
Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease




Abstract
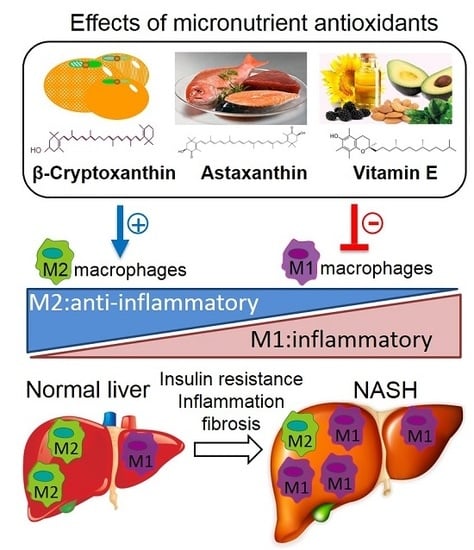
:
1. Introduction
2. Pathogenesis of Nonalcoholic Fatty Liver Disease (NAFLD)
2.1. Obesity
2.2. Diabetes
2.2.1. Diabetes to NAFLD
2.2.2. NAFLD to Diabetes
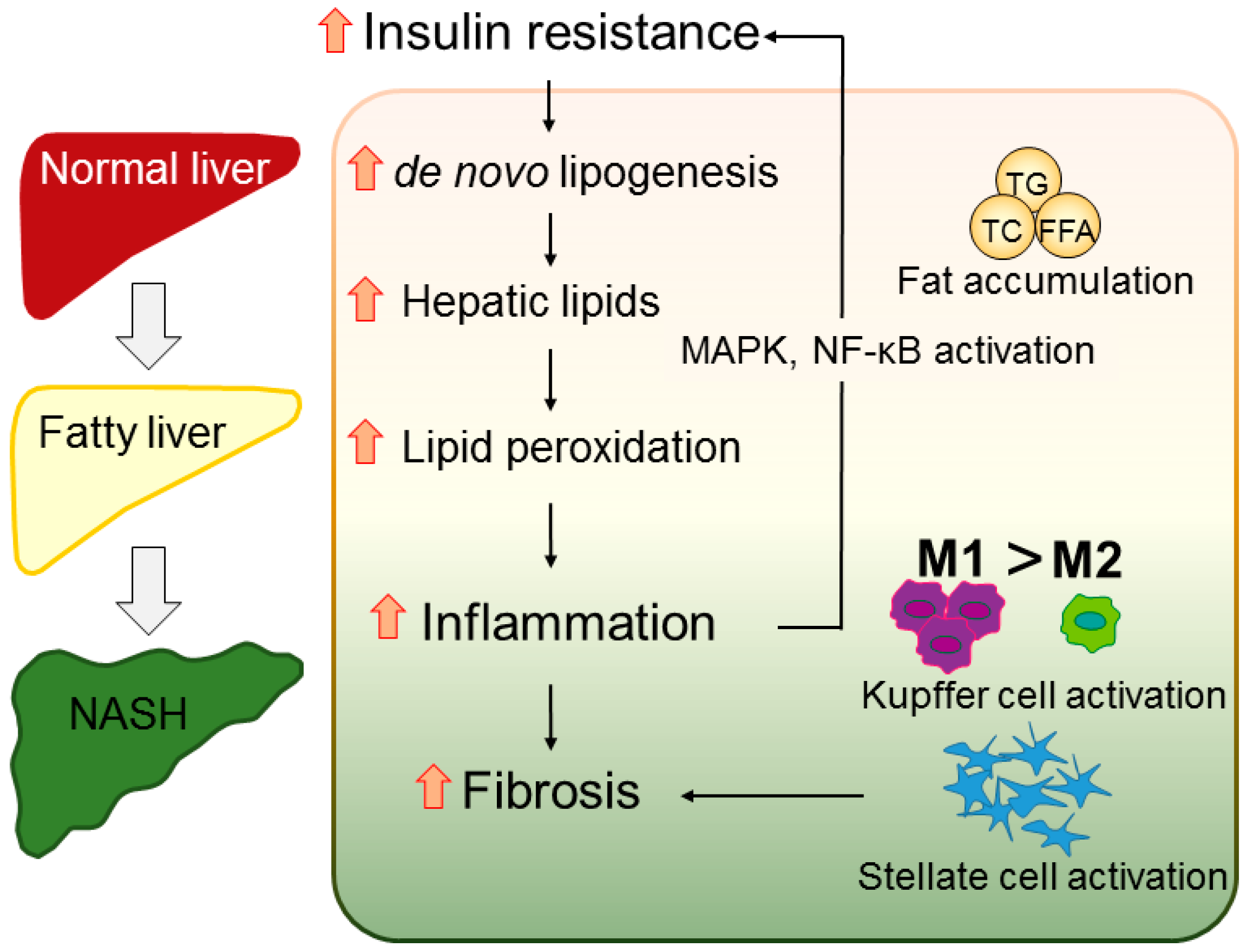
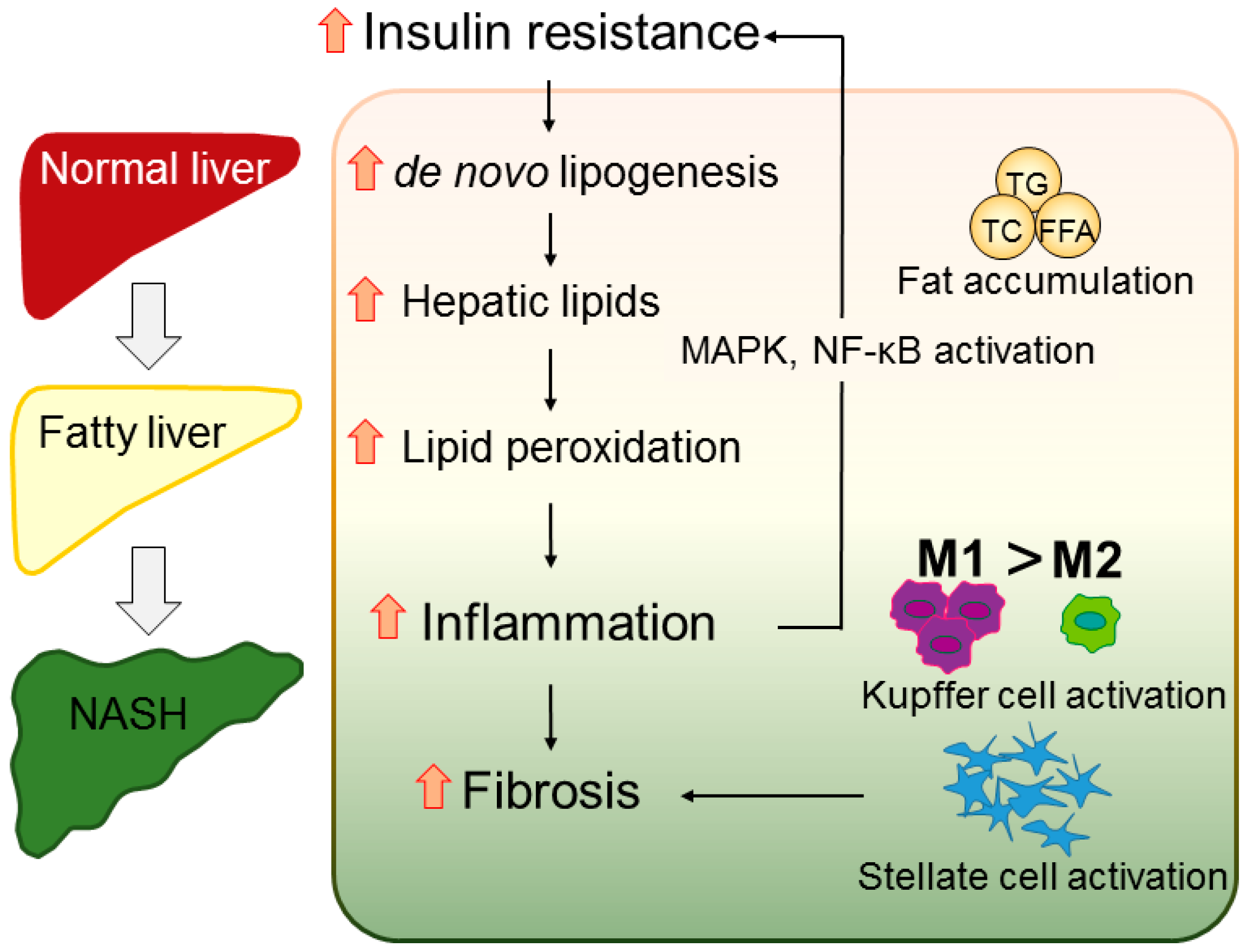
2.3. Inflammation
2.4. Fibrosis
3. Micronutrients and NAFLD/Nonalcoholic Steatohepatitis (NASH)
3.1. Vitamin D
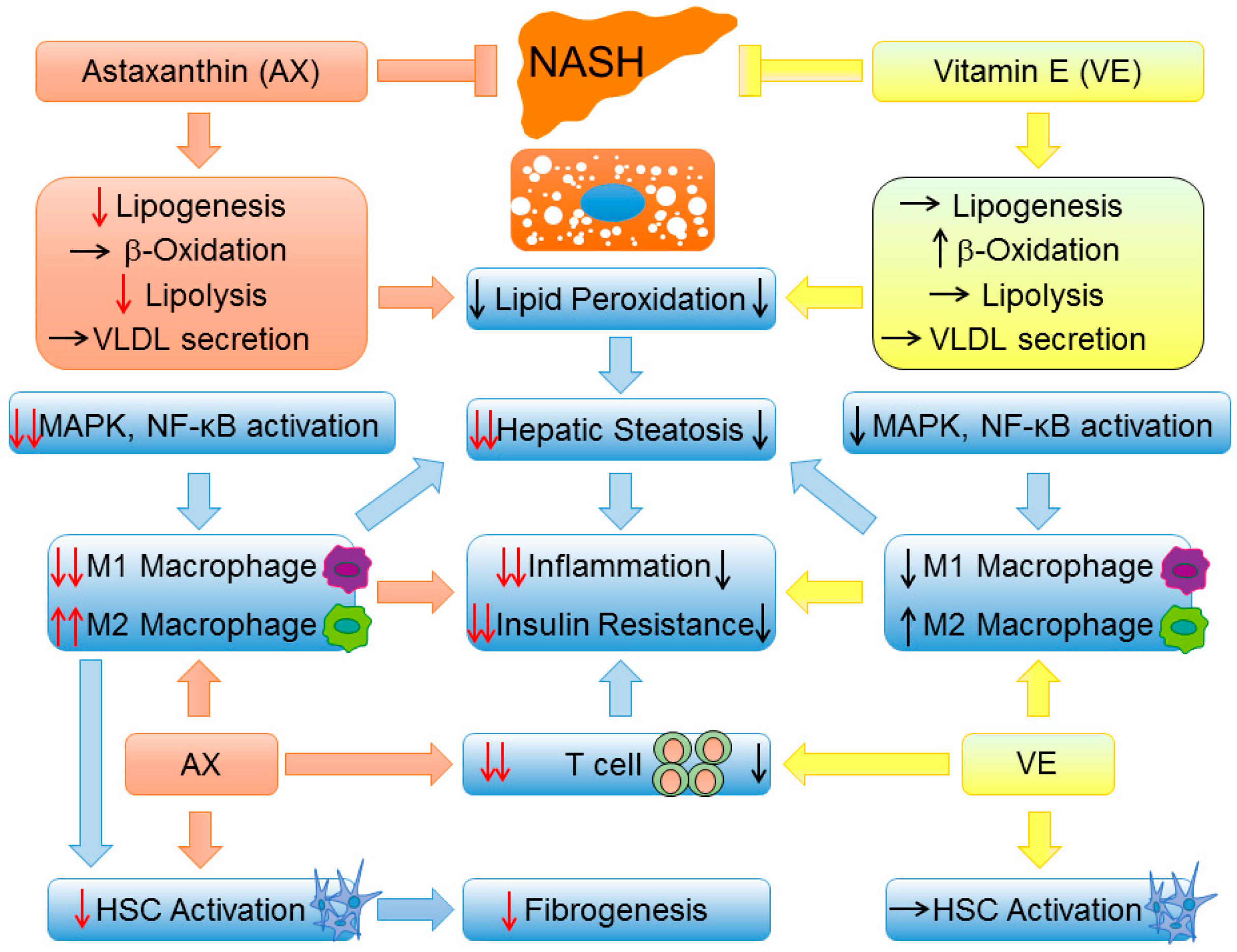
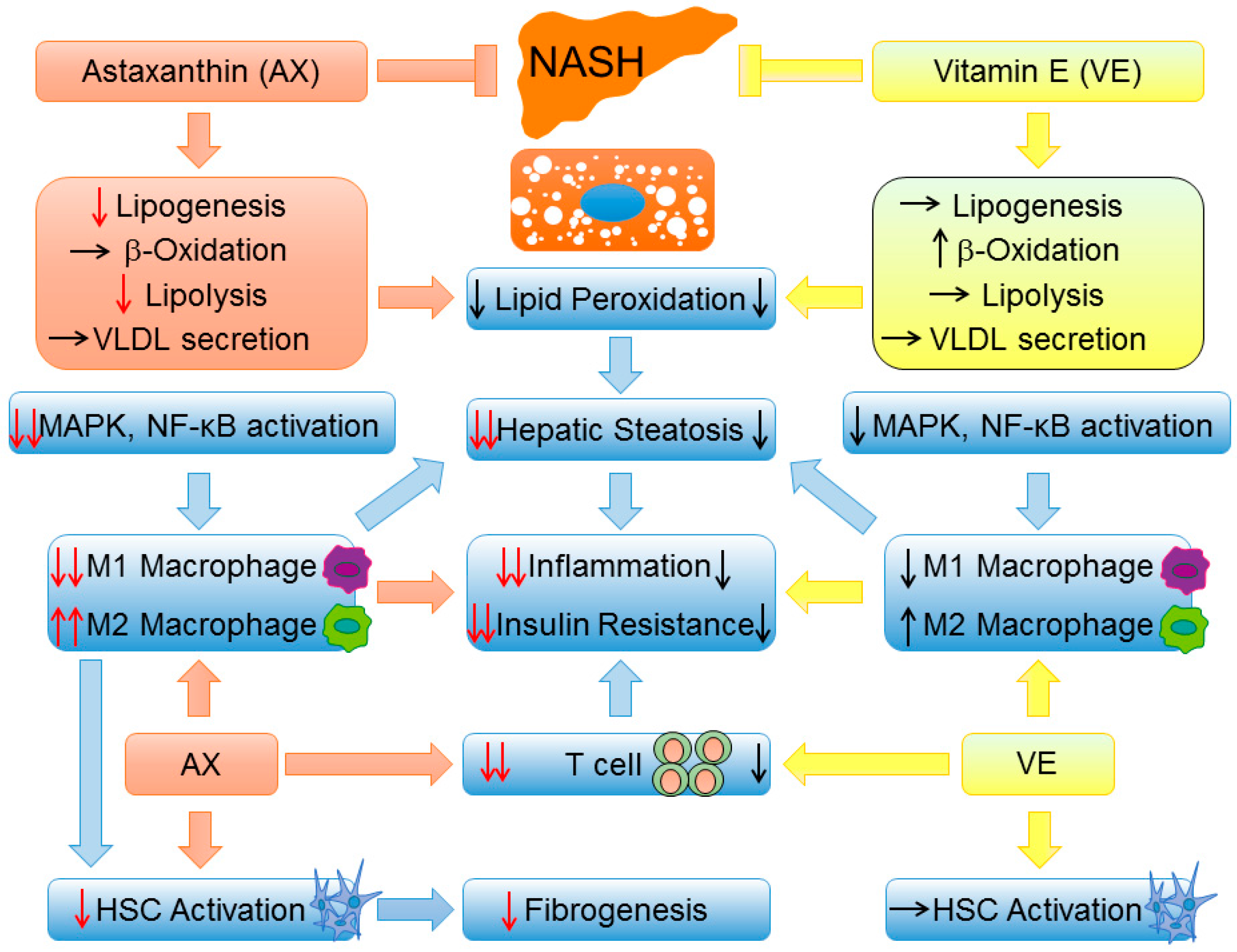
3.2. Vitamin E
3.3. Astaxanthin
3.4. Other Micronutrients
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of non-alcoholic fatty liver disease–meta-analytic assessment of prevalence, incidence and outcomes. Hepatology 2015, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Z.; Dai, Y.N.; Wang, Y.M.; Zhou, Q.Y.; Yu, C.H.; Li, Y.M. Prevalence of nonalcoholic fatty liver disease and economy. Dig. Dis. Sci. 2015, 60, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J. Prevalence and risk factors for non-alcoholic fatty liver disease in Asian people who are not obese. J. Gastroenterol. Hepatol. 2012, 27, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Aga technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Wong, V.W.; Chitturi, S. Nafld in Asia—As common and important as in the west. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Jia, J.D.; Li, Y.M.; Wang, B.Y.; Lu, L.G.; Shi, J.P.; Chan, L.Y. Guidelines for the diagnosis and management of nonalcoholic fatty liver disease. J. Dig. Dis. 2011, 12, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Nayak, S.; Swain, M.; Rout, N.; Mallik, R.; Agrawal, O.; Meher, C.; Rao, M. Prevalence of nonalcoholic fatty liver disease in coastal eastern india: A preliminary ultrasonographic survey. Trop. Gastroenterol. 2003, 25, 76–79. [Google Scholar]
- Amarapurkar, D.N.; Hashimoto, E.; Lesmana, L.A.; Sollano, J.D.; Chen, P.J.; Goh, K.L. How common is non-alcoholic fatty liver disease in the Asia-Pacific region and are there local differences? J. Gastroenterol. Hepatol. 2007, 22, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Onyekwere, C.A.; Ogbera, A.O.; Balogun, B.O. Non-alcoholic fatty liver disease and the metabolic syndrome in an urban hospital serving an African community. Ann. Hepatol. 2011, 10, 119–124. [Google Scholar] [PubMed]
- Marcuccilli, M.; Chonchol, M. Nafld and chronic kidney disease. Int. J. Mol. Sci. 2016, 17, 562. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the united states. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.; Argo, C. The natural history of non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kitade, H.; Ni, Y.; Ota, T. Roles of chemokines and chemokine receptors in obesity-associated insulin resistance and nonalcoholic fatty liver disease. Biomolecules 2015, 5, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Saibara, T.; Chitturi, S.; Kim, B.I.; Sung, J.J.; Chutaputti, A. What are the risk factors and settings for non-alcoholic fatty liver disease in Asia-Pacific? J. Gastroenterol. Hepatol. 