987, 20, 1174–1176. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Silk Fraction in Sample (%) | β-Sheet Crystallinity (B) | α-Helix & Random Coils (A + R) | Turns (T) | Side Chains (S) |
|---|---|---|---|---|---|
| SKS0 | 0 | 25.5 | 44.3 | 26.5 | 3.7 |
| SKS10 | 10 | 28.3 | 43.1 | 24.4 | 4.2 |
| SKS20 | 20 | 30.5 | 43.8 | 24.1 | 1.6 |
| SKS30 | 30 | 34.2 | 41.9 | 22.3 | 1.7 |
| SKS50 | 50 | 35.3 | 41.2 | 21.9 | 1.6 |
| SKS70 | 70 | 43.2 | 31.5 | 21.6 | 4.3 |
| SKS80 | 80 | 48.8 | 27.1 | 20 | 4.1 |
| SKS90 | 90 | 50.2 | 22.8 | 19.2 | 7.8 |
| SKS100 | 100 | 54.5 | 22.9 | 19.3 | 3.2 |
| Sample | Silk Fraction in Sample (%) | β-Sheet Crystallinity (B) | α-Helix & Random Coils (A + R) | Turns (T) | Side Chains (S) |
|---|---|---|---|---|---|
| SKN0 | 0 | 22.6 | 45.7 | 25.6 | 6.1 |
| SKN10 | 10 | 24.8 | 44.5 | 29.1 | 1.5 |
| SKN30 | 30 | 32.4 | 41.3 | 24.3 | 2.1 |
| SKN50 | 50 | 41.5 | 36.3 | 21.8 | 0.3 |
| SKN70 | 70 | 45.7 | 32.4 | 21.4 | 0.5 |
| SKN90 | 90 | 51.1 | 28.6 | 19.5 | 0.7 |
| SKN100 | 100 | 52.4 | 28.5 | 18.6 | 0.4 |
| Sample | Glass Transition Temperature (°C) | ∆Cp (J·g−1·°C−1) | Degradation Peak (°C) |
|---|---|---|---|
| SKS0 | 153.5 1 | 0.159 | 274.3 |
| SKS10 | 154.0 3 | 0.189 | 293.3 |
| SKS20 | 155.1 7 | 0.200 | 296.3 |
| SKS30 | 159.7 8 | 0.202 | 299.6 |
| SKS50 | 162.7 8 | 0.215 | 276.1 |
| SKS70 | 164.8 0 | 0.222 | 275.1 |
| SKS80 | 171.0 0 | 0.243 | 273.9 |
| SKS100 | 183.9 0 | 0.250 | 273.1 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, T.; Xue, Y.; Vuong, T.; Erbe, M.; Bennet, C.; Palazzo, B.; Popielski, L.; Rodriguez, N.; Hu, X. Comparative Study of Ultrasonication-Induced and Naturally Self-Assembled Silk Fibroin-Wool Keratin Hydrogel Biomaterials. Int. J. Mol. Sci. 2016, 17, 1497. https://doi.org/10.3390/ijms17091497
Vu T, Xue Y, Vuong T, Erbe M, Bennet C, Palazzo B, Popielski L, Rodriguez N, Hu X. Comparative Study of Ultrasonication-Induced and Naturally Self-Assembled Silk Fibroin-Wool Keratin Hydrogel Biomaterials. International Journal of Molecular Sciences. 2016; 17(9):1497. https://doi.org/10.3390/ijms17091497
Chicago/Turabian StyleVu, Trang, Ye Xue, Trinh Vuong, Matthew Erbe, Christopher Bennet, Ben Palazzo, Lucas Popielski, Nelson Rodriguez, and Xiao Hu. 2016. "Comparative Study of Ultrasonication-Induced and Naturally Self-Assembled Silk Fibroin-Wool Keratin Hydrogel Biomaterials" International Journal of Molecular Sciences 17, no. 9: 1497. https://doi.org/10.3390/ijms17091497
APA StyleVu, T., Xue, Y., Vuong, T., Erbe, M., Bennet, C., Palazzo, B., Popielski, L., Rodriguez, N., & Hu, X. (2016). Comparative Study of Ultrasonication-Induced and Naturally Self-Assembled Silk Fibroin-Wool Keratin Hydrogel Biomaterials. International Journal of Molecular Sciences, 17(9), 1497. https://doi.org/10.3390/ijms17091497