2007, 22, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Aykut, U.E.; Akyuz, U.; Yesil, A.; Eren, F.; Gerin, F.; Ergelen, R.; Celikel, C.A.; Yilmaz, Y. A comparison of fibrometer nafld score, nafld fibrosis score, and transient elastography as noninvasive diagnostic tools for hepatic fibrosis in patients with biopsy-proven non-alcoholic fatty liver disease. Scand. J. Gastroenterol. 2014, 49, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. GI epidemiology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef]
- Clark, J.M.; Diehl, A.M. Nonalcoholic fatty liver disease: An underrecognized cause of cryptogenic cirrhosis. JAMA 2003, 289, 3000–3004. [Google Scholar] [CrossRef] [PubMed]
- Saviano, M.C.; Brunetti, F.; Rubino, A.; Franzese, A.; Vajro, P.; Argenziano, A.; Puzziello, A.; Iannucci, M.P. Liver involvement in obese children (ultrasonography and liver enzyme levels at diagnosis and during follow-up in an italian population). Dig. Dis. Sci. 1997, 42, 1428–1432. [Google Scholar] [CrossRef]
- Tominaga, K.; Kurata, J.H.; Chen, Y.K.; Fujimoto, E.; Miyagawa, S.; Abe, I.; Kusano, Y. Prevalence of fatty liver in Japanese children and relationship to obesity. Dig. Dis. Sci. 1995, 40, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis—New insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Arrese, M.; Zamora-Valdes, D.; Uribe, M. Current concepts in the pathogenesis of nonalcoholic fatty liver disease. Liver Int. 2007, 27, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease a feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. NAFLD, obesity, and bariatric surgery. Gastroenterology 2006, 130, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein ACRP30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Federico, A.; Abenavoli, L.; Loguercio, C.; Persico, M. Non alcoholic fatty liver: Epidemiology and natural history. Rev. Recent Clin. Trials 2014, 9, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Pagotto, U.; Manini, R.; Vanni, E.; Gastaldelli, A.; de Iasio, R.; Gentilcore, E.; Natale, S.; Cassader, M.; Rizzetto, M.; et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J. Clin. Endocrinol. Metab. 2005, 90, 3498–3504. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Soardo, G.; Esposito, W.; Fallo, F.; Basan, L.; Donnini, D.; Federspil, G.; Sechi, L.A.; Vettor, R. Plasma adiponectin is decreased in nonalcoholic fatty liver disease. Eur. J. Endocrinol. 2005, 152, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Savvidou, S.; Hytiroglou, P.; Orfanou-Koumerkeridou, H.; Panderis, A.; Frantzoulis, P.; Goulis, J. Low serum adiponectin levels are predictive of advanced hepatic fibrosis in patients with nafld. J. Clin. Gastroenterol. 2009, 43, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.S. Obesity, visceral fat, and nafld: Querying the role of adipokines in the progression of nonalcoholic fatty liver disease. ISRN Gastroenterol. 2011, 2011, 592404. [Google Scholar] [CrossRef] [PubMed]
- Anania, F.A. Adiponectin and alcoholic fatty liver: Is it, after all, about what you eat? Hepatology 2005, 42, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Chiba, S.; Tatsukawa, H.; Yasuda, T.; Noguchi, H.; Seike, M.; Yoshimatsu, H. Adiponectin protects LPS-induced liver injury through modulation of TNF-α in KK-AY obese mice. Hepatology 2004, 40, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Matsumoto, H.; Tamura, S.; Fukushima, J.; Kiso, S.; Fukui, K.; Igura, T.; Maeda, N.; Kihara, S.; Funahashi, T. Hypoadiponectinemia accelerates hepatic tumor formation in a nonalcoholic steatohepatitis mouse model. J. Hepatol. 2007, 47, 556–564. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Considine, R.V.; Leone, T.C.; Kelly, D.P.; Crabb, D.W. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 2005, 42, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Marra, F. Leptin and liver fibrosis: A matter of fat. Gastroenterology 2002, 122, 1529–1532. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, K.; Honda, H.; Yoshikawa, M.; Hirose, M.; Kitamura, T.; Takei, Y.; Sato, N. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology 2001, 34, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kakuma, T.; Lee, Y.; Higa, M.; Wang, Z.-W.; Pan, W.; Shimomura, I.; Unger, R.H. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc. Natl. Acad. Sci. USA 2000, 97, 8536–8541. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Lipotoxic diseases. Annu. Rev. Med. 2002, 53, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Liver injury in the setting of steatosis: Crosstalk between adipokine and cytokine. Hepatology 2004, 40, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y. Tumour necrosis factor α signalling through activation of kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Rich, A.S.; Rhoades, B.; Shapiro, J.S.; Obici, S.; Rossetti, L.; Lazar, M.A. Abnormal glucose homeostasis due to chronic hyperresistinemia. Diabetes 2004, 53, 1937–1941. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Nguyen, M.A.; Miles, P.D.; Imamura, T.; Usui, I.; Olefsky, J.M. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J. Clin. Investig. 2004, 114, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Obici, S.; Scherer, P.E.; Rossetti, L. Adipose-derived resistin and gut-derived resistin-like molecule-β selectively impair insulin action on glucose production. J. Clin. Investig. 2003, 111, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Papatheodoridis, G.V.; Archimandritis, A.J. Adipokines in nonalcoholic steatohepatitis: From pathogenesis to implications in diagnosis and therapy. Mediators Inflamm. 2009, 2009, 831670. [Google Scholar] [CrossRef] [PubMed]
- El-Assal, O.; Hong, F.; Kim, W.-H.; Radaeva, S.; Gao, B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell. Mol. Immunol. 2004, 1, 205–211. [Google Scholar] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Roberts, E.A. Nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 2000, 30, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Manton, N.D.; Lipsett, J.; Moore, D.J.; Davidson, G.P.; Bourne, A.J.; Couper, R.T. Non-alcoholic steatohepatitis in children and adolescents. Med. J. Aust. 2000, 173, 476–479. [Google Scholar] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of nafld to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Jimba, S.; Nakagami, T.; Takahashi, M.; Wakamatsu, T.; Hirota, Y.; Iwamoto, Y.; Wasada, T. Prevalence of non-alcoholic fatty liver disease and its association with impaired glucose metabolism in Japanese adults. Diabet. Med. 2005, 22, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Ryysy, L.; Häkkinen, A.-M.; Goto, T.; Vehkavaara, S.; Westerbacka, J.; Halavaara, J.; Yki-Järvinen, H. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes 2000, 49, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Lopez, C.; Lomonaco, R.; Orsak, B.; Finch, J.; Chang, Z.; Kochunov, V.G.; Hardies, J.; Cusi, K. Prevalence of prediabetes and diabetes and metabolic profile of patients with nonalcoholic fatty liver disease (NAFLD). Diabetes Care 2012, 35, 873–878. [Google Scholar] [CrossRef] [PubMed]
- de Marco, R.; Locatelli, F.; Zoppini, G.; Verlato, G.; Bonora, E.; Muggeo, M. Cause-specific mortality in type 2 diabetes. The verona diabetes study. Diabetes Care 1999, 22, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Harmsen, S.; St Sauver, J.L.; Charatcharoenwitthaya, P.; Enders, F.B.; Therneau, T.; Angulo, P. Nonalcoholic fatty liver disease increases risk of death among patients with diabetes: A community-based cohort study. Am. J. Gastroenterol. 2010, 105, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Ryu, S.; Sung, E.; Woo, H.Y.; Oh, E.; Cha, K.; Jung, E.; Kim, W.S. Nonalcoholic fatty liver disease predicts chronic kidney disease in nonhypertensive and nondiabetic Korean men. Metabolism 2008, 57, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Rodella, S.; Zoppini, G.; Lippi, G.; Day, C.; Muggeo, M. Non-alcoholic fatty liver disease is independently associated with an increased prevalence of chronic kidney disease and proliferative/laser-treated retinopathy in type 2 diabetic patients. Diabetologia 2008, 51, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Mansouri, A.; Fromenty, B.V. Mitochondrial dysfunction in steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G193–G199. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and FAS-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-α expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Diehl, A.M. Cytokines in alcoholic and nonalcoholic steatohepatitis. N. Engl. J. Med. 2000, 343, 1467–1476. [Google Scholar] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bengmark, S.; Qu, S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 2010, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Mottaran, E.; Vidali, M.; Reale, E.; Saksena, S.; Occhino, G.; Burt, A.D.; Day, C.P. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut 2005, 54, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. Cd8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Soloski, M.J.; Diehl, A.M. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology 2005, 42, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.J.; Leon, P.; Ryan, J.C. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology 2008, 48, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E. Chemokine receptors: Multifaceted therapeutic targets. Nat. Rev. Immunol. 2002, 2, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Conductier, G.; Blondeau, N.; Guyon, A.; Nahon, J.L.; Rovere, C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 2010, 224, 93–100. [Google Scholar] [CrossRef] [PubMed]
- De Waard, V.; Bot, I.; de Jager, S.C.; Talib, S.; Egashira, K.; de Vries, M.R.; Quax, P.H.; Biessen, E.A.; van Berkel, T.J. Systemic MCP1/CCR2 blockade and leukocyte specific MCP1/CCR2 inhibition affect aortic aneurysm formation differently. Atherosclerosis 2010, 211, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.-I.; Ginsberg, H.N. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.; Benyon, R.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Investig. 1998, 102, 538. [Google Scholar] [CrossRef] [PubMed]
- Takehara, T.; Tatsumi, T.; Suzuki, T.; Rucker, E.B.; Hennighausen, L.; Jinushi, M.; Miyagi, T.; Kanazawa, Y.; Hayashi, N. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 2004, 127, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Otogawa, K.; Kinoshita, K.; Fujii, H.; Sakabe, M.; Shiga, R.; Nakatani, K.; Ikeda, K.; Nakajima, Y.; Ikura, Y.; Ueda, M.; et al. Erythrophagocytosis by liver macrophages (Kupffer cells) promotes oxidative stress, inflammation, and fibrosis in a rabbit model of steatohepatitis: Implications for the pathogenesis of human nonalcoholic steatohepatitis. Am. J. Pathol. 2007, 170, 967–980. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Inokuchi, S.; Taura, K.; Miyai, K.; van Rooijen, N.; Schwabe, R.F.; Brenner, D.A. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009, 50, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory GR1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Candia, R.; Zapata, R.; Munoz, C.; Arancibia, J.P.; Poniachik, J.; Soza, A.; Fuster, F.; Brahm, J.; Sanhueza, E.; et al. Management of nonalcoholic fatty liver disease: An evidence-based clinical practice review. World J. Gastroenterol 2014, 20, 12182–12201. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Review article: Current management of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2008, 28, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The tonic randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wen, T.; Chen, X.Y.; Wu, H. Protective effects of pirfenidone on D-galactosamine and lipopolysaccharide-induced acute hepatotoxicity in rats. Inflamm. Res. 2008, 57, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Kaibori, M.; Yanagida, H.; Yokoigawa, N.; Kwon, A.H.; Okumura, T.; Kamiyama, Y. Pirfenidone prevents endotoxin-induced liver injury after partial hepatectomy in rats. J. Hepatol. 2004, 40, 94–101. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.M.; Rinella, M.E. The role of diet and nutrient composition in nonalcoholic fatty liver disease. J. Acad. Nutr. Diet. 2012, 112, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Lanti, C.; Riso, P.; Valenti, L. Nutritional therapy for nonalcoholic fatty liver disease. J. Nutr. Biochem. 2016, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eliades, M.; Spyrou, E.; Agrawal, N.; Lazo, M.; Brancati, F.L.; Potter, J.J.; Koteish, A.A.; Clark, J.M.; Guallar, E.; Hernaez, R. Meta-analysis: Vitamin D and non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013, 38, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J. Vitamin D insufficiency. N. Engl. J. Med. 2011, 364, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.-J.; Kim, M.K.; Park, S.E.; Park, C.-Y.; Baek, K.H.; Lee, W.-Y.; Kang, M.I.; Park, S.-W.; Kim, S.-W.; Oh, K.W. High serum vitamin D levels reduce the risk for nonalcoholic fatty liver disease in healthy men independent of metabolic syndrome. Endocr. J. 2013, 60, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Maximos, M.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Subbarayan, S.; Correa, M.; Lo, M.; Suman, A.; Cusi, K. Relationship of vitamin D with insulin resistance and disease severity in non-alcoholic steatohepatitis. J. Hepatol. 2015, 62, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Giorgio, V.; Liccardo, D.; Bedogni, G.; Morino, G.; Alisi, A.; Cianfarani, S. Vitamin D levels and liver histological alterations in children with nonalcoholic fatty liver disease. Eur. J. Endocrinol. 2014, 170, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Angelico, F.; del Ben, M.; Baroni, M.G.; Pozzilli, P.; Morini, S.; Cavallo, M.G. Strong association between non alcoholic fatty liver disease (NAFLD) and low 25(OH) vitamin D levels in an adult population with normal serum liver enzymes. BMC Med. 2011, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.M.; Torres, D.M.; Harrison, S.A. Vitamin D and nonalcoholic fatty liver disease (NAFLD): Is it more than just an association? Hepatology 2013, 58, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cordero, P.; Nguyen, V.; Oben, J.A. The role of vitamins in the pathogenesis of non-alcoholic fatty liver disease. Integr. Med. Insights 2016, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; van Natta, M.L.; Kleiner, D.E.; Clark, J.M.; Kowdley, K.V.; Loomba, R.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; Tonascia, J. Vitamin E and changes in serum alanine aminotransferase levels in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2013, 38, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E. Vitamin E treatment of nonalcoholic steatohepatitis in children: A pilot study. J. Pediatr. 2000, 136, 734–738. [Google Scholar] [CrossRef]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.; Ward, J.; Schenker, S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2003, 98, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (select). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—a review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, M.; Okimasu, E.; Inoue, M.; Utsumi, K. Inhibition of oxidative injury of biological membranes by astaxanthin. Physiol. Chem. Phys. Med. NMR 1989, 22, 27–38. [Google Scholar]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kim, S.; Kim, H. Effect of astaxanthin on the hepatotoxicity, lipid peroxidation and antioxidative enzymes in the liver of CCL4-treated rats. Methods Find. Exp. Clin. Pharmacol. 2001, 23, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Koyama, T.; Takahashi, J.; Yazawa, K. Effects of astaxanthin in obese mice fed a high-fat diet. Biosci. Biotechnol. Biochem. 2007, 71, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, M.; Nishida, Y.; Ishibashi, H.; Wada, T.; Fujisaka, S.; Takikawa, A.; Urakaze, M.; Sasaoka, T.; Usui, I.; Tobe, K. Impact of divergent effects of astaxanthin on insulin signaling in L6 cells. Endocrinology 2013, 154, 2600–2612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bae, M.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Astaxanthin prevents and reverses the activation of mouse primary hepatic stellate cells. J. Nutr. Biochem. 2016, 29, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kim, B.; Park, Y.K.; Koo, S.I.; Lee, J.Y. Astaxanthin prevents TGFβ1-induced pro-fibrogenic gene expression by inhibiting SMAD3 activation in hepatic stellate cells. Biochim. Biophys. Acta 2015, 1850, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef] [PubMed]
- Khaire, A.; Rathod, R.; Kale, A.; Joshi, S. Vitamin B12 and ω-3 fatty acids together regulate lipid metabolism in wistar rats. Prostaglandins Leukot. Essent. Fatty Acids 2015, 99, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, U.; Katre, P.; Yajnik, C.S. Influence of maternal vitamin B12 and folate on growth and insulin resistance in the offspring. In Maternal and Child Nutrition: The First 1000 Days; Karger Publishers: Basel, Switzerland, 2013; Volume 74, pp. 145–156. [Google Scholar]
- Haegele, A.D.; Gillette, C.; O’Neill, C.; Wolfe, P.; Heimendinger, J.; Sedlacek, S.; Thompson, H.J. Plasma xanthophyll carotenoids correlate inversely with indices of oxidative DNA damage and lipid peroxidation. Cancer Epidemiol. Biomark. Prev. 2000, 9, 421–425. [Google Scholar]
- Ni, Y.; Nagashimada, M.; Zhan, L.; Nagata, N.; Kobori, M.; Sugiura, M.; Ogawa, K.; Kaneko, S.; Ota, T. Prevention and reversal of lipotoxicity-induced hepatic insulin resistance and steatohepatitis in mice by an antioxidant carotenoid, β-cryptoxanthin. Endocrinology 2015, 156, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Ni, Y.; Takahashi, Y.; Watanabe, N.; Sugiura, M.; Ogawa, K.; Nagashimada, M.; Kaneko, S.; Naito, S.; Ota, T. β-Cryptoxanthin alleviates diet-induced nonalcoholic steatohepatitis by suppressing inflammatory gene expression in mice. PLoS ONE 2014, 9, e98294. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Sanyal, A.J. NAFLD in 2014: Genetics, diagnostics and therapeutic advances in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Wang, H. Silymarin attenuated hepatic steatosis through regulation of lipid metabolism and oxidative stress in a mouse model of nonalcoholic fatty liver disease (NAFLD). Am. J. Transl. Res. 2016, 8, 1073. [Google Scholar] [PubMed]
- Abenavoli, L.; Milic, N.; Peta, V.; Alfieri, F.; De Lorenzo, A.; Bellentani, S. Alimentary regimen in non-alcoholic fatty liver disease: Mediterranean diet. World J. Gastroenterol. 2014, 20, 16831–16840. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L. Non-alcoholic fatty liver disease and beneficial effects of dietary supplements. World J. Hepatol. 2015, 7, 1723. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Adipokines | Function |
|---|---|
| Adiponectin | Anti-inflammatory, improve insulin sensitivity, prevent lipid accumulation, attenuate fibrosis, inhibit tumor necrosis factor (TNF-α) synthesis and/or release [36,37,38,39,40] |
| Leptin | Prevent lipid accumulation, amplify inflammation, induce fibrosis, increase TNF-α concentration [41,42,43,44] |
| TNF-α | Promote inflammation, induce lipid accumulation and insulin resistance, pro-fibrotic effect [45,46,47,48] |
| Resistin | Cause insulin resistance, reduce interleukin 6 (IL-6) secretion, participate in liver fibrogenesis [49,50,51,52,53] |
| IL-6 | Suppress oxidative stress and prevent mitochondrial dysfunction [54,55,56] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.; Ni, Y.; Nagata, N.; Xu, L.; Ota, T. Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 1379. https://doi.org/10.3390/ijms17091379
Chen G, Ni Y, Nagata N, Xu L, Ota T. Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2016; 17(9):1379. https://doi.org/10.3390/ijms17091379
Chicago/Turabian StyleChen, Guanliang, Yinhua Ni, Naoto Nagata, Liang Xu, and Tsuguhito Ota. 2016. "Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 17, no. 9: 1379. https://doi.org/10.3390/ijms17091379
APA StyleChen, G., Ni, Y., Nagata, N., Xu, L., & Ota, T. (2016). Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 17(9), 1379. https://doi.org/10.3390/ijms17091